Effect of salt in the mobile phase on the critical conditions of poly(ethylene glycol) in liquid chromatography-mass spectrometry coupling
Marion
Girod
a,
Emmanuel
Beaudoin
b and
Laurence
Charles
*a
aUniversités Aix-Marseille I, II et III – CNRS, UMR 6264: Laboratoire Chimie Provence, Spectrométries Appliquées à la Chimie Structurale, F-13397, Marseille, France. E-mail: laurence.charles@univ-provence.fr; Fax: +33 4 91 28 28 97; Tel: +33 4 91 28 86 78
bUniversités Aix-Marseille I, II et III – CNRS, UMR 6264: Laboratoire Chimie Provence, Chimie Radicalaire, Organique et Polymères de Spécialité, F-13397, Marseille, France
First published on 10th September 2009
Abstract
The nature of salts introduced in the chromatographic mobile phase to promote on-line electrospray ionization was shown to be a key parameter in the optimization of LCCC-MS couplings, both using non-polar (Si–C18) and polar (Si–NH2) stationary phases. The critical conditions of poly(ethylene glycol), which reflect a compensation process between exclusion and interaction effects, were strongly modified when changing the size of the cation in the eluent. This phenomenon could be attributed to interactions between the cation and water molecules from the mobile phase, in the case of the non-polar stationary phase, giving rise to a salting out effect due to a lowered solvent quality of the eluent; or between the cation and amino-modified silanols of the polar stationary phase, inducing a decrease of the surface adsorptivity. Accordingly, increasing the size of the cation in the mobile phase caused the polymer molecules to be eluted according to the adsorption mode using non-polar adsorbent and according to the exclusion mode using polar stationary phase.
Introduction
The synthesis of block-, random or grafted-copolymers usually generates a series of distribution owing to the presence of polymer chains with different functionalities, chemical compositions and architectures, yielding very complex samples. As a result, implementation of separation techniques prior to specific detection methods has attracted much interest for the analysis of polymeric mixtures. In this context, combination of liquid chromatography at critical conditions (LCCC) with mass spectrometry (MS) was reported to be a powerful analytical tool.1–7Using a porous stationary phase, critical conditions are defined as the experimental settings at which effects due to size exclusion and interaction mechanisms compensate each other. As a result, the retention of homopolymer molecules becomes independent of their molecular weight.8–14LCCC has often been employed for the analysis of block copolymers 15–23 since, by making one block chromatographically “invisible”, retention time of co-oligomer molecules is mainly dictated by the size of the second block. To allow unambiguous identification of separated species, LCCC has been coupled with mass spectrometry using an off-line approach,1,3–5 where LCCC is performed in a preparative mode and the so-collected fractions further mass analyzed in a second step using matrix-assisted laser desorption/ionization (MALDI), or on-line via an electrospray ionization (ESI) source as the interface.2,6,7 Post-column addition of the cationization agent required to promote electrospray ionization is usually performed, although this analytical configuration may not always ensure a proper mixing of the chromatographic effluent (and thus of the analytes) with the solution containing the salt and also induces dilution effects. Alternatively, the cationization agent can be added directly in the mobile phase of the on-line LCCC-ESI-MS coupling. A careful choice of this salt24–27 allows informative structural data to be reached by further submitting electrosprayed co-oligomer adducts to collision-induced dissociation (CID). For example, we recently optimized critical conditions for poly(ethylene oxide) using a water–methanol eluent containing a lithium salt to characterize a poly(ethylene oxide)/polystyrene block copolymer (PEO-b-PS).6 Using these conditions, co-oligomer molecules were mainly separated according to the PS block size and mass spectra extracted from each chromatographic peak typically consisted of PEO-like distributions. Tandem mass spectrometry (MS/MS) spectra of lithiated co-oligomers from these distributions further allowed us to confirm the PS block size and provided detailed information about the co-oligomer end-groups. However, one main drawback of the developed method was the detectability of large co-oligomers. In particular, the ion signal was observed to rapidly decrease as the PS block size increases.
Concentration and type of the cationizing agent are key parameters for optimal production of ionic adducts in electrospray. Adjusting lithium salt concentration in the mobile phase allowed different PEO-b-PS cationic adducts to be produced (and thus complementary structural information to be reached in MS/MS)7 but did not sufficiently improve ionization of the largest molecules. Higher molecular weight co-oligomer ions are expected to be more stable when associated with bigger cations.28 PEO homopolymers can readily be ionized using alkali cations since any oxygen atoms in the polymeric chain are potential binding sites for these cations27,29,30 while cationization of PS is usually enhanced using a silver salt in the electrosprayed solution.31 However, while testing mobile phases containing different cations on the chromatographic behaviour of PEG homopolymers, it was found that changing the nature of the cation in the mobile phase dramatically affects the critical conditions initially established for PEG using a lithium salt. This unexpected phenomenon was further scrutinized, using both non-polar and polar stationary phases.
Experimental
Chemicals
HPLC grade methanol (MeOH) and water (purity 99.9%) were purchased from SDS (Peypin, France). Lithium chloride, sodium iodide, potassium chloride, rubidium iodide, cesium iodide and silver trifluoroacetate (AgTFA) were from Sigma-Aldrich (St. Louis, MO). The salt was added at a 1.0 mM concentration in the mobile phase. Poly(ethylene glycol) (PEG) standards (PSS, Mainz, Germany), ranging from Mw = 200 to 1000 g mol−1, were dissolved in the eluent mixture at a concentration of 100 µg mL−1.Liquid chromatography at critical conditions
LCCC experiments were performed on an Agilent 1000 series system (Agilent Technology, Santa Clara, CA, USA) equipped with a degasser, a binary pump, an auto-sampler and an oven. Chromatographic columns used in reversed-phase (octadecyl-silica EQUISIL, 150 × 4 mm, pore diameter: 100 Å, particle size: 5 µm) and normal-phase (amino-silica NUCLEOSIL, 250 × 4.6 mm, pore diameter: 100 Å, particle size: 5 µm) experiments were purchased from C.I.L. Cluzeau (Sainte Foy La Grande, France) and were maintained at a constant temperature of 25 °C. For all experiments, the injection volume was 20 µL and the flow rate was 1 mL min−1. Prior to entering the electrospray source, the chromatographic effluent was split down to 50 µL min−1 using a zero-dead-volume tee connector.Mass spectrometry
Mass spectrometry was performed with a 3200 Q-TRAP mass spectrometer (Applied Biosystems SCIEX, Concord, ON, Canada) equipped with an electrospray ionization source operated in positive mode. The capillary voltage was set at 5500 V and the cone voltage at 80 V. In this hybrid instrument, ions were measured using a quadrupole mass analyzer. Air was used as the nebulizing gas (20 psi) and the curtain gas was nitrogen (20 psi). Analyst software version 1.4.1 (Applied Biosystems SCIEX) was used for instrument control, data acquisition and data processing for both chromatography and mass spectrometry.Rheology
Intrinsic viscosity measurements were performed with a ThermoRheo RS600 rheometer (Thermo Haake GmbH, Karlsruhe, Germany) using a double-gap geometry. Solutions were thermostated at 25 °C and their volume was 6.3 mL. Viscosity was measured within 1–100 s−1 shear speed range.Results and discussion
Using the non-polar C18 stationary phase, the critical composition of the MeOH–water eluent, first established for PEG standards (Mw = 200 to 1000 g mol−1) using 1 mM LiCl concentration, was determined at 88.8:11.2 (%, v/v) and allows all PEG chains to have the same elution volume (Ve = 2.33 mL), independently of their molecular mass.6 Changing the nature of the salt, while keeping the mobile phase composition constant, PEG elution volume was observed to increase as a function of both the molecule and the cation size (Fig. 1). Although statistically significant, this effect is quite weak for the lowest molecular weight oligomers: the elution volume of the smallest PEG chains (Mw = 200 g mol−1) was observed to increase from 2.33 mL to 2.37 mL (standard deviation σ ∼ 0.01 for n = 3 experiments) when increasing the size of the cation present in the mobile phase (Table 1). However, as the polymer molecules become larger, changing the cationization agent from Li+ to Cs+ has a much stronger effect on PEG elution volume, as exemplified by the 20% increase measured for oligomers with Mw = 1000 g mol−1 in Fig. 1. Typically, increasing the size of the cation in the mobile phase induces the PEG molecules to be separated according to the interaction mode. | ||
| Fig. 1 Elution volume of poly(ethylene)glycol standards (Mw = 200 to 1000 g mol−1) in a 88.8:11.2 (%, v/v) MeOH–water binary mixture containing 1 mM of (□) LiCl, (●) AgTFA, (△) NaI, (■) KCl, (○) RbI, and (▲) CsI. Stationary phase: octadecyl-silica, T = 25 °C, flow-rate: 1 mL min−1, detection ESI-MS. Data result from a set of four injections (20 µL). | ||
A completely opposite effect was observed using a polar (Si–NH2) stationary phase. For useful comparison, the critical composition of the MeOH–water eluent was first determined for PEG standards using a 1 mM LiCl concentration. For the selected system, critical conditions were obtained with a 93.5:6.5 (%, v/v) MeOH–water eluent, as indicated by the same elution volume (Ve = 4.15 mL) measured for all PEG chains (Fig. 2). In this system involving a polar adsorbent, methanol is an adsorli (i.e., a liquid promoting adsorption) and water is a desorli, with regards to PEG molecules. Then, as expected, increasing the methanol content caused the polymer molecules to be separated according to the adsorption mode (as exemplified in Fig. 2 using 97.3% MeOH), whereas elution volumes decreased with PEG size in a classical exclusion mode using eluents with less than 93.5% methanol. The LCCC-ESI-MS coupling was then performed using a 93.5:6.5 (%, v/v) MeOH–water eluent and different added salts. As the size of the cation in the mobile phase increases, PEG elution volumes were observed to decrease and this effect becomes stronger as the size of oligomers increases (Fig. 3). When comparing data obtained with Li+ to Cs+, variation of elution volumes was smaller as compared to the case of the non-polar adsorbent but was found to be significant, ranging from −1.4% to −4.1% for PEG with Mw = 200 g mol−1 and Mw = 1000 g mol−1, respectively. In other words, separation of PEG molecules according to the exclusion mode becomes preponderant as the size of the cation in the mobile phase increases, when using a polar adsorbent.
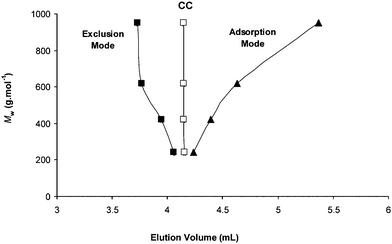 | ||
| Fig. 2 Elution volume of poly(ethylene)glycol standards (Mw = 200 to 1000 g mol−1) in a binary mixture of MeOH–water containing LiCl (1 mM) of different composition (%, v/v): 70.0:30.0 (■), 93.5:6.5 (□) and 97.3:2.3 (▲). Stationary phase: amino-silica, T = 25 °C, flow-rate: 1 mL min−1, detection ESI-MS. Data result from a set of four injections (20 µL). | ||
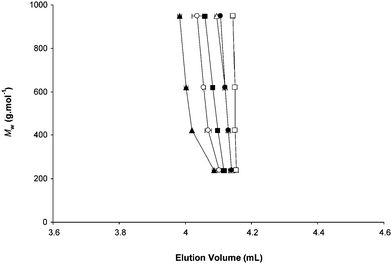 | ||
| Fig. 3 Elution volume of poly(ethylene)glycol standards (Mw = 200 to 1000 g mol−1) in a 93.5:6.5 (%, v/v) MeOH–water binary mixture containing 1 mM of (□) LiCl, (●) AgTFA, (△) NaI, (■) KCl, (○) RbI, and (▲) CsI. Stationary phase: octadecyl-silica, T = 25 °C, flow-rate: 1 mL min−1, detection ESI-MS. Data result from a set of four injections (20 µL). | ||
This last phenomenon could have been attributed to variations of PEG chain conformation as a function of the nature of the cation. Indeed, conformational studies have shown that the oligomer chain is quite flexible and warps itself around the cation, which is thus coordinated to as many oxygen atoms as possible.27,29,30,32 Considering a given n-mer, the hydrodynamic volume of the oligomer–cation system would thus increase with the cation size, inducing stronger exclusion effects. Rheological measurements were then performed to monitor any conformational changes of polymer in solutions which composition matches the established critical conditions and containing different added salts. However, experimental intrinsic viscosities were identical, within the range of experimental errors, for all tested PEGs (for example, [η] = 9.3 mL g−1 for PEG 2000 in the presence of any of the tested salts).
Alternatively, the cation could also interact with the mobile or the stationary phase, depending on the polarity of the stationary phase. Using the C18 stationary phase, electrostatic interactions between the cation and water molecules in the eluent become stronger as the cation size increases.33–36 Moreover, the concentration of available cations in solution increases as the salt dissociation energy decreases. A salting-out effect37,38 could thus occur towards PEG molecules which would increase concentration in the stationary phase, leading to the observed interaction mode elution which becomes more pronounced as the size of the polymer molecules increases. In other words, the mobile phase containing 1 mM CsI would act as a poorer solvent towards PEGs as compared to the same eluent containing 1 mM LiCl. The liquid C18 phase becomes a better solvent and partition of PEG molecules between this aliphatic phase and the eluent is more pronounced, resulting in the observed interaction mode elution. This result is consistent with data from Table 1, which indicate a lower dissociation energy for the salt containing the bigger cation. In contrast, a polar adsorbent such as the amino–silica stationary phase has a surface which can bind cations from the mobile phase. As a result, less sites in the stationary phase would be available to interact with polymer chains, which would thus elute according to the exclusion mode.
Finally, despite our efforts, critical conditions could not be determined for PEG, with one or the other column, using any of the tested cations but lithium. As a result, the use of a bigger cation in the mobile phase to promote the formation of large molecules in electrospray would require a different stationary phase to be used for LCCC-ESI-MS of the targeted PEO-b-PS copolymers .
Conclusion
The nature of salts added in the chromatographic mobile phase to promote on-line electrospray ionization was shown to strongly affect the critical conditions of PEG molecules. This phenomenon was shown to occur both using non-polar and polar stationary phases. Polymer molecules would be evicted from the mobile phase due to a salting out effect when a non-polar C18 stationary phase is used whereas a decrease of adsorption of polymer chains onto the solid surface would occur in the case of the polar NH2stationary phase, both phenomena being driven by specific interactions involving the cation. In any case, these findings imply that, when using a mobile phase containing the cationizing agent to avoid drawbacks associated with its post-column addition, LCCC methods involving different salts cannot be simply derived from each others by slightly altering solvent composition. In contrast, the cationizing agent should first be carefully selected based on the nature of the block copolymers to be studied and an appropriate chromatographic system is then found to allow critical conditions to be established for one of the blocks.Acknowledgements
This work was supported by the French Research Agency (ANR-06-JCJC-0112). L. Charles acknowledges support from Spectropole, the Analytical Facility of Aix-Marseille University, by allowing a special access to the instruments purchased with European Funding (FEDER OBJ2142-3341). E. Beaudoin acknowledges Pr Dusan Berek for fruitful discussions.References
- H. Pasch and K. Rode, J. Chromatogr., A, 1995, 699, 21–29 CrossRef CAS.
- M. W. F. Nielen and F. A. Buijtenhuijs, Anal. Chem., 1999, 71, 1809–1814 CrossRef CAS.
- H. Lee, W. Lee, T. Chang, S. Choi, D. Lee, H. Ji, W. K. Nonidez and J. W. Mays, Macromolecules, 1999, 32, 4143–4146 CrossRef CAS.
- D. Y. Cho, S. Park, K. Kwon and T. Y. Chang, Macromolecules, 2001, 34, 7570–7572 CrossRef CAS.
- S. Trimpin, S. M. Weidner, J. Falkenhagen and C. N. McEwen, Anal. Chem., 2007, 79, 7565–7570 CrossRef CAS.
- M. Girod, T. N. T. Phan and L. Charles, Rapid Commun. Mass Spectrom., 2008, 22, 3767–3775 CrossRef CAS.
- M. Girod, T. N. T. Phan and L. Charles, Rapid Commun. Mass Spectrom., 2009, 23, 1476–1482 CrossRef CAS.
- D. Berek, Macromol. Symp., 1996, 110, 33–56 CAS.
- D. Berek, Macromolecules, 1998, 31, 8517–8521 CrossRef CAS.
- D. Berek, M. Janco and G. R. Meira, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 1363–1371 CrossRef CAS.
- A. Gorbunov, A. Skvortsov, B. Trathnigg, M. Kollroser and M. Parth, J. Chromatogr., A, 1998, 798, 187–201 CrossRef CAS.
- S. L. Phillips, L. Ding, M. Stegemiller and S. V. Olesik, Anal. Chem., 2003, 75, 5539–5543 CrossRef CAS.
- B. Trathnigg, C. Rappel, S. Fraydl and A. Gorbunov, J. Chromatogr., A, 2005, 1085, 253–261 CrossRef CAS.
- A. V. Gorshkov, I. A. Tarasova, V. V. Evreinov, M. M. Savitski, M. L. Nielsen, R. A. Zubarev and M. V. Gorshkov, Anal. Chem., 2006, 78, 7770–7777 CrossRef CAS.
- R. Murgasova, I. Capek, E. Lathova, D. Berek and S. Florian, Eur. Polym. J., 1998, 34, 659–663 CrossRef CAS.
- H. D. Bijsterbosch, M. A. C. Stuart, G. J. Fleer, P. van Caeter and E. J. Goethals, Macromolecules, 1998, 31, 7436–7444 CrossRef CAS.
- J. Falkenhagen, H. Much, W. Stauf and A. H. E. Muller, Macromolecules, 2000, 33, 3687–3693 CrossRef CAS.
- W. Lee, D. Y. Cho, T. Y. Chang, K. J. Hanley and T. P. Lodge, Macromolecules, 2001, 34, 2353–2358 CrossRef CAS.
- W. H. Jiang, S. Khan and Y. M. Wang, Macromolecules, 2005, 38, 7514–7520 CrossRef CAS.
- B. Trathnigg, Polymer, 2005, 46, 9211–9223 CrossRef CAS.
- E. Beaudoin, P. E. Dufils, D. Gigmes, S. Marque, C. Petit, P. Tordo and D. Bertin, Polymer, 2006, 47, 98–106 CrossRef CAS.
- H. F. Gao, K. Min and K. Matyjaszewski, Macromol. Chem. Phys., 2006, 207, 1709–1717 CrossRef CAS.
- B. Trathnigg and A. Gorbunov, Macromol. Symp., 2006, 237, 18–27 CrossRef CAS.
- R. P. Lattimer, J. Am. Soc. Mass Spectrom., 1992, 3, 225–234 CrossRef CAS.
- R. P. Lattimer, J. Am. Soc. Mass Spectrom., 1994, 5, 1072–1080 CrossRef CAS.
- T. L. Selby, C. Wesdemiotis and R. P. Lattimer, J. Am. Soc. Mass Spectrom., 1994, 5, 1081–1092 CrossRef CAS.
- M. Girod, Y. Carissan, S. Humbel and L. Charles, Int. J. Mass Spectrom., 2008, 272, 1–11 Search PubMed.
- J. H. Scrivens, A. T. Jackson, H. T. Yates, M. R. Green, G. Critchley, J. Brown, R. H. Bateman, M. T. Bowers and J. Gidden, Int. J. Mass Spectrom. Ion Processes, 1997, 165, 363–375 CrossRef.
- G. Vonhelden, T. Wyttenbach and M. T. Bowers, Science, 1995, 267, 1483–1485.
- J. L. Wu, M. J. Polce and C. Wesdemiotis, J. Am. Chem. Soc., 2000, 122, 12786–12794 CrossRef CAS.
- T. Gruendling, G. Hart-Smith, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Macromolecules, 2008, 41, 1966–1971 CrossRef CAS.
- T. Wyttenbach, G. von Helden and M. T. Bowers, Int. J. Mass Spectrom. Ion Processes, 1997, 165, 377–390 CrossRef.
- T. A. Graber, M. E. Taboada, J. K. Asenjo and B. A. Andrews, J. Chem. Eng. Data, 2001, 46, 765–768 CrossRef CAS.
- M. E. Taboada, O. A. Rocha, T. A. Graber and B. A. Andrews, J. Chem. Eng. Data, 2001, 46, 308–311 CrossRef CAS.
- M. E. Taboada, J. A. Asenjo and B. A. Andrews, Fluid Phase Equilib., 2001, 180, 273–280 CrossRef CAS.
- M. E. Taboada, H. R. Galleguillos and T. A. Graber, J. Chem. Eng. Data, 2005, 50, 264–269 CrossRef CAS.
- F. E. Bailey and R. W. Callard, J. Appl. Polym. Sci., 1959, 1, 56–65 CrossRef CAS.
- M. Ataman, Colloid Polym. Sci., 1987, 265, 19–25.
- J. E. Hukee, E. A. Keiter, R. L. Keiter, A. Pousse and J. Fisher, in Chimie Inorganique, De Boeck Université, Paris, 1st edn., 1996 Search PubMed.
| This journal is © The Royal Society of Chemistry 2009 |