Determination of total selenium by multicommutated-flow hydride generation atomic absorption spectrometry. Application to cow's milk and infant formulae
Mariela
Pistón
,
Javier
Silva
,
Ramiro
Pérez-Zambra
and
Moisés
Knochen
*
Universidad de la República (UdelaR), Facultad de Química, Cátedra de Química Analítica. Av. Gral. Flores 2124, Casilla 1157, 11800, Montevideo, Uruguay. E-mail: mknochen@fq.edu.uy
First published on 6th October 2009
Abstract
A multicommutated flow system was designed and evaluated for the determination of selenium by hydride generation atomic absorption spectrometry (HG-AAS). It was applied to the determination of total selenium in samples of cow's milk (fluid and powder) and infant formulae. Linearity was satisfactory in the range up to 27.5 µg L−1 (h = 0.082 C + 0.0033, h = peak-height, absorbance, C = concentration in µg L−1, r2 = 0.999). Detection (3s) and quantification (10s) limits in solution were LD = 0.08 µg L−1 and LQ = 0.27 µg L−1, corresponding to LD = 3.2 µg kg−1 and LQ = 10.8 µg kg−1 in solid samples, and to LD = 0.8 µg L−1, LQ = 2.7 µg L−1 in fluid milk samples. Trueness was verified by analysis (n = 5) of two reference materials (NIST 1549, Non-fat Milk Powder and NIST 1846 Infant Formula). At the 95% significance level, results were statistically equivalent to the certified values. Instrumental precision (sr(%), n = 5) was in the range 1.4% to 11.7%, analytical precision (sr(%), n = 5) being 4.2 and 9.3% respectively for the determination of the above mentioned reference materials. The sampling frequency of the system was 160 hour−1.
Introduction
In the last few decades considerable interest has arisen concerning the role of some trace elements in relation to nutrition and health. It has been proposed that selenium deficiencies are related to some medical conditions such as the Keshan Disease (a heart affection), the Kashin-Beck Disease (an osteoarthropathy) and Myxedematous Endemic Cretinism.1,2 Selenium is an essential component of enzymes such as glutathione peroxidase which is found in human tissues.3The RDA (Recommended Dietary Allowance) accepted value in the USA ranges from 20 µg/day for children under 3 up to 55 µg/day for adults. These values can be increased to 60 µg/day for pregnant women and 70 µg/day during lactation.1 These nutritional requirements should be satisfied by food and water ingested by the individual. Milk is especially important among foods because of its contribution in the nutrition of infants and young children.
Selenium content in cow's milk samples is in the µg L−1 range. For instance, an investigation involving milk from 15 countries produced values ranging from 3 to 40 µg L−1 with a mean value of 10 µg L−1.4
The determination of total selenium in milk at the trace and ultra trace levels is often carried out by electrothermal atomic absorption spectrometry (ET-AAS).5,6 Another popular technique is hydride-generation atomic absorption spectrometry (HG-AAS).7 This technique is preferable since it provides a degree of separation of the analyte from the matrix thus reducing the effects of a number of interferences. Hydride generation (HG) can also be coupled to atomic fluorescence spectrometry (AFS) when lower detection limits are required, however AFS detection systems are less readily available in many laboratories.
Hydride generation can be carried out batch-wise, or alternatively mechanised or even automated employing different flow techniques. Continuous-flow analysis employing a multichannel peristaltic pump has been used in several commercial hydride-generation accessories. Its main advantage is its inherent simplicity, but it also suffers from a high consumption of sample and reagents, which are fed continuously as long as the pump is on. However it is routinely used by many researchers.8 Flow injection analysis (FIA)9,10 is a useful automation technique that can be used for hydride generation. Several papers have been published dealing with the use of FIA for the generation of selenium hydride in HG-AAS,11,12HG-AFS13 and to a lesser extent to atomic emission spectrometry (HG-ICP-OES).14,15 The subject of interfacing FIA and other flow techniques to different atomic spectrometric techniques, some of them involving hydride generation has also been the subject of two books.16,17
Multicommutated flow analysis (MCFA)18–21 is an emerging flow technique based on flow networks built around solenoid valves which can be commutated independently under computer control in order to perform specific tasks. The literature reflects the usefulness of the technique that has been applied to the determination of different analytes in a range of matrixes.22–28
However the authors so far have not found evidence of previous work on the application of multicommutated flow analysis for the generation of selenium hydride. Given the advantages presented by this technique, an analytical system was designed for the determination of selenium by hydride generation atomic absorption spectrometry. The method was validated and applied to the determination of samples of milk, milk powder and milk-based infant formula.
Experimental
Reagents
Sodium tetrahydroborate (Hydride-Generation grade) was obtained from Fluka. A 0.5 % (w/v) solution was prepared daily by dissolving the solid in 0.05% (w/v) NaOH. All other reagents were of analytical reagent grade.Purified water (ASTM Type I) was obtained from a Millipore (São Paulo, Brazil) Simplicity 185 purifier fed with glass-distilled water.
A 1000 mg L−1 selenium standard solution was prepared from selenium metal (Aldrich, 99.99%) dissolved in nitric acid and made up to volume with 10% (v/v) hydrochloric acid. An intermediate standard solution (0.8 mg L−1) was prepared daily by stepwise dilution with 1.5% (v/v) hydrochloric acid. Calibration solutions were prepared by dilution of the intermediate solution.
Samples
Samples of fluid milk, milk powder and infant formulae representative of the local market were obtained locally.Materials
All glassware was soaked overnight in 10% (v/v) nitric acid and then rinsed exhaustively with distilled water.The flow system (Fig. 1) was based upon a Gilson (Villiers-le-Bel, France) Minipuls 2 multichannel peristaltic pump fitted with either Tygon or Viton tubing.
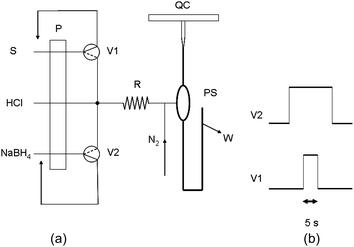 | ||
| Fig. 1 a: Flow system for the determination of total Se by multicommutated-flow HG-AAS. P: peristaltic pump. V1, V2: solenoid valves. R: reactor coil. PS: phase separator. QC: quartz cell. W: waste. S: sample, 7.2 mL min−1. HCl: 5% (v/v) HCl solution, 3.2 mL min−1. NaBH4: 0.5% (w/v) solution in 0.05% (w/v) NaOH, 1.7 mL min−1. b: Time sequence of activation of the solenoid valves V1 and V2 for one measurement cycle. | ||
Two 3-way 12 V solenoid valves (NResearch, West Caldwell, NJ, USA, model 161T031), were used for fluid control. These valves were driven by a lab-made interface built around a ULN-2803 integrated circuit which in turn was controlled by a personal computer via the LPT1 parallel port.
Connections and doubly-helical mixing coils were made from 0.8-mm internal diameter Teflon PFA tubing.
A U-shaped glass gravitational gas-liquid phase separator was built in the laboratory.
Nitrogen (dried and purified by a combined Drierite/molecular sieve trap) was used as carrier gas.
Measurements were carried out with a Perkin-Elmer (Norwalk, CT, USA) model 5000 atomic absorption spectrometer fitted with a 10-cm burner (air-acetylene flame) and a T-shaped quartzatomisation cell (Precision Glassblowing, Centennial, CO, USA) and operated at the 196.0 nm analytical line. The light source was a Photron (Narre Warren, Australia) Superlamp intensified emission hollow cathode lamp operated as recommended by the manufacturer.
The analytical signal (absorbance) was obtained from the analogue output connector of the spectrometer (1 V full scale) and digitised via a Measurement Computing, (Norton, MA, USA) 12-bit analogue to digital interface (model USB 1208LS) connected to a USB port and operated at a sampling rate of 1 s−1.
For the operation of the system, a programme was compiled in Visual Basic 6.0 (Microsoft) using the Softwire 3.1 (Measurement Computing) graphic programming environment. The programme controlled the timing and the activation of the solenoid valves using the parameters set up before the beginning of the analysis. This programme also handled the data acquisition. Analytical signals were presented on-screen in real time, scaled and saved to hard disk in ASCII format. Raw-data files were later processed with the Peak Simple programme (SRI, Torrance, CA, USA), which provided signal smoothing, baseline correction, peak-height measurement and hard copy printout.
Operation of the system (Fig. 1b) begins with the activation of V2 (NaBH4) for a total period of 15 s. 5 s after the activation of V2, V1 is activated for 5 s in order to introduce the sample segment and then turned off. Thus it is ensured that the NaBH4 solution flows before the beginning of the sample segment and after its end so that excess NaBH4 is available. When V2 is turned off, an additional period (7 s) with both valves off is allowed for the signal to return to the baseline. The total length of the analytical cycle is 22 s. The 5% (v/v) hydrochloric acid solution used to provide the acidic pH circulates continuously.
Calibration
Calibration solutions in the range 1.0 to 10.0 µg L−1 were prepared by accurately measuring 0.05, 0.1, 0.2, 0.3 and 0.4 mL aliquots of the 0.8 mg L−1 intermediate standard solution, to which 20 mL of water and 10 mL of concentrated hydrochloric acid was added. The mixture was heated on a hot plate for 1 hour at gentle boiling to carry out the pre-reduction of Se(VI) to Se(IV), then cooled down to room temperature and diluted to 30.0 mL with water.Sample preparation
Samples of milk powder or infant formula were prepared as follows: 0.50 g of milk powder was accurately weighed in a 30-mL screw-capped Teflon PFA vessel (Savillex, Minnetonka, MN, USA). Then 6 mL of concentrated nitric acid was added, the vessel was loosely capped, placed in a modified polypropylene “fast cooker” and heated in a household microwave oven (Ariston model MO991B). The microwave energy pattern of the oven was determined in previous work described elsewhere.29 The cooker was modified in order to vent all acid vapours and other gasesvia a piece of tubing to a flask containing sodium hydroxide solution which acted as a trap for acidic vapours. The oven was programmed to heat for 5 minutes at 30% and then for 3 minutes at 40% of the maximum power. It was then cooled down to room temperature, 1 mL of 30% hydrogen peroxide was added and the vial (loosely capped) was heated again for 2 minutes at 40% power. Afterwards the contents of the vial was transferred quantitatively to a 50-mL Erlenmeyer flask containing 10 mL of 10% (w/v) sulfamic acid solution and 10 mL of concentrated HCl, the pre-reduction step was carried out by heating at gentle boiling on a hot plate for 1 hour, and then cooled down to room temperature and diluted with water to 20.0 mL.Samples of fluid milk (2.0 mL) were mixed in a 50-mL Erlenmeyer flask with 5 mL of aqua regia (HCl/HNO3 3 + 1 by volume), diluting to 10.0 mL with water and sonicating for 20 minutes. Afterwards pre-reduction was carried out by adding 10 mL concentrated HCl, 0.5 mL of butanol (antifoam agent), 10 mL of 10% (w/v) sulfamic acid solution and heating the mixture at gentle boiling on a hot plate for 1 hour, then cooling down to room temperature and diluting with water to 20.0 mL. The resulting solution was filtered by a 25-mm diameter, 0.45-µm PTFE membrane filter.
Standard additions on fluid-milk samples were carried out on 2.0-mL aliquots of the sample by adding 0.1 mL of a 0.8-mg L−1 selenium standard solution, 5 mL of aqua regia and diluting with water to a final volume of 10.0 mL. The mixture was then processed as described in the preceding paragraph.
In order to estimate the contamination introduced by the sample preparation process, reagent blanks were measured alongside the samples in all mineralisation and pre-reduction steps.
Results and discussion
Optimisation
The influence of three variables (concentration of HCl and NaBH4 and length of the mixing coil R) was studied by means of a three-level central composite design. Maximum sensitivity (calibration slope), good precision and linearity were found when using 5% (v/v) HCl, 0.5% (w/v) NaBH4 and a 100-cm coil.Optimisation of flow rates was carried out by a second experimental design where the influence of 2 variables (NaBH4 and HCl flow rates) was studied at 3 levels. Optimum values were 1.7 mL min−1 (NaBH4 solution) and 3.2 mL min−1 (HCl solution).
The influence of the carrier gas flow rate was studied in the range 0.21 to 1.6 L min−1 (Fig. 2). A value of 0.32 L min−1 was adopted as it provided maximum signal and precision.
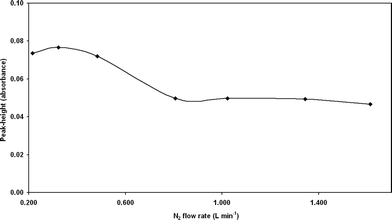 | ||
| Fig. 2 Signal (peak height, absorbance) variation with nitrogen flow rate. | ||
Sample volume was selected by varying the sampling time. Response (peak height, absorbance) increased from 0.061 to 0.106 when increasing the sample size from 0.24 to 0.60 mL, and did not vary significantly when increasing the sample volume to 1.20 and 2.40 mL. Thus a sample volume of 0.6 mL (corresponding to a sampling time of 5 seconds) was chosen because it allowed the highest sampling frequency, which was, under the final operating conditions, 160 hour−1.
In order to minimise the carryover when changing samples, a purge routine was devised using software.
Typical signals (calibration curve, blank and samples) can be seen in Fig. 3.
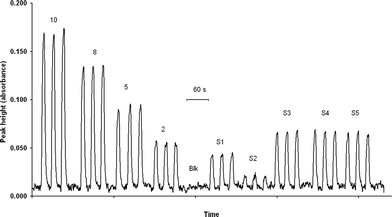 | ||
| Fig. 3 Typical signal (absorbance) obtained for triplicate injections of a calibration curve (10, 8, 5 and 2 µg L−1), blank (Blk) and 5 samples (S1 to S5). | ||
Interferences
Transition metals, especially those belonging to the fourth period may interfere with the determinations of Se by HG-AAS. The influence of concomitants affects either the efficiency of the hydride generation or the release of the hydride. Besides, concomitants are usually in large excess with respect to the analyte.Of the potentially interfering elements, only copper, iron and zinc can be found at significant levels in milk and infant formulae. An experiment was carried out to verify possible interferences.
Two different Se (VI) standard solutions (1 and 10-µg L−1) were prepared with and without the potential interferents (Cu 0.13 µg L−1, Fe 1.6 µg L−1, Zn 1.5 µg L−1), prereduced and injected in the system. These concentrations correspond to those produced by typical samples of Infant Formula processed according to the procedure used in this work and were chosen because this is the most unfavourable conditions set, as fluid milk and milk powder have lower concentrations of the three metals.
The experiment was repeated with 10 times higher concentrations of Cu, Fe and Zn. Reagent blanks were also run. Variations in net signal amplitude were in all cases less than 8% compared to the standard Se solutions. This figure is non-significant when compared to the repeatability precision of the method, thus no evidence of this kind of interference was found.
Validation
For the evaluation of linearity, a blank and 8 standard solutions in the range 1–27.5 µg L−1 were measured (n = 5) and the results were plotted as a function of the concentration. Linearity of the resulting curve (h = 0.082 C + 0.0033, h = peak-height, absorbance, C = concentration in µg L−1, r2 = 0.999) was confirmed by visual inspection of the plot and analysis of residuals.Detection (LD, 3s) and quantification (LQ, 10s) limits were estimated by measuring (n = 10) the dispersion of the blank signal and referring the measurements to the calibration curve. Estimated limits were LD = 0.08 µg L−1 and LQ = 0.27 µg L−1. These values correspond to LD = 3.2 µg kg−1, LQ = 10.8 µg kg−1 in solid samples and LD = 0.8 µg L−1, LQ = 2.7 µg L−1 in fluid milk samples.
In order to establish the trueness and precision of the method, two different standard reference materials, namely NIST 1549 (Non-Fat Milk Powder) and NIST 1846 (Infant Formula) as well as 7 samples of fluid milk were analysed 5 times each. Results obtained for the reference materials (Table 1) were statistically equivalent to the certified values.
Since reference materials or alternative reference values were not available for the fluid milk samples, it was decided to investigate the possible existence of multiplicative interferences by comparing the slope of the calibration curve with that of the standard additions curve by means of statistical hypothesis testing. It was found that some of the samples showed significant differences in the slopes, suggesting the existence of interferences. For this reason it was decided that for all fluid milk samples without exception the calibration would be made by means of the standard additions method.
Having decided this issue, the trueness of the method was verified for fluid milk samples by a spike/recovery approach. Each one of seven samples of fluid milk was spiked with Se(VI) additions at two different levels (equivalent to about 30 and 60 µg L−1 in the sample), processed as explained under “Sample preparation” above and injected 5 times in the flow system. Percent recoveries (100 × found/added) were calculated. Mean percent recovery ± standard deviation for the 7 samples was 97.8 ± 5.2% for the first spike and 102.2 ± 5.3% for the second one. These values were compared with the nominal value of 100% by means of the Student's t-test, finding t-values of 1.112 and −1.116 for the two spike levels. The value of t(0.05,6) is 2.447, thus it may be concluded that at the 95% significance level recoveries do not differ significantly from 100% and the trueness of the method is ensured for fluid milk samples.
Precision (sr(%)) was estimated by both instrumental and analytical repetition. Repeatability (n = 5) for the various levels of a calibration curve (1.4–27.5 µg L−1) was in the range 1.4% (for the highest concentration) to 11.7% (for the lowest one).
Analytical repeatability (sr(%)) for the analysis of the reference materials (n = 5) was 4.2% for NIST 1549 and 9.3% for NIST 1846.
With the operating conditions chosen the sampling frequency was 160 hour−1.
Sample mineralisation
For the digestion of solid samples two approaches were investigated, namely heating using a dry block heater and using a microwave oven.For the first approach, 1 g of sample was digested with 6 mL HNO3 in a Kjeldahl tube placed in a dry block heater set at 120 °C. When brown fumes appeared, 2 mL of H2O2 were added and digestion continued until a clear digest was obtained. This process required about 6 hours. The sample was then ready for pre-reduction.
Digestion in a microwave oven was carried out as described under “Sample preparation” above.
Both procedures allowed recoveries close to 100% to be attained but the use of microwave heating was much faster, hence it was adopted for the digestion of solid samples.
Given the low Se concentrations, none of these procedures was appropriate for mineralisation of fluid milk samples, as they would require inordinately large amounts of sample and reagents, producing projections and other problems during the digestion. For this reason a different procedure using ultrasound, based on work by Cava-Montesinos et al.13 was used for fluid milk samples. This procedure is described under “Sample preparation”.
The literature30 mentions the interference on the generation of hydrides produced when using HNO3 during the digestion step. This interference has been attributed either to the HNO3 itself, or more recently to nitrogen oxides generated during the digestion step. The existence of this interference was confirmed in this work and reflected in poor recoveries. With the aim to reduce that interference, the addition of sulfamic acid as proposed by Lopes Nunes et al.30 was investigated. Recoveries close to 100% were obtained, thus this reagent was adopted for use during sample preparation.
Application to commercial samples
The method was applied to the determination of selenium in samples of fluid milk (10 samples), milk powder (17 samples) and infant formulae (6 samples). Mean values for fluid milk were 19.6 µg L−1 (range 11.5–34.1 µg L−1) and for milk powder 73.6 µg kg−1 (range 31–130 µg kg−1) expressed on the dry basis.Commercial samples of milk-based infant formulae of six different brands representative of the Uruguayan market were purchased locally, four of them for infants from 0–6 months and 6–12 months and two generic ones. Results obtained for these samples ranged from 42 to 138 µg kg−1 (dry basis) and are presented in Table 2. These results were in agreement within 10% with the label claims.
| Total Se (µg kg−1) | Sample | |||||
|---|---|---|---|---|---|---|
| A | B | C | D | E | F | |
| Label claim | 150 | 150 | ND | ND | 40 | 120 |
| Found ± s | 138 ± 10 | 135 ± 3 | 84 ± 6 | 56 ± 14 | 42 ± 2 | 116 ± 3 |
| Relative difference (%) | 8 | 10 | — | — | 4.8 | 3.3 |
Comparison with other methods
The proposed method based on the multicommutated flow system was compared with other popular methods/techniques used for the determination of selenium in milk, namely ET-AAS, continuous flow HG-AAS and FIA-HG-AAS.Detection limits in whole milk (fluid) found in this work (0.8 µg L−1) are similar or better with those found by ET-AAS5,6 (2.5 µg L−1, 0.35–0.70 µg L−1) and HG-AAS8 (0.95 µg L−1). Analytical throughput (sampling frequency = 160 samples per hour) is much higher than that attainable by ET-AAS and comparable to FIA-HG-AAS. The method can be run in a flame AAS spectrometer that is less expensive and more easily found in laboratories than a graphite furnace (ET-AAS) spectrometer.
When compared with continuous flow HG-AAS and FIA-HG-AAS, the use of multicommutation provided several advantages such as low reagent and sample consumption (0.05 mL concentrated HCl, 2.5 mg NaBH4 and 0.6 mL of sample per measurement) and less generation of chemical residues.
Conclusion
Multicommutated flow analysis could be successfully used for the generation of selenium hydride and applied to the determination of selenium by HG-AAS in samples of cow's milk (fluid and powder) and infant formulae. The method based on such a system was validated and found to be accurate and precise enough for the determination of total selenium content in that type of sample.This technique is very fast, and flexible since several modifications can be carried out simply by changing parameters in the control software without physical modifications of the system. For example, the sample volume can be easily changed by modifying the time a given solenoid valve is energised thus allowing for different concentration ranges.
The sample preparation procedures developed were fast and simple.
The system was applied to the determination of total selenium in samples of cow's milk (fluid and powder) from the Uruguayan market. Results found were in agreement with those published in the literature for samples from other countries. The method could be used for the quality control of infant formulae.
Acknowledgements
The authors thank UdelaR-CSIC for a research grant, PEDECIBA-Química for partial support and Prof. Dr. Luis Barros (UdelaR-Faculty of Veterinary) for providing samples of fluid milk.References
- National Institutes of Health (USA), Office of Dietary Supplements. “Dietary Supplement Fact Sheet: Selenium”. http://dietary-supplements.info.nih.gov/factsheets/selenium.asp (accessed November 27th, 2008) Search PubMed.
- C. D. Thomson, Eur. J. Clin. Nutr., 2004, 58, 391–402 CrossRef CAS.
- F. Fordyce, in Essentials of Medical Geology. Impacts of the Natural Environment on Public Health, ed. O. Selinus, B. Alloway, J. A. Centeno, R. B. Finkelman, R. Fuge, U. Lindh, P. Smedley, Elsevier, Burlington, MA, USA, 2005, ch. 15, pp. 373–415 Search PubMed.
- A. Flynn and K. Cashman, in Advanced Dairy Chemistry, ed. P. F. Fox, Chapman & Hall, London, 2nd edition, 1997, vol. 3, ch. 7, pp. 280–283 Search PubMed.
- P. C. Aleixo and J. A. Nóbrega, Food Chem., 2003, 83, 457–462 CrossRef CAS.
- A. Paiva Oliveira, J. A. Gomes Neto, J. Araújo Nóbrega, P. R. Miranda Correia and P. V. Oliveira, Food Chem., 2005, 93, 355–360 CrossRef CAS.
- J. Dedina, in Flow Analysis with Atomic Spectrometric Detectors, ed. A. Sanz-Medel, Elsevier, Amsterdam, 1999, ch. 8, pp. 237–273 Search PubMed.
- P. Van Dael and D. V. Barclay, Food Chem., 2006, 96, 512–518 CrossRef CAS.
- J. Ruzicka and E. Hansen, Flow Injection Analysis, Wiley, New York, 2nd edn., 1989 Search PubMed.
- M. Trojanowicz, Flow Injection Analysis – Instrumentation and Applications, World Scientific, Singapore, 2000 Search PubMed.
- O. Muñiz-Naveiro, R. Domínguez-González, A. Bermejo-Barrera, J. A. Cocho, J. M. Fraga and P. Bermejo-Barrera, Anal. Bioanal. Chem., 2005, 381, 1145–1151 CrossRef CAS.
- Y. Zhang and S. B. Adeloju, Talanta, 2008, 76, 724–730 CrossRef CAS.
- P. Cava-Montesinos, M. L. Cervera, A. Pastor and M. De la Guardia, Talanta, 2004, 62, 175–184 CrossRef CAS.
- S. Moyano, J. A. Gásquez, E. Marchevsky, R. Olsina and L. D. Martinez, At. Spectrosc., 1997, 18, 152–155 CAS.
- N. Etxebarria, R. Antolín, G. Borge, T. Posada and J. C. Raposo, Talanta, 2005, 65, 1209–1214 CrossRef CAS.
- Flow Injection Atomic Spectroscopy, ed. J. L. Burguera, Dekker, New York, 1989 Search PubMed.
- Flow Analysis with Atomic Spectrometric Detectors, ed. A. Sanz-Medel, Elsevier, Amsterdam, 1999 Search PubMed.
- B. F. Reis, M. F. Giné, E. A. G. Zagatto, J. L. C. Lima and R. A. Lapa, Anal. Chim. Acta, 1994, 293, 129–138 CrossRef CAS.
- F. R. P. Rocha, B. F. Reis, E. A. G. Zagatto, J. L. F. C. Lima, R. A. S. Lapa and J. L. M. Santos, Anal. Chim. Acta, 2002, 468, 119–131 CrossRef CAS.
- M. Catalá Icardo, J. V. García Mateo and J. Martínez Calatayud, TrAC, Trends Anal. Chem., 2002, 21, 366–378 CrossRef CAS.
- M. A. Feres, P. R. Fortes, E. A. G. Zagatto, J. L. M. Santos and J. L. F. C. Lima, Anal. Chim. Acta, 2008, 618, 1–17 CrossRef CAS.
- F. R. P. Rocha and B. F. Reis, Anal. Chim. Acta, 2000, 409, 227–235 CrossRef CAS.
- J. A. V. Prior, J. L. M. Santos and J. L. F. C. Lima, J. Pharm. Biomed. Anal., 2003, 33, 903–910 CrossRef CAS.
- B. F. Reis, M. Knochen, G. Pignalosa, N. Cabrera and J. Giglio, Talanta, 2004, 64, 1220–1225 CrossRef CAS.
- M. Knochen, M. Pistón, L. Salvarrey and I. Dol, J. Pharm. Biomed. Anal., 2005, 37, 823–828 CrossRef CAS.
- M. Pistón, I. Dol and M. Knochen, J. Autom. Methods Manage. Chem., 2006 DOI:10.1155/JAMMC/2006/47627.
- C. Pons, R. Forteza, A. O. S. S. Rangel and V. Cerdà, TrAC, Trends Anal. Chem., 2006, 25, 583–588 CrossRef CAS.
- A. Sixto and M. Knochen, Talanta, 2009, 77, 1534–1538 CrossRef CAS.
- G. Pignalosa, N. Cabrera, A. Mollo, I. Portillo, V. Rouco and L. Vázquez, Spectrochim. Acta, Part B, 2001, 56, 1995–1999 CrossRef.
- D. Lopes Nunes, E. Pereira dos Santos, J. Smanioto Barin, S. R. Mortari, V. L. Dressler and E. M. de Moraes Flores, Spectrochim. Acta, Part B, 2005, 60, 731–736 CrossRef.
- E. Ródenas-Torralba, P. Cava-Montesinos, A. Morales-Rubio, M. L. Cervera and M. de la Guardia, Anal. Bioanal. Chem., 2004, 379, 83–89 CAS.
| This journal is © The Royal Society of Chemistry 2009 |