Raman spectroscopic prediction of the solid fat content of New Zealand anhydrous milk fat
C. M.
McGoverin
a,
A. S. S.
Clark
b,
S. E.
Holroyd
c and
K. C.
Gordon
*a
aDepartment of Chemistry and MacDiarmid Institute for Advanced Materials and Nanotechnology, University of Otago, Dunedin, New Zealand. E-mail: kgordon@chemistry.otago.ac.nz; Fax: +64 3479 7906; Tel: +64 3479 7599
bDepartment of Mathematics and Statistics, University of Otago, Dunedin, New Zealand
cFonterra Research Centre, Palmerston North, New Zealand
First published on 8th September 2009
Abstract
The functionality of anhydrous milk fat (AMF) is determined from solid fat content (SFC) and triacylglycerol (TG) profiles, parameters traditionally measured using nuclear magnetic resonance and high pressure liquid chromatography respectively. Raman spectroscopy coupled with partial least squares (PLS) analysis has been assessed as an alternative method for SFC and TG class quantification. Sample temperature at which the Raman spectra were collected, method of spectral preprocessing and type of PLS analysis were all investigated and found to significantly affect the resulting calibrations (as parameterized by root mean square error of cross validation). Physically heterogeneous AMF samples held at 20 °C were shown to allow reliable SFC predictions on the basis of collected Raman spectra. In contrast to SFC calibrations, physically homogenous samples in a liquid form were ideal for TG class concentration predictions, however, not all TG classes could be reliably predicted.
Introduction
Interest in the adaptation of vibrational spectroscopic techniques to the measurement of sample properties (e.g. solid fat content (SFC) and the concentrations of certain triacylglycerols (TGs)) has been motivated by a desire for more convenient and non-destructive, rapid methods of analysis. Anhydrous milk fat (AMF) (99.8% milk fat, 0.2% water),1 the commercially important separated fat component of milk, is characterized primarily by the solid fat content (SFC) parameter. The SFC is a measure of the solid fraction within a fat sample at a specified temperature under reference conditions, expressed as a percentage weight for weight (% w/w).2,3 Solid fat content at various temperatures is indicative of the physical nature of the sample. Specific characterization of AMF samples is possible through analysis of certain triacylglycerol (TG) class concentrations (Table 1). Currently SFC and TG class concentrations are determined by low resolution nuclear magnetic resonance (NMR)2,4 and reverse-phase high pressure liquid chromatography (HPLC).5| TG class | Abbreviation | Definition |
|---|---|---|
| a These two groups are distinguished by stage of crystallisation: HSS TGs crystallise earlier in commercial fractionation than SS TGs. | ||
| Hard long saturates | HLS | Triacylglycerols with three saturated fatty acids, all of which are of length C10 or greater |
| Hard short saturatesa | HSS | Triacylglycerols with three saturated fatty acids, two of which are long (C14 or greater), the third is short (mostly C4 or C6) |
| Long monounsaturated | LM | Triacylglycerols with two saturated fatty acids, both of which are C8 or greater, and a cis-monoenoic fatty acid (mostly C18:1) |
| Short monounsaturated | SM | Triacylglycerols with two saturated fatty acids, one long (C12 or greater), the other short (mostly C4 or C6) and a cis-monoenoic fatty acid (mostly C18:1) |
| c-dienes | CD | Triacylglycerols with two cis-monoenoic fatty acids (mostly C18:1), and a saturated fatty acid of any length |
| t-monounsaturated | TM | Triacylglycerols with two saturated fatty acids, both of which are C12 or greater, and a trans-monoenoic fatty acid (mostly trans-C18:1) |
| c,t-dienes | CTD | Triacylglycerols with two monoenoic fatty acids (both mostly C18:1), one cis-, the other trans-. The third fatty acid is saturated and of length C12 or greater. |
| Short saturateda | SSat | Triacylglycerols with three saturated fatty acids, two of which are C8 or greater, the third is short (mostly C4 or C6) |
The aim of the current study was to determine the utility of Raman spectroscopy in the measurement of industrially relevant TG classes and SFC of AMF samples sourced from normal dairy production within New Zealand. The typical factory supply of New Zealand milk has a fat content of 4.4–4.8% in July (the beginning of the milking season) which increases to 5.8–6.0% in April (the end of the season).2 Furthermore, seasonal fluctuations are observed in milk fat characteristics.2,6 The samples examined covered a milking season to represent annual variation within AMF and were sourced from the Fonterra Kauri milk processing plant which has a collection range over 300 km from north to south.
The quantification of TG classes using vibrational spectroscopy has not been previously reported in the literature. The quantification of SFC (or the closely related parameter solid fat index) has however previously been investigated using Raman, MIR and NIR spectroscopies.2,3,7,8 The Raman spectroscopic study of Beattie et al.7 quantified SFC in clarified butter. Clarified butter samples were held at 55 °C and predictive models were calculated for SFC levels from 5 to 25 °C in 5 °C increments. Prediction of the lower temperature SFCs was more accurate than the higher temperature SFCs; ratio of prediction to deviation (RPD)9 ranged from 2.46 for SFC5 to 1.27 for SFC25. It was proposed by Beattie et al.7 that the Raman spectroscopic predictions could be improved by recording the spectral data at lower temperatures, allowing the information regarding physical state to be included. The study presented here examines this proposal in further detail. Raman spectra were recorded from AMF samples at various temperatures. Partial least squares (PLS) analyses were carried out to determine the optimum temperature for collecting data for the purpose of quantifying SFC or industrially important classes of TGs. In addition, the effect of different methods of preprocessing and PLS analysis on calibration performance has been assessed.
Materials and methods
Anhydrous milk fat samples
Seventeen AMF samples were supplied from the Fonterra Kauri milk processing plant near Whangarei, New Zealand. The samples spanned a single production season, August 2004 to March 2005. The extraction procedure used to obtain the AMF samples is outlined by MacGibbon and McLennan.10Briefly, butter was melted at 50 °C with the fat layer centrifuged and the separated fat phase filtered and collected. The samples were supplied with SFC values, as determined by low resolution pulsed NMR, and concentrations of various TG classes (Table 1), as measured by reversed-phase HPLC. The method of MacGibbon and McLennan10 was followed for the SFC measurements. Samples were tempered at 0 °C overnight and subsequently measured at 5 °C increments (up to 35 °C) between 45 min equilibration periods. Duplicates were recorded for each sample, the mean of which was the taken as the reference SFC value. The method of Robinson et al.5 was followed for the TG class concentration measurements. A binary solvent system was used to separate TGs into distinct peaks, based on chain length and the number of double bonds. The various TG group concentrations were only supplied for 15 of the standard AMF samples; Tables 2 and 3 list the relevant parameters relating to reference values for the SFC values and TG class concentrations respectively.
| SFCa | Minimumb | Maximumb | Medianb | Meanb | Standard deviationb |
|---|---|---|---|---|---|
| a Units: °C. b Units: % w/w. | |||||
| 0 | 63.11 | 69.91 | 66.56 | 66.68 | 2.26 |
| 5 | 59.35 | 66.31 | 62.85 | 63.00 | 2.35 |
| 10 | 52.41 | 59.28 | 55.37 | 55.77 | 2.62 |
| 15 | 37.05 | 44.28 | 39.32 | 40.21 | 3.04 |
| 20 | 19.03 | 25.48 | 20.80 | 21.55 | 2.42 |
| 25 | 10.22 | 13.69 | 11.18 | 11.67 | 1.39 |
| 30 | 4.26 | 6.79 | 5.00 | 5.33 | 0.95 |
| 35 | 0.08 | 1.68 | 0.44 | 0.65 | 0.49 |
| TG classa | Minimumb | Maximumb | Medianb | Meanb | Standard deviationb |
|---|---|---|---|---|---|
| a Class definitions are in Table 1. b Units: % w/w. | |||||
| HLS | 13.44 | 19.73 | 16.48 | 16.55 | 1.96 |
| HSS | 16.12 | 20.09 | 17.32 | 17.67 | 1.32 |
| LM | 16.92 | 20.13 | 18.26 | 18.50 | 0.82 |
| SM | 14.14 | 17.46 | 15.76 | 15.95 | 1.08 |
| CD | 6.53 | 9.61 | 7.81 | 7.99 | 1.01 |
| TM | 2.35 | 4.27 | 3.00 | 3.19 | 0.58 |
| CTD | 1.44 | 4.53 | 2.30 | 2.61 | 0.95 |
| SSat | 7.87 | 11.04 | 10.46 | 10.12 | 0.93 |
Raman spectroscopy
Raman spectra of the AMF samples were recorded using 752 nm excitation, which was generated by a continuous-wave Innova I-302 krypton-ion laser (Coherent Inc.). Power at the sample was ∼225 mW. Raman scattering was collected in a 135° backscattering geometry. Scattered light was focused via matched aperture lens through a notch filter (Kaiser Optics), on to the 100 µm entrance slit of an Acton Research SpectraPro 500i spectrograph. The collected light was dispersed in the horizontal plane by a 300 groove mm−1 diffraction grating. Diffracted photons were detected by a liquid-nitrogen cooled back-illuminated Spec-10:100B charge coupled device (CCD) controlled by a ST-133 controller and Winspec/32 (version 2.5.8.1) software (Roper Scientific, Princeton Instruments).11,12 Each day of measurement the laser was allowed to warm up for at least an hour to ensure laser power stability and was operated with power control activated. The spectral region recorded was 35–2500 cm−1 and all spectra were the sum of 30 one second scans. Throughout the experiment the wavenumber scale was accurate to ±1 cm−1.Raman spectra of each sample were recorded at temperatures from 0 to 50 °C in 10 °C increments (Temperature of Data Collection: TDC0, TDC10…TDC50). AMF samples were pipetted into small glass tubes of 8 mm internal diameter and height of 40 mm. The samples were held at a specific TDC for 45–60 minutes before beginning Raman measurements. The sample was contained in a water bath (Fig. 1) designed to permit in situ Raman measurements.
 | ||
| Fig. 1 The Raman variable temperature measurement system utilised in this study used a pump/heater to cycle water through a small water bath (shown) in which the fat sample was placed. Each division of the scale bar is 1 cm. | ||
The temperature of the sample water bath was controlled by water pumped (at a rate of ca. 100 mL min−1) from a larger resevoir (4.5–5.0 L) which was held at a constant temperature. The temperature of the water bath and AMF samples was monitored using a Fluke 52 K/J dual channel digital thermometer and K-type thermocouple. Samples were maintained within one degree of the specified temperature throughout the course of the measurement, except for TDC0, which ranged ±1.5 °C. A random sample order was used for the Raman measurements, and five spectra were collected from each sample at each TDC. At TDC0 condensation would collect on the front window of the sample waterbath within the 30 s acquisition time, and was cleared between each measurement. The Raman data were acquired over several days.
Two samples were selected from which to measure repeatability (10 consecutive Raman spectral measurements without sample replacement) and reproducibility (10 Raman spectral measurements with sample replacement). A spectrum of dimethylformamide (DMF) was recorded between each sample spectrum in order to monitor the performance of the Raman system, except during the repeatability runs. DMF and cyclohexane were used to monitor the wavenumber stability of the system (±1 cm−1). Repeatability and reproducibility were parameterized as average relative standard deviation (RSD), a value calculated from the median smoothed data of 760–1786 cm−1. The focus of the laser was optimized for a liquid sample, creating a bias towards such samples: spectra collected from liquid samples were more intense than those collected from solid samples.
Quantification model development
AMF spectral data were imported into The Unscrambler® (version 9.7, Camo Technologies Inc.). The spectral data set was first analyzed for outliers by visual inspection and principal components analysis (PCA). One spectrum was eliminated from one sample set at TDC0. At TDC30 one sample had to be removed due to an erroneous background. The five spectra collected (or four when a spectrum was deleted) for each sample at each TDC were averaged. All spectra were preprocessed with a median smooth of five points to remove or reduce the effect of errant spectral features, i.e. cosmic spikes. Several methods of spectral transformation for baseline and sample volume correction were applied to the data at each TDC; standard normal variate (SNV), first derivative Norris Gap 3 points (1stD), second derivative Norris Gap 3 points (2ndD), area normalization (AN), and baseline correction both alone (BL) and coupled with either standard normal variate (BL-SNV) or area normalization (BL-AN). The median smoothed data were imported into OPUS (version 5.5, Bruker) for rubberband correction with three baseline points.13,14 The data set was then imported back into The Unscrambler®.The SFC and TG class concentration calibrations were treated separately, and were formed using three PLS methods: PLS1, PLS1 with modified jack-knifing (PLS1-JK) and PLS2. All calibrations were validated using full cross validation. A full factorial design was followed to examine six different TDCs, seven different methods of preprocessing and three methods of analysis for all eight SFC and all eight TG class parameters.15 The calibrations were parameterized by root mean square error of cross validation16,17 (RMSECV) and the square root of RMSECV. The RMSECV values were transformed by calculating the square root, enabling the treatments to exhibit a normal distribution and thereby allowing the application of an analysis of variance (ANOVA). An ANOVA treats the SFC levels or TG classes as pseudo-replicates, which permits the investigation of possible interactions between preprocessing technique, method of analysis and TDC. Ranking of the mean square root of RMSECV for each treatment and ranking of mean square error (MSE) were used to assess which calibration treatments gave the best predictive performance. Mean square error of an estimator (MSE(![[small straight theta, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e12d.gif) )) (eqn (1)) is equal to the sum of the variance (V()) and the square of the bias (B()), here the estimator () was the square root of RMSECV. The optimal value of RMSECV, and therefore the square root of RMSECV, was zero, hence the bias associated with a treatment was equivalent to the mean of the square root of RMSECV. Bias (eqn (2)) is equal to the difference between the mean of the estimator (E()) and the expected value of this estimator (θ). The MSE parameter allowed the spread and average square root RMSECV value across the SFC levels or TG classes for a specific treatment to be considered concurrently.
)) (eqn (1)) is equal to the sum of the variance (V()) and the square of the bias (B()), here the estimator () was the square root of RMSECV. The optimal value of RMSECV, and therefore the square root of RMSECV, was zero, hence the bias associated with a treatment was equivalent to the mean of the square root of RMSECV. Bias (eqn (2)) is equal to the difference between the mean of the estimator (E()) and the expected value of this estimator (θ). The MSE parameter allowed the spread and average square root RMSECV value across the SFC levels or TG classes for a specific treatment to be considered concurrently.
| MSE() = V() + (B())2 | (1) |
| B() = E() − θ | (2) |
Results and discussion
Temperature dependent Raman spectra
Raman spectra of AMF samples are dependent both on composition and physical state. Fig. 2 illustrates the raw averaged data collected from an AMF sample. In general, AMFs exhibited visible signs of softening at 20 °C and were completely liquid at 40 °C. The gross spectral changes exhibited by a sample upon transition from a solid to liquid were similar. All AMFs exhibit an increase in the wavenumber of the ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)7,18,24,25 mode of approximately 6 cm−1, centered around 1745 cm−1. The two shoulders of the 1442 cm−1 peak [(δ(CH2)sc]19,24,25 at ca. 1460 and 1420 cm−1 decreased in relative intensity while the δ(CH2)tw band upshifts ∼6 cm−1 (centred about 1300 cm−1).19,25,26 The δ(=CH)ipcis mode (1262 cm−1)19,25,27 broadened to become more of a shoulder on the δ(CH2)tw band as the sample became more liquid. The 1178 cm−1 peak decreased in relative intensity, while the 1159 cm−1 band increased. The most apparent spectral alterations upon the solid to liquid phase transition were below 1150 cm−1. The spectral changes in the 1000–1150 cm−1 regions were similar to those of stearic and palmitic acid upon transition from solid to liquid.26 The ν(C–C)ip mode (centered ca. 1124 cm−1) exhibited a wavenumber downshift of approximately 8 cm−1 and a decrease in relative intensity concurrent with the decrease in relative intensity of the out-of-phase ν(C–C) mode (1063 cm−1).28 The 1080 cm−1 band that increased in relative intensity with the transition to a liquid was due to gaucheν(C–C). The complex ν(C–C), CH3,rk, ν(C–O) region below 920 cm−1 was characterized by a reduction in band intensity at 920 and 890 cm−1 and increases at 870 and 846 cm−1. Band shape of the ν(CC) peak (1656 cm−1ν(CC)cis, 1670 cm−1ν(CC)trans)19,24,25,29 was largely indifferent to physical state.
O)7,18,24,25 mode of approximately 6 cm−1, centered around 1745 cm−1. The two shoulders of the 1442 cm−1 peak [(δ(CH2)sc]19,24,25 at ca. 1460 and 1420 cm−1 decreased in relative intensity while the δ(CH2)tw band upshifts ∼6 cm−1 (centred about 1300 cm−1).19,25,26 The δ(=CH)ipcis mode (1262 cm−1)19,25,27 broadened to become more of a shoulder on the δ(CH2)tw band as the sample became more liquid. The 1178 cm−1 peak decreased in relative intensity, while the 1159 cm−1 band increased. The most apparent spectral alterations upon the solid to liquid phase transition were below 1150 cm−1. The spectral changes in the 1000–1150 cm−1 regions were similar to those of stearic and palmitic acid upon transition from solid to liquid.26 The ν(C–C)ip mode (centered ca. 1124 cm−1) exhibited a wavenumber downshift of approximately 8 cm−1 and a decrease in relative intensity concurrent with the decrease in relative intensity of the out-of-phase ν(C–C) mode (1063 cm−1).28 The 1080 cm−1 band that increased in relative intensity with the transition to a liquid was due to gaucheν(C–C). The complex ν(C–C), CH3,rk, ν(C–O) region below 920 cm−1 was characterized by a reduction in band intensity at 920 and 890 cm−1 and increases at 870 and 846 cm−1. Band shape of the ν(CC) peak (1656 cm−1ν(CC)cis, 1670 cm−1ν(CC)trans)19,24,25,29 was largely indifferent to physical state.
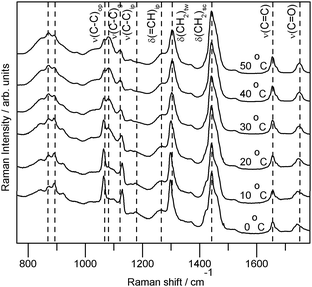 | ||
| Fig. 2 The averaged raw spectra of one standard AMF sample collected at 0–50 °C, showing relative peak intensity changes. The intensity of each spectrum is scaled for presentation. The spectra collected at higher temperatures are more intense than those collected at lower temperatures. Vibrational mode assignments after ref. 8,18–23. Mode assignment subscripts: g—gauche, op—out-of-phase, ip—in-phase for ν(C–C) and in-plane for δ(=CH). | ||
The repeatability and reproducibility measurements indicated that data collected at 40 °C were the least variable; when samples were liquid the variation inherent to the measurements was smallest. In quantification there are two competing effects, the increased variability within the spectral measurements and the increased spectral information when using a physically heterogeneous sample.
Solid fat content quantification
Temperature at which data were collected (TDC), preprocessing type, method of analysis, and SFC value all significantly influenced the square root of RMSECV; the interactions between method of analysis and preprocessing, method of analysis and TDC, and preprocessing and TDC were also significant.From the TDC-preprocessing interaction plot (Fig. 3) it was observed that 1stD and 2ndD methods of preprocessing both performed optimally when the samples were liquid. Derivative preprocessing methods amplify the low frequency noise within a spectrum. Spectra recorded from solid samples were of lower signal to noise than spectra collected from liquid samples. The noise amplification of differentiation would therefore have had a greater effect on the spectra of more solid samples, reducing the efficacy of calibrations formed on the basis of these spectra. This was supported by the observation that application of PLS1-JK offered the largest improvement when derivative methods were used, and was a function of the removal of irrelevant variables. The normalization preprocessing methods, SNV and AN, exhibited a trend similar to that of the overall trend (Fig. 3). When combining baselining and normalization, BL-SNV and BL-AN, the trend across TDCs changed appreciably; while TDC10 was still the worst, TDC20 was better than any other TDC. The TDC-preprocessing interaction plot indicated that effective preprocessing was dependent on the physical nature of the sample. Normalization methods were necessary when the sample was heterogeneous (as at TDC20). The application of baselining alone did not compensate for signal variability coupled to physical heterogeneity (TDC40 and TDC50 were better than TDC20 when BL was used), however, the use of both baselining and normalization lead to more effective use of spectral signatures due to the solid to liquid transition (TDC20 was better than TDC40 and TDC50 when BL-SNV or BL-AN was used).
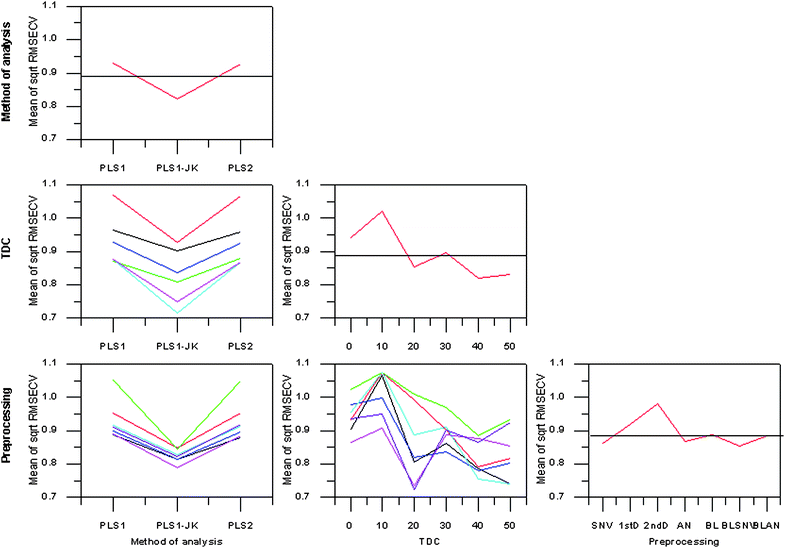 | ||
| Fig. 3 Main effects and interactions plot for SFC calibrations. TDC0—black, TDC10—red, TDC20—green, TDC30—blue, TDC40—cyan, TDC50—magenta. Standard normal variate—black, 1stD—red, 2ndD—green, area normalisation—blue, baselining—cyan, baselining and standard normal variate—magenta, baselining and area normalisation—purple. | ||
Ranking of the calibration treatments by mean square root RMSECV and the mean square error of square root RMSECV were near coincident. Predictably, calibration treatments involving TDC20, TDC40 and TDC50 were the only ones present in the upper 20 rankings. The top 10 calibration treatments were all PLS1-JK based. All three methods of analysis were in the top 20 rankings when TDC20 and BL-SNV or BL-AN were used.
Calibrations based on PLS1-JK coupled with BL-AN preprocessing and utilizing data collected at 20 or 40 °C rated highly in terms of prediction accuracy. Comparison of the two TDCs indicated lower RMSECV values (and better predicted versus reference regression line parameters) generally resulted from the TDC40 prediction model (Table 4). The TDC40 calibrations required a greater number of factors though and were based on fewer variables. The TDC20 results were generally a better compromise between fit and prediction.
| SFC | TDC | RMSEC | R 2 | RMSECV | RPD | Number of factors |
|---|---|---|---|---|---|---|
| a Abbreviations: SFC—solid fat content (at temperature (°C) indicated), TDC—temperature of data collection (°C), RMSEC—root mean squared error of calibration (% w/w), RMSECV—root mean squared error of cross validation (% w/w), RPD—ratio of prediction to deviation. R2 is a parameter of the validation predicted versus reference curve. | ||||||
| 0 | 20 | 0.42 | 0.92 | 0.65 | 3.49 | 3 |
| 5 | 20 | 0.44 | 0.92 | 0.67 | 3.51 | 3 |
| 10 | 20 | 0.49 | 0.93 | 0.73 | 3.59 | 3 |
| 15 | 20 | 0.50 | 0.95 | 0.69 | 4.39 | 3 |
| 20 | 20 | 0.41 | 0.95 | 0.57 | 4.23 | 3 |
| 25 | 20 | 0.25 | 0.94 | 0.35 | 4.02 | 3 |
| 30 | 20 | 0.19 | 0.93 | 0.26 | 3.65 | 3 |
| 35 | 20 | 0.17 | 0.77 | 0.24 | 2.03 | 3 |
| 0 | 40 | 0.14 | 0.93 | 0.62 | 3.63 | 8 |
| 5 | 40 | 0.40 | 0.90 | 0.77 | 3.04 | 6 |
| 10 | 40 | 0.14 | 0.95 | 0.59 | 4.48 | 8 |
| 15 | 40 | 0.30 | 0.96 | 0.65 | 4.66 | 5 |
| 20 | 40 | 0.11 | 0.98 | 0.31 | 7.82 | 6 |
| 25 | 40 | 0.25 | 0.88 | 0.50 | 2.76 | 5 |
| 30 | 40 | 0.11 | 0.96 | 0.19 | 4.92 | 5 |
| 35 | 40 | 0.07 | 0.93 | 0.13 | 3.65 | 6 |
Fig. 4 shows the regression coefficients for all SFC levels as calculated from the TDC20 data set. In both TDC20 and TDC40 calibrations the ν(CC) region (1665 cm−1) was predominantly negatively weighted with some sign splitting of this region in the TDC40/BL-AN/PLS1-JK calibrations; ∼1668 cm−1 coupled with negative regression coefficients and ∼1662 cm−1 with positive. This may be attributable to a cis/trans splitting of the spectral data in this region. The 1060 and 1130 cm−1 bands (in-phase and out-of-phase ν(C–C) characteristic of solid fat) were positively loaded in the TDC20 regression coefficients. Significant bands in the 1000–1180 cm−1 region of the TDC20 regression coefficients were positively loaded for SFC0, SFC5 and SFC10; beyond this level 1000–1180 cm−1 becomes more complex with the introduction of negative coefficients. The 1000–1180 cm−1 region exhibits extensive spectral changes with the phase transition from solid to liquid, it is therefore unsurprising that variables within this region were significant to quantification and had large coefficients relative to other bands. Within the TDC40 regression coefficients were significant variables in the 1000–1180 cm−1 region. However, unlike the TDC20 regression coefficients, the 1060 cm−1 (out-of-phase ν(C–C)) band was non-significant and the liquid band of 1088 cm−1 (ν(C–C)gauche) was significant. The 1130 cm−1ν(C–C)ip was, despite the absence of the out-of-phase stretch, still significant in all regression coefficients. The other major region of spectral change, below 1000 cm−1, was largely insignificant in the determination of SFC0 or SFC5 for TDC20. In TDC40 there were significant bands in this region for all SFCs. In TDC20 the 900–1000 cm−1 region was positive and the <900 cm−1 area was negative when significant. In TDC40 this trend was typically followed with the exception of the 800–840 cm−1 spectral area which was associated with positive coefficients. In TDC20 the methylene deformations (1300 and 1420 cm−1) were generally split between positive and negative coefficients. The in-plane olefinic hydrogen bend (δ(=CH)ip ∼ 1272 cm−1) was associated with negative coefficients for all levels of SFC except SFC25. The carbonyl stretch (1750 cm−1) was not significant to the quantification of SFC0 and SFC5 when TDC20 was used. In TDC40 calibrations the carbonyl stretch was present only in the SFC20, SFC35 and SFC35 calibrations. Negative regression coefficients were associated with bands attributable to unsaturation and positive coefficients with saturated modes. Note that unsaturated lipids have lower melting points, thereby increasing liquid fat content, hence the opposing weighting of saturated and unsaturated bands.
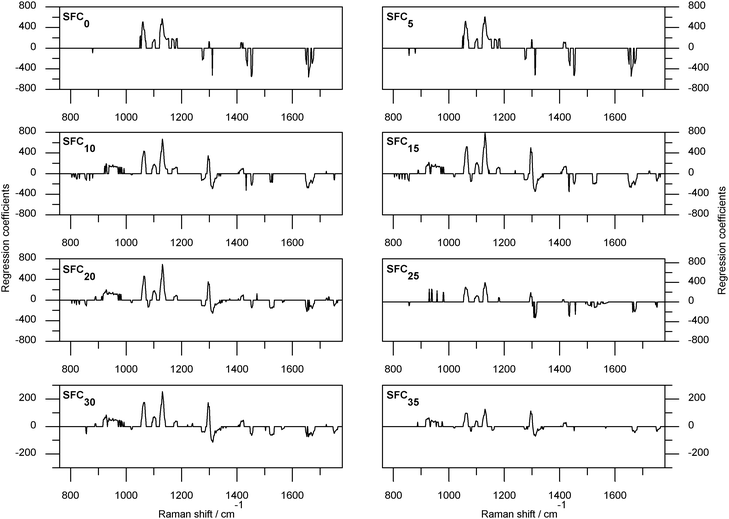 | ||
| Fig. 4 Regression coefficients for solid fat content prediction from SFC0 (the fraction of anhydrous milk fat that is solid when the sample is held at 0 °C) to SFC35 (the fraction of anhydrous milk fat that is solid when the sample is held at 35 °C) as calculated from data collected at 20 °C, preprocessed with baselining and area normalisation, and using partial least squares one with modified jack-knifing. | ||
The RPDs of the aforementioned calibration treatments exceeded two and as such were reliable prediction models (Table 4).30 The NIR spectroscopic study of Meagher et al.2 was based on samples from the same population as the samples utilized in the present study. At lower SFC levels the NIR study out-performed the Raman results presented here, at high SFC levels the opposite was true (RPDs of the NIR study: SFC0 4.43, SFC5 6.19, SFC10 5.28, SFC15 5.37, SFC20 4.79, SFC25 4.57, SFC30 3.52 and SFC35 1.08).
The aforementioned calibrations based on TDC20, while not offering the smallest RMSECV values for this data set, were perhaps more stable as evidenced by the more comparable RMSEC and RMSECV values and the presence of all three methods of analyses in combination with BL-SNV or BL-AN in the top 20 rankings. These calibrations required fewer factors and the regression coefficients were easily interpreted in terms of bands associated with the liquid and solid states. Further investigations with a more extensive data set should be carried out at 20 °C.
Triacylglycerol class quantification
As with the SFC quantification models all calibration parameters (the identity of the TG group, the method of analysis, the TDC and preprocessing type) made significant contributions to the analyzed parameter square root of RMSECV. The interaction between TDC and method of preprocessing was also significant.The PLS1-JK procedure was the optimum method of analysis (Fig. 5), in agreement with the observations of the SFC predictive models. In contrast to the SFC predictive model results TDC20 was not an optimum parameter while TDC30 was when quantifying TG class concentration. The TDC-preprocessing interaction plot of TG class concentration calibration was more complex than that of the SFC predictive models. Similar methods of preprocessing did not follow the same trends to the degree observed in the SFC example. Overall, all preprocessing methods lead to smaller RMSECVs with increasing TDC.
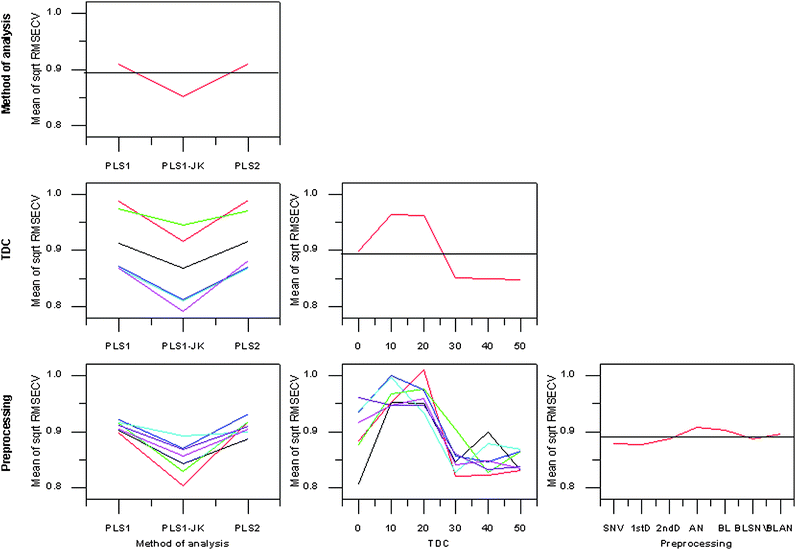 | ||
| Fig. 5 Main effects and interactions plot for TG class concentration calibrations. TDC0—black, TDC10—red, TDC20—green, TDC30—blue, TDC40—cyan, TDC50—magenta. Standard normal variate—black, 1stD—red, 2ndD—green, area normalisation—blue, baselining—cyan, baselining and standard normal variate—magenta, baselining and area normalisation—purple. | ||
It was not possible to calculate a legitimate quantification model for all treatments of each TG class. If the concentration residual validation variance curve of a calibration increased from the initial starting value, then the predictive model was deemed invalid. Accurate prediction results in small residuals, a constant increase in residual validation variance indicates the model is not predictive.16 Each of the methods of analyses had a similar number of these invalid models, which was not the case for TDC or preprocessing method. TDC10 and TDC20 had the most invalid predictive models of the TDCs; such poor performance was in agreement with the TDC profile. In terms of preprocessing BL followed by 2ndD was the worst.
Ranking of treatments on the basis of square root of RMSECV and the MSE of square root RMSECV were again largely coincident and indicated the treatment TDC40/2ndD/PLS1-JK resulted in the lowest RMSECV values. In agreement with the SFC study, the PLS1-JK method of analysis dominated the top 20 rankings. No individual preprocessing method dominated the top 20 rankings; TDC30, TDC40, and TDC50 did dominate the top 20 rankings as suggested by the TDC profile.
Parameters relevant to the TDC40/2ndD/PLS1-JK predictive models are shown in Table 5. From this table it was apparent that just three TG classes, hard long saturates (HLS), c-dienes (CD), and c,t-dienes (CTD), were modelled in a reliable predictive manner (RPD exceeded two) by this calibration treatment. Due to the lack of reliable predictive models for each group, the individual rankings for each of the TG classes were examined (Table 6). A reliable predictive model could be calculated for all but the long monounsaturated (LM), short monounsaturated (SM), and short saturated (SSat) groups.
| TG class | RMSEC | R 2 | RMSECV | RPD | Number of factors |
|---|---|---|---|---|---|
| a See Table 1 for TG class definitions and Table 4 for abbreviations. | |||||
| HLS | 0.50 | 0.89 | 0.68 | 2.88 | 3 |
| HSS | 0.68 | 0.56 | 0.90 | 1.46 | 2 |
| LM | 0.41 | 0.44 | 0.64 | 1.29 | 3 |
| SM | 0.51 | 0.59 | 0.71 | 1.51 | 3 |
| CD | 0.20 | 0.90 | 0.34 | 2.99 | 5 |
| TM | 0.16 | 0.66 | 0.35 | 1.67 | 5 |
| CTD | 0.17 | 0.81 | 0.43 | 2.20 | 5 |
| SSat | 0.31 | 0.71 | 0.52 | 1.79 | 4 |
| TG class | Calibration treatment | RMSEC | R 2 | RMSECV | RPD | Number of factors |
|---|---|---|---|---|---|---|
| a See Table 1 for TG class definitions and Table 4 for abbreviations. | ||||||
| HLS | TDC30/BL-SNV/PLS1-JK | 0.25 | 0.94 | 0.51 | 3.79 | 4 |
| HSS | TDC10/BL-AN/PLS1-JK | 0.35 | 0.88 | 0.47 | 2.83 | 3 |
| LM | TDC0/SNV/PLS2 | 0.40 | 0.62 | 0.53 | 1.56 | 3 |
| SM | TDC0/SNV/PLS1-JK | 0.36 | 0.69 | 0.62 | 1.75 | 4 |
| CD | TDC40/2ndD/PLS1-JK | 0.20 | 0.90 | 0.34 | 2.99 | 5 |
| TM | TDC0/SNV/PLS1-JK | 0.07 | 0.88 | 0.20 | 2.85 | 6 |
| CTD | TDC50/BL-AN/PLS1-JK | 0.11 | 0.95 | 0.22 | 4.40 | 7 |
| SSat | TDC0/SNV/PLS1-JK | 0.30 | 0.54 | 0.58 | 1.59 | 5 |
The regression coefficients calculated using TDC40/2ndD/PLS1-JK for HLS, CD, and CTD are shown in Fig. 6. Examination of the ν(CC) spectral region was instructive in determining the chemical relevancy of the selected variables within the TDC40/2ndD/PLS1-JK calibration as this group is comprised of TGs with saturated fatty acids which should have no Raman activity in this region. The concentrations of the HLS TG class are negatively correlated with the concentrations of the CD, TM and CTD TG classes (Pearson correlation −0.75, −0.82 and −0.80 respectively). These correlations may account for the presence of significant variables in the ν(CC) spectral region. Large coefficients were observed in the δ(CH2)sc region, the remaining variables were in regions of ν(C–C) modes. The regression coefficients for CD prediction assign positive coefficients to variables in the transν(CC) and conjugated ν(CC) regions, and negative coefficients to cisν(CC) regions. The CD group is characterized by TGs with two cis-monoenoic fatty acids and a saturated fatty acid. The sign separation of ν(CC) region correlates well with this chemical distinction. For CTD there was less separation of the ν(CC) region than seen in the CD regression coefficients—this was expected as the CTD group contains two monoenoic fatty acids, one cis and one trans. Negative coefficients were assigned to the 1655 cm−1 variable, which may be a result of other TGs with cis CC functionality.
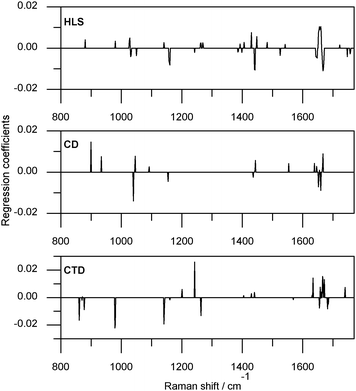 | ||
| Fig. 6 Regression coefficients of HLS, CD, and CTD prediction models as calculated using data collected at 40 °C, preprocessed by calculating the second derivative and using partial least squares one with modified jack-knifing. | ||
The optimum model regression coefficients (as determined by individual rankings) for each TG group are shown in Fig. 7 (except for the regression coefficients of CD which are in Fig. 6). Unlike the previous CTD regression coefficients, the cis and transν(CC) spectral regions were assigned opposing signs. The 1330 cm−1 region was associated with large negative coefficients, examination of Fig. 2 indicates this was not an intense region of Raman activity within the AMF samples, however, the 1330 cm−1 band has been utilized as a lipid specific band in Raman studies of cells.31 Other major coefficients were at 1272 and 1261 cm−1, in the δ(CH2)tw region. In the HLS TDC30/BL-SNV/PLS1-JK calibration regression coefficients variables in the ν(CC) region were significant, as observed for the TDC40/2ndD/PLS1-JK calibration, though in the TDC30/BL-SNV/PLS1-JK calibration this region was not as dominant. The HLS TDC30/BL-SNV/PLS1-JK calibration was based on a greater number of variables than the HLS TDC40/2ndD/PLS1-JK calibration. The largest structure within the HLS TDC30/BL-SNV/PLS1-JK regression coefficients plot (Fig. 7) was associated with the ν(C–C) spectral region, perhaps a function of the saturated nature of this group. The δ(CH2) modes were of importance (the bending and twisting modes more so than the scissoring mode) as expected for saturated TGs. The regression coefficients of the hard short saturates (HSS) TDC10/BL-AN/PLS1-JK calibration were not interpretable in terms of chemical specificity. The opposing ends of the 760–1800 cm−1 region used to form the calibration had several variables with significant coefficients, each end having opposing signs. The only other significant variables were in the 1072 and 1255 cm−1 regions. The HSS regression coefficients may be partially interpreted in terms of physical specificity, both the positive regression coefficient regions exhibit an intensity increase with the solid to liquid transformation, while 1255 cm−1 exhibits a decrease. The significance of the below 835 cm−1 region was difficult to determine; this region is the low wavenumber side of a broad band of AMF. The last of the reliable prediction models was for the TG class TM using TDC0/SNV/PLS1-JK. The TM TDC0/SNV/PLS1-JK predictive model was questionable, with areas lacking Raman activity (such as 1565–1615 cm−1) associated with reasonably large negative coefficients. Despite quantifying a TG class characterised by the presence of trans-monoenoic fatty acids the transν(CC) region had no variables of significance, instead the 1682–1720 cm−1 spectral area had several variables with negative coefficients, another region lacking Raman activity. The model was not a good compromise between fit and prediction (RMSEC was just 35% of RMSECV) further evidence of the questionable nature of this predictive model. The quantification of TM was further investigated by examining the calibration treatment ranked second in the individual TG class listings, TDC40/1stD/PLS1-JK. The regression coefficients of this calibration were chemically meaningful; variables within the transν(CC) region had negative coefficients; other regions with negative coefficients were assignable to ν(C–C) and δ(CH2) modes.
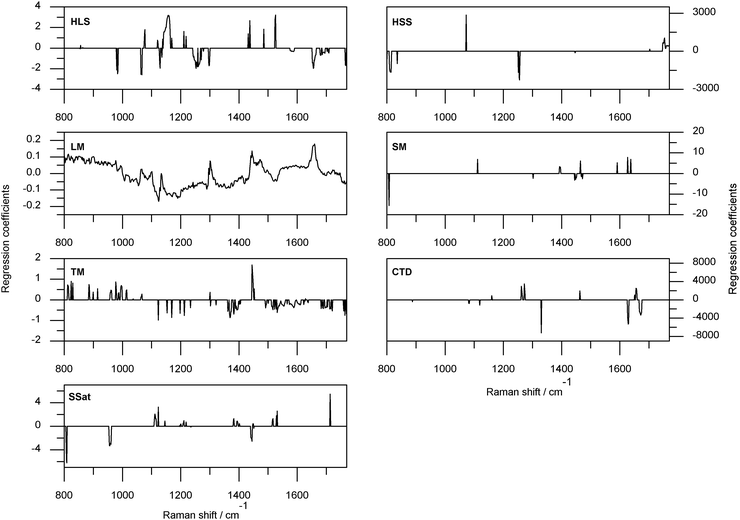 | ||
| Fig. 7 Regression coefficients for the highest ranked calibration treatments when sorted individually for each triacylglycerol class. See Table 6 for details of the calibration treatment. | ||
Regression coefficient analysis of the SSat, SM and LM calibrations did not explain the unreliable nature of predictions associated with these calibrations. The SSat TDC0/SNV/PLS1-JK regression coefficients are depicted in Fig. 7. In keeping with the saturated nature of the TG class spectral regions associated with CC functionality were free of significant variables, the remaining significant coefficients were associated with low intensity/shoulder regions of the AMF sample spectra. The TDC0/SNV/PLS1-JK regression coefficients for SM had large negative coefficients associated with variables about 807 cm−1, and coefficients in δ(CH2)tw, δ(CH2)sc, ν(C–C) and conjugated ν(CC) regions. Of the TG classes only LM ranked a method of analysis other than PLS1-JK as best. The calibration treatment TDC0/SNV/PLS2 regression coefficients have positive coefficients with the cisν(CC), δ(CH2)tw and δ(CH2)sc modes, and the solid state regions about 1060, 1100 1130 cm−1 were distinguishable.
Conclusions
It was shown that solid fat content (SFC) of anhydrous milk fat may be reliably quantified from physically heterogeneous samples; samples do not need to be heated so that they are completely liquid. Reliable quantification using a single method of calibration (temperature of data collection, preprocessing and partial least squares method) was possible for all SFC levels. In contrast, reliable quantification of all triacylglycerol (TG) classes considered was not possible using a single method of calibration and indeed reliable prediction was not possible using Raman spectral data for all examined TG classes. In general, quantification of TG class concentrations was optimal when the samples were liquid.Raman spectroscopy is a viable alternative to NMR and HPLC for SFC and some TG class concentration measurement. In the case of SFC measurement, Raman spectroscopy has the added appeal that it may be applied to samples at room temperature with little sample preparation.
Acknowledgements
The authors wish to thank Lucy Meagher and Susan Lane of Fonterra Research Centre for the provision of samples and related data. Cushla McGoverin wishes to thank the Tertiary Education Commission for a Top Achiever Doctoral scholarship. The authors also wish to thank the reviewers for their insightful comments.References
- H. Abbas, M. M. Hossain and X. D. Chen, Sep. Purif. Technol., 2006, 48, 167–175 CrossRef CAS.
- L. P. Meagher, S. E. Holroyd, D. Illingworth, F. van de Ven and S. Lane, J. Agric. Food Chem., 2007, 55, 2791–2796 CrossRef CAS.
- J. C. Rodrigues, A. C. Nascimento, A. Alves, N. M. Osorio, A. S. Pires, J. H. Gusmao, M. M. R. da Fonseca and S. Ferreira-Dias, Anal. Chim. Acta, 2005, 544, 213–218 CrossRef CAS.
- M. C. M. Gribnau, Trends Food Sci. Technol., 1992, 3, 186–190 CrossRef CAS.
- N. P. Robinson, M. Y. Blair and A. K. H. MacGibbon, NZDRI Report FC95R16, Palmerston North, 1995 Search PubMed.
- M. J. Auldist, B. J. Walsh and N. A. Thomson, J. Dairy Res., 1998, 65, 401–411 CrossRef CAS.
- J. R. Beattie, S. E. J. Bell, C. Borgaard, A. M. Fearon and B. W. Moss, Lipids, 2004, 39, 897–906 CrossRef CAS.
- F. R. van de Voort, K. P. Memon, J. Sedman and A. A. Ismail, J. Am. Oil Chem. Soc., 1996, 73, 411–416 CAS.
- P. Williams, 13th Australian Near Infrared Spectroscopy Conference, Hamilton, Victoria, Australia, 2008 Search PubMed.
- A. K. H. MacGibbon and W. D. McLennan, New Zeal. J. Dairy Sci., 1987, 22, 143–156 Search PubMed.
- S. L. Howell, K. C. Gordon and J. J. McGarvey, J. Phys. Chem. A, 2005, 109, 2948–2956 CrossRef CAS.
- S. L. Howell, B. J. Matthewson, M. I. J. Polson, A. K. Burrell and K. C. Gordon, Inorg. Chem., 2004, 43, 2876–2887 CrossRef CAS.
- M. Pirzer and J. Sawatzki, US Pat., 7 359 815, 2006.
- M. Pirzer and J. Sawatzki, US Pat., 0 212 275, 2006.
- G. R. Flaten and A. D. Walmsley, Analyst, 2003, 128, 935–943 RSC.
- K. H. Esbensen, Multivariate data analysis - in practice, CAMO Process AS, Oslo, 5th edn, 2004, ch. 6, pp. 137–145, ch.14, pp. 327–332 Search PubMed.
- K. R. Beebe, R. J. Pell and M. B. Seasholtz, Chemometrics: A Practical Guide, 1998, ch. 4, pp. 93–94 Search PubMed.
- J. R. Beattie, S. E. J. Bell, C. Borgaard, A. Fearon and B. W. Moss, Lipids, 2006, 41, 287–294 CrossRef CAS.
- H. Sadeghi-Jorabchi, P. J. Hendra, R. H. Wilson and P. S. Belton, J. Am. Oil Chem. Soc., 1990, 67, 483–486 CrossRef CAS.
- G. L. Johnson, R. M. Machado, K. G. Freidl, M. L. Achenbach, P. J. Clark and S. K. Reidy, Org. Process Res. Dev., 2002, 6, 637–644 Search PubMed.
- R. C. Barthus and R. J. Poppi, Vib. Spectrosc., 2001, 26, 99–105 CrossRef CAS.
- S. F. Parker, S. M. Tavender, N. M. Dixon, H. Herman, K. P. J. Williams and W. F. Maddams, Appl. Spectrosc., 1999, 53, 86–91 CrossRef CAS.
- L. Rimai, R. G. Kilponen and D. Gill, J. Am. Chem. Soc., 1970, 92, 3824–3825 CrossRef CAS.
- G. F. Bailey and R. J. Horvat, J. Am. Oil Chem. Soc., 1972, 49, 494–498 CrossRef CAS.
- H. Sadeghi-Jorabchi, R. H. Wilson, P. S. Belton, J. D. Edwards-Webb and D. T. Coxon, Spectrochim. Acta, Part A, 1991, 47, 1449–1458 CrossRef.
- G. Zerbi, G. Conti, G. Minoni, S. Pison and A. Bigotto, J. Phys. Chem., 1987, 91, 2386–2393 CrossRef CAS.
- Y.-M. Weng, R.-H. Weng, C.-Y. Tzeng and W. Chen, Appl. Spectrosc., 2003, 57, 413–418 CrossRef CAS.
- H. Susi, J. Sampugna, J. W. Hampson and J. S. Ard, Biochemistry, 1979, 18, 297–301 CrossRef CAS.
- B. Chmielarz, K. Bajdor, A. Labudzinska and Z. Klukowska-Majewska, J. Mol. Struct., 1995, 348, 313–316 CrossRef CAS.
- R. Karoui, A. M. Mouazen, E. Dufour, L. Pillonel, E. Schaller, J. De Baerdemaeker and J.-O. Bosset, Int. Dairy J., 2006, 16, 1211–1217 CrossRef CAS.
- M. Gniadecka, P. A. Philipsen, S. Sigurdsson, S. Wessel, O. F. Nielsen, D. H. Christensen, J. Hercogova, K. Rossen, H. K. Thomsen, R. Gniadecki, L. K. Hansen and H. C. Wulf, J. Invest. Dermatol., 2004, 122, 443–449 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |