High throughput single molecule detection for monitoring biochemical reactions
Paul I.
Okagbare
a and
Steven A.
Soper
*abc
aDepartment of Chemistry, Louisiana State University, Baton Rouge, LA 70803, USA
bDepartment of Mechanical Engineering, Louisiana State University, Baton Rouge, LA 70803, USA
cCenter for BioModular Multi-Scale Systems, Louisiana State University, Baton Rouge, LA 70803, USA
First published on 24th November 2008
Abstract
The design, performance and application of a novel optical system for high throughput single molecule detection (SMD) configured in a continuous flow format using microfluidics is reported. The system consisted of a microfabricated polymer-based multi-channel fluidic network situated within the optical path of a laser source (λex = 660 nm) with photon transduction accomplished using an electron-multiplying charge coupled device (EMCCD) operated in a frame transfer mode that allowed tracking single molecules as they passed through a large field-of-view (FoV) illumination zone. The microfluidic device consisted of 30 microchannels possessing dimensions of 30 µm (width) × 20 µm (depth) with a 25 µm pitch. Individual molecules were electrokinetically driven through the fluidic network and excited within the wide-field illumination area with the resulting fluorescence collected via an objective and imaged onto the EMCCD camera. The detection system demonstrated sufficient sensitivity to detect single DNA molecules labeled with a fluorescent tag (AlexaFluor 660) identified through their characteristic emission wavelength and the burst of photons produced during their transit through the excitation volume. In its present configuration and fluidic architecture, the sample processing throughput was ∼4.02 × 105 molecules s−1, but could be increased dramatically through the use of narrower channels and a smaller pitch. The system was further evaluated using a single molecule-based fluorescence quenching assay for measuring the population differences between duplexed and single-stranded DNA molecules as a function of temperature for determining the duplex melting temperature, Tm.
Introduction
The ability to process large amounts of information for high throughput applications has become a primary ambition due in part to the large number of samples that must be interrogated and the complexity of those samples. An example where high throughput data processing is significant is in the drug discovery pipeline (i.e., high throughput screening) where combinatorial libraries consisting of millions of potential small molecule drug candidates are screened for their interaction with therapeutic targets using fluorescence spectroscopy as the readout modality.1–6An approach toward increasing the data production rate for processing biochemical reactions is to increase the number of reactions run in parallel, which allows for analyzing a large number of targets simultaneously. This parallel processing is commonly carried out in titer plates, with increases in the well density used to increase data throughput. For example, titer plates can be configured to run 96–9600 reactions in parallel on a 3″ × 5″ plate.7,8 With increases in well density also comes reductions in well volume, placing severe demands on the readout hardware to provide parallel detection to further increase throughput while at the same time providing high detection sensitivity and favorable limits of detection in spite of the reduced well volume. Other applications that require the ability to transduce large numbers of targets quickly include capillary array electrophoresis,9 DNA microarrays,10 small-molecule arrays,11,12 and protein microarrays.13 In addition, micro-Total Analysis Systems (µ-TAS) have been employed as a tool for increasing throughput in many applications due to their exquisite characteristics conducive to high sampling rates brought about by reductions in analysis times and the parallelization of processing units.14–17
Another analytical technology, which facilitates the realization of high throughput biochemical data processing, is single molecule detection (SMD). SMD provides the ability to eliminate many sample processing steps associated with multi-step assays, significantly reducing assay time. This has recently been demonstrated by Wabuyele et al., where the overhead processing time associated with PCR amplification of target DNAs was eliminated by detecting single nucleotide polymorphisms (SNPs) in genomic DNA using single-pair FRET (spFRET).18 Using spFRET, the authors were able to detect SNPs in a processing time less than 5 min. Another advantage of SMD is the detailed information revealed when monitoring biochemical reactions. For example, it can yield insight into complex fluctuation phenomena and hidden heterogeneity that cannot be observed in ensemble techniques.19
However, the challenge associated with many SMD assays is their intrinsic low throughput, especially for flow-based SMD experiments where a laser focused to a diffraction-limited spot is used to interrogate material as it traverses through the excitation volume.20,21 To realize the full processing potential of SMD for high throughput applications, the readout hardware needs to be configured in a parallel format without sacrificing sensitivity.
The common approach for realizing high single molecule sensitivity is, as stated above, the utilization of ultra-small probe volumes, which reduces background interferences resulting from the sample matrix emanating from autofluorescence or scattering.22,23 A common method for achieving ultra-small probe volumes is the use of confocal optics,24–27 in which the laser beam is focused to a diffraction-limited area producing sampling volumes in the sub-picoliter regime. However, these confocal sampling volumes are usually designed with single element detectors and, thus, generate small sample processing throughput rates.
This limitation can be addressed by operating the readout system in a multi-channel configuration, such that single molecules are transported through a series of fluidic processors that are interrogated using wide-field illumination with the resulting fluorescence imaged onto an array detector for parallel readout. An array detector that can be employed for fluorescence imaging appropriate for single molecule high throughput processing is the charge coupled device (CCD).28 Advancements in CCD technology has made available ultra-sensitive cameras appropriate for realizing detection of single fluorescence molecules in flow-based measurements where the signal is transitory in nature.29–32 For example, Emory and Soper33 recently described a CCD-based single molecule analysis system in which a flowing sample composed of double-stranded DNA molecules labeled with an intercalating dye (TOTO-3) was imaged onto a 1340 × 100 pixel CCD camera operated in a time-delayed integration (TDI) mode.33 The TDI mode was used to collect photo-generated signal from single multiply-labeled DNA molecules into a potential well of the CCD array as the molecules traversed the field-of-view (FoV), thereby enhancing the signal-to-noise ratio (SNR) and at the same time increasing the duty cycle of the detection system. Van Orden et al. also demonstrated the ability to image single DNA molecules labeled with an intercalating dye using a CCD operated in a snap-shot mode.34
While the aforementioned works are excellent examples of increasing the sample throughput for flow-based SMD experiments, the limitations are that the single molecules were double-stranded DNAs loaded with multiple fluorophores. In the case of λ-DNA (∼48![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 bp), each DNA molecule had a load of approximately 9700 dye molecules. Therefore, high throughput flow-based SMD systems have yet to demonstrate the ability to detect single fluorophore molecules.
500 bp), each DNA molecule had a load of approximately 9700 dye molecules. Therefore, high throughput flow-based SMD systems have yet to demonstrate the ability to detect single fluorophore molecules.
Another viable operational modality for a CCD camera directed toward SMD is the frame transfer mode (FTM) when the CCD contains an electron-multiplying stage (EMCCD), which allows for high speed imaging.35,36 For example, Li and Yeung utilized a frame transfer EMCCD to monitor dynamic conformational changes of flavin adenine dinucleotides (FADs) in different media on a cover slip in which the molecules were immobile. Their results demonstrated the ability to monitor the real-time dynamics of the photoproduct formation between FAD molecules at the single molecule level.37
The availability of FTM EMCCDs has created the capability to study biochemical reactions at the single molecule level utilizing flow-based measurements in which the single molecules are dynamically transported through the illumination zone. These EMCCDs are equipped with a full frame storage region and a charge multiplication register that generates secondary electrons via an impact-ionization process. The net gain on these cameras can be greater than 1000-fold. Although, the resulting gain has a complex relationship with the applied voltage, it can be estimated using the expression;
| G = (1 + g)N | (1) |
| St = S × QE × G | (2) |
In this paper, we combined the advantages of microfluidics with a wide-field fluorescence imaging system and present, for the first time, the ability to detect single chromophore molecules in a multi-channel flow-through configuration and monitor biochemical reactions at the single molecule/fluorophore level. The system reported herein provides high throughput single molecule processing capabilities using an ultra-sensitive epi-fluorescence imaging system consisting of an FTM EMCCD. The system possessed a large FoV (200 µm, diameter, generated by a 40×/0.75 microscope objective) capable of imaging a series of parallel fluidic channels simultaneously with an optical irradiance that was near saturation as opposed to our previous report using a TDI mode of operation for the CCD.33 As an example of the utility of this system to monitor biomolecular reactions at the single molecule level at relatively high processing rates, a melt analysis of double-stranded DNA (dsDNA) using a resonance energy transfer reporting system was used. The dsDNA contained a construct labeled on the 5′-end of the sense strand with a dye and a quencher on the 3′-end of the anti-sense strand. The number of photon burst events arising from melted duplexes at different temperatures could be used to determine the melt profile of this double-stranded complex.
Experimental
Optical system
The optical set-up for the flow-based multi-channel SMD is shown in Fig. 1a. An HL6535MG laser diode (Thorlabs, Newton, NJ, USA) provided excitation (660 nm) with a peak power of 30 mW. The laser diode was mounted on an LDM21 (Thorlabs) laser mount connected to a laser driver (LDC 205, Thorlabs) and a temperature controller unit (TED 200, Thorlabs). The laser output was collimated using an aspheric lens (C230TM-B, Thorlabs) and passed through a 660 nm laser line filter (660DF10, Omega Optical, Brattleboro, VT, USA) that was placed in the optical path of the excitation source. A dichroic mirror was used to direct the laser into a microscope objective (40×, 10× or 5×, Zeiss, Thornwood, NY, USA) and then, onto the sample after being shaped using a plano-convex lens (LA1509, focal length = 100 mm, Thorlabs). The shaping lens was used to generate a collimated light output from the objective with a diameter determined by the aperture of the objective; the diameter of the collimated light, which determined the FoV of the imaging system, was different for each microscope objective used in our experiments (0.2 mm for a 40×, NA = 0.75 objective; 0.8 mm for a 10×, NA = 0.55 objective; and 1.6 mm for a 5×, NA = 0.12 objective). The fluorescence emission from the sample was collected with the same objective from a PMMA fluidic chip and directed through the dichroic filter, then a band pass filter (719AF40, Omega Optical), and finally a long pass filter (690ALP, Omega Optical) to spectrally isolate the emission from scattered radiation. The isolated emission was then directed onto a reflecting surface of a mirror, which was placed at a 45° angle of incidence with respect to the optical path to direct the fluorescence onto the CCD camera. A plano-convex lens (AC254-100, focal length = 100 mm) was placed between the mirror and EMCCD camera, which generated a fluorescence image onto the CCD.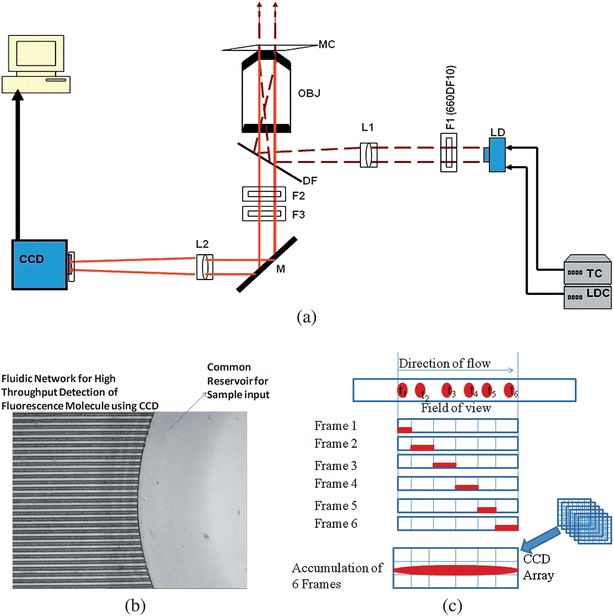 | ||
| Fig. 1 (a) Optical set-up of the imaging system using epi-illumination and employing a large FoV. The beam from the diode laser (λ = 660 nm) was isolated using a laser line filter (F1) and then shaped with a plano-convex lens (L1), which was used to focus the laser beam behind the input aperture of the microscope objective (OBJ). Following beam shaping, it was directed by a dichroic filter (DF) into the OBJ with the collimated laser beam impinging upon a multi-channel microchip (MC). The fluorescence signal generated from the chip was collected by this same objective, passed through the DF and spectrally selected using a long pass filter (F2) and an interference band pass filter (F3). A mirror (M) was used to steer the fluorescence signal onto a CCD after passing it through a lens (L2), which focused the radiation onto the photoactive area of the CCD. The total magnification of the system was 40×. (b) An optical micrograph showing a section of the multi-channel microfluidic chip; the fluidic network consisted of microchannels with dimensions of 30 µm wide × 20 µm deep and a pitch of 25 µm. All channels had a common sample input reservoir. The driving electric field was applied at this reservoir and another on the opposite end of the fluidic network (not shown) to drive the sample electrokinetically through the imaging area. (c) Diagram showing the operation of the CCD in a frame transfer mode with image accumulation occurring during single molecule travel within the FoV of the microscope. The images of single molecules produced streaks on the CCD due to the molecular transit time being greater than the CCD exposure time and multiple frames summed to produce the final image. | ||
The EMCCD camera (Cascade 1K, Photometrics, Tuscon, AZ, USA) was a frame transfer EMCCD equipped with signal enhancement via on-chip gain and possessed 8 µm × 8 µm pixels configured in a 1004 × 1002 front illumination format. The camera had a 10 MHz digital converter and a 16-bit digitization range. The frame transfer capability of the EMCCD allowed near 100% duty cycle when single molecules were tracked along their flow path within the illumination field. The CCD camera was thermoelectrically cooled to −30 °C to minimize dark noise and operated at the optimal gain to maximize the SNR in the measurement.
Multi-channel illumination
The optical system was configured in an epi-illumination format (see Fig. 1a) with beam shaping optics that generated a collimated output of the excitation source through the microscope objective for wide-field illumination as opposed to our previous work, which used a collimated laser beam of 200 µm diameter irradiating the fluidic chip from the side.33 The collimated beam was used to irradiate a series of parallel microchannels. The system was operated with a laser power of ∼25 mW generating an irradiance of 1.9 × 1020 photons cm−2 s−1 for the 40× objective. For a dye such as AlexaFluor 660 that possesses an absorption cross-section of 1.82 × 10−16 cm2 (molar absorptivity = 110000 cm−1 M−1) and a fluorescence lifetime of ∼1 ns, optical saturation occurs at an irradiance of ∼5.4 × 1024. Therefore, the system is currently being operated below optical saturation.
Microfluidic design
A PMMA microchip was designed for high throughput single molecule data processing that consisted of a series of flow channels tightly packed to provide the ability to image a number of single molecule experiments within the microscope's FoV (see Fig. 1b). The microfluidic devices were fabricated via direct milling of the fluidic vias into a PMMA wafer using a Kern MMP2522 micromilling machine (KERN micro-und Feinwerktechnick, GmbH & CO, Murnau Westried, Germany). The device consisted of a series of microchannels with dimensions of 30 µm in width, 20 µm in depth and a pitch of 25 µm. All channels possessed a common sample reservoir (see Fig. 1b). Following micromilling, the device was sonicated for 1 min in IPA and rinsed thoroughly with deionized water. A PMMA cover plate (125 µm thickness) was placed on the device to enclose the microfluidic channels. The chip assembly was clamped between two glass plates and annealed in a GC oven at 107 °C (slightly above the Tg of PMMA) for 22 min.Chemicals and reagents
Stock solutions of Tris-Taps-EDTA (TTE) buffer (10×) were diluted to obtain 1× TTE solutions (pH = 8.7) using nanopure water obtained from a Barnstead NANOpure System (Model D8991, Dubuque, IA). Fluorescence reagents used included AlexaFluor 660 and Syto-63 (Molecular Probes, Eugene, Oregon, USA). λ-DNA (48.5 kbp) was obtained from Molecular Probes and diluted in 1× TTE buffer to the required concentration after being labeled with Syto-63 at a molar ratio of 5 : 1 (bp : dye). Single-stranded DNA (ssDNA, 60 bases in length) were 5′-labeled with AlexaFluor 660 (Molecular Probes) and a stock solution (80 nM) was prepared using a 1× TTE buffer. Double-stranded DNA (dsDNA, 15 bp in length) was obtained from Molecular Probes as well; the duplex had the sense strand labeled with AlexaFluor 660 at its 5′-end while the complementary strand (anti-sense strand) was labeled with Black Hole Quencher-3 (BHQ-3) at is 3′-end. Stock solutions (0.4 µM) of the duplex were prepared in Tris/HCl buffer containing 20 mM Tris-HCl, 10 mM MgCl2 and 10 mM KCl at pH 7.5. Dilutions from this stock solution to the desired concentrations required for the single molecule experiments was accomplished with the same buffer. In all cases, buffer solutions were filtered through 0.2 µm filters prior to sample preparation.Stock solutions of λ-DNA molecules were labeled with an intercalating dye (Syto-63) and diluted in 1× TTE buffer to 10 pM. This concentration was selected to keep the single molecule occupancy probability low to reduce the double occupancy probability. With the system possessing a probe volume of 0.8 fL (CCD pixel size = 8 µm × 8 µm), a sample concentration of 100 pM produced a single molecule occupancy probability of 0.01 (probability of double occupancy = 0.0001).
Single molecule tracking via frame transfer EMCCD operation
The system was optimized for its single molecule sensitivity when operated in the frame transfer mode using λ-DNA (48.5 kpb) labeled with Syto-63 diluted in 1× TTE buffer. Single λ-DNA molecules were loaded into the common sample reservoir of the PMMA wafer and driven through the microfluidic channels by electrokinetic pumping using a high voltage power source (Spellman CZE1000R, Hauppauge, NY, USA). The movement of the molecules through the illumination zone generated streak images for each single DNA molecule when the system was operated in the frame transfer mode and multiple images could be combined. A schematic representation of this process (frame transfer with image accumulation) is shown in Fig. 1c. A streak image resulted from the collection of fluorescence signal from the same molecule into potential wells along a single column of the EMCCD as the molecule migrated through the FoV.The single fluorophore detection capability of the system was evaluated using DNA molecules that were end-labeled with a single AlexaFluor 660 dye molecule. In this case, only a single image frame was used for the presented data due to the short bleaching lifetimes associated with the single chromophore assemblies.
Bulk fluorescence measurements
Emission spectra of the duplexed DNA/AlexaFluor 660 constructs used for the bulk melt analyses were acquired using a FLUOROLOG-3 spectrofluorometer (Horiba Jobin Yvon, Edison, NJ). The spectrometer was equipped with a 450 W xenon lamp and a cooled Hamamatsu R928 extended-red photomultiplier that was operated at 900 V in the photon-counting mode. The cuvette was thermostatically controlled in order to provide the required temperatures for the melt analysis.Results and discussion
Multi-channel illumination and system optimization
The optical system was operated using epi-illumination (see Fig. 1a) with beam shaping of the laser source to generate a collimated light output from the microscope objective for wide-field illumination to provide the simultaneous excitation of fluorophores traversing through a series of channels contained within the objective's FoV. The channel dimensions of the fluidic network were 30 µm (width) × 20 µm (depth), with the depth selected to provide sufficient single molecule sampling efficiency (SE), which was determined by the depth of focus of the microscope objective used for collecting the fluorescence (1.4 µm for 40× objective; 6.6 µm for 10× objective; and 60.1 µm for the 5× objective), and the percentage of molecules traveling through the illumination zone, which in this case is near 100%. The 40× objective (NA = 0.75) was used for single fluorophore measurements due to its high collection efficiency in spite of its smaller depth of focus. Therefore, the SE for our system for single fluorophores was approximately 7% (100% × 1.4/20). However, we are not using a pinhole aperture in the secondary image plane of the relay objective, which will allow for a larger depth of focus for the optical system than that calculated above and as such, the SE determined above should be considered a minimum estimation only.The 40× objective generated a FoV of 200 µm, which allowed imaging five microchannels with the current fluidic architecture. Changing the imaging objective to a lower magnification would allow for the processing of more fluidic channels (see Table 1). For example, the use of a 5× objective would provide the ability to image 30 channels with the channel architecture used herein due to its larger FoV. However, the cost for this higher throughput is reductions in the collection efficiency of the resulting fluorescence emission, which could potentially reduce single molecule sensitivity. Clearly, reductions in the channel width and pitch could significantly improve the number of channels we could image. For example, using a microfluidic chip with a 1 µm wide channel and a pitch of 1 µm, which can be fabricated using optical lithography, would permit the ability to image 100 such channels for a FoV of 200 µm.
| Total magnification (magnification/numerical aperture) | Effective field-of-view (FoV) | Number of channels within the FoV (30 µm × 20 µm; 25 µm pitch) | Single molecule SNR for λ-DNA stained with Syto-63 | Theoretical single molecule throughput |
|---|---|---|---|---|
| 40×/0.75 | 200 µm | 5 | 810 | 6.7 × 104 |
| 10×/0.5 | 800 µm | 17 | 510 | 2.28 × 105 |
| 5×/0.25 | 1.6 mm | 30 | 314 | 4.02 × 105 |
Detection of single λ-DNA molecules
Experiments were first carried out at different electric field strengths to establish the transport velocity that would yield optimal signal-to-noise ratio (SNR) for the single molecules, which in this case were λ-DNA molecules stained with Syto-63, since the molecular transit time should be approximately equal to the photobleaching time to provide the highest SNR.38 Results from 10 different measurements were averaged and plotted versus the applied field strength, which generated an optimal field strength of ∼80 V/cm (see Fig. 2a). At this field strength, the linear velocity was 0.01 cm/s, which was based on the applied field strength and the electrophoretic mobility of λ-DNA loaded with Syto-63. At this linear velocity and FoV, the molecular transit time was estimated to be ∼1.6 s. We note that the processing rate is directly related to the flow velocity for flow-based measurements and as such, higher field strengths would result in higher system throughput. Even at field strengths of 300 V/cm (0.037 cm/s), we were able to generate sufficient SNR to detect single λ-DNA molecules.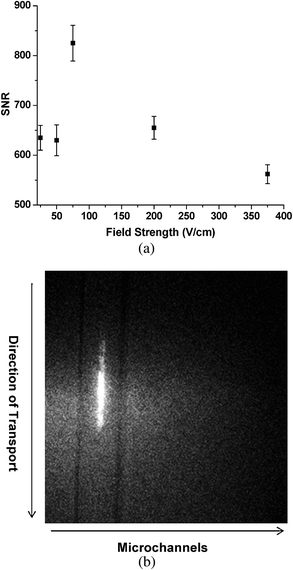 | ||
| Fig. 2 (a) Plot of the signal-to-noise ratio (SNR) at different field strengths used to transport single molecules through the irradiation zone of the imaging system. The SNR values were obtained from an average of 10 different measurements. (b) Fluorescence image from the CCD for a single molecule tracked along several pixels of the CCD, which was accumulated from two image frames that were acquired using 100 ms exposure time per frame with the fluorescence collected using a 40× objective (NA = 0.75). | ||
To demonstrate the ability to track single dsDNA molecules traveling electrokinetically through the irradiation zone, experiments were next carried out using single and multiple microchannels. Fig. 2b shows a fluorescence image of a single DNA molecule tracked along the FoV of the imaging system by accumulating two image frames with a 100 ms exposure time per frame. From the length of the streak (∼60 µm) and the image accumulation time (2 frames at 100 ms per frame), we estimated the linear velocity to be 0.03 cm/s, very close to that determined above at a field strength of 300 V/cm.
To evaluate the sample throughput (ST) capabilities of the system, a 100 pM solution of λ-DNA was electrokinetically driven through the system at a field strength of 300 V/cm and the fluorescence image was collected using the 10× objective (NA = 0.5); the FoV of the system was 800 µm, which allowed imaging 17 channels. With the system operated in a frame transfer mode, the SMD sensitivity was improved by using 3× binning (3 × 3) with the multiplication gain of the EMCCD set at 3700. Fig. 3a shows one image frame from a series of images acquired for single λ-DNA molecules stained with Syto-63 traveling through the illumination zone. Each fluorescent streak represents an individual DNA molecule migrating through the chip due to the low probability of double occupancy per imaging pixel at this DNA concentration. The intensity distribution of this image was constructed by transforming the image in Fig. 3a to a 3-D image (see Fig. 3b).
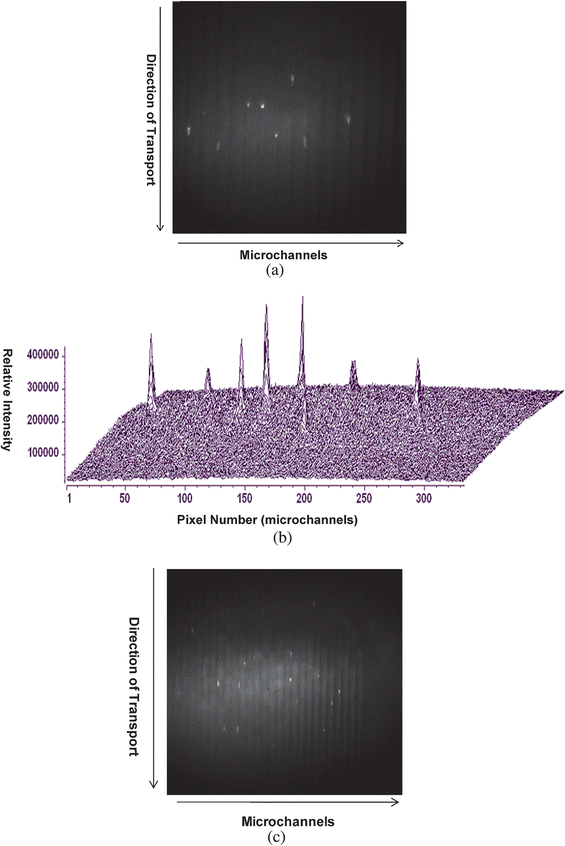 | ||
| Fig. 3 (a) Image of single λ-DNA molecules migrating through a series of microchannels (17 channels are shown). The image was acquired using a 10× (NA = 0.5) objective at an exposure time of 100 ms with the CCD multiplication gain set at 3700 (controller gain set at 1). The sample solution (100 pM) was electrokinetically pumped through the fluidic channels with a field strength of 300 V/cm (linear velocity = 0.037 cm/s). Each fluorescent spot represents a single λ-DNA molecule. (b) 3-D image showing the intensity distribution of λ-DNA molecules shown in (a). (c) Single λ-DNA molecules migrating through series of 25 microchannels. Image was acquired using a 5× objective (NA = 0.25). In all cases, the λ-DNA was stained with Syto-63 at a dye to base pair ratio of 5 : 1. | ||
To further demonstrate the ability to increase the throughput of the multi-channel fluidic device, a 5× objective (NA = 0.25) was incorporated into the imaging system and the same sample concentration of λ-DNA as that use above (100 pM) was analyzed. Fig. 3c presents a single image frame showing photon bursts from single λ-DNA molecules in 25 different microchannels. This objective generated a FoV of 1.6 mm, which allowed imaging 30 channels using our present fluidic architecture, but only 25 are shown in Fig. 3c.
For flow-based single molecule detection experiments in which the single molecule events are dynamically transported through a fixed interrogation (sampling) zone, the sample throughput (ST) can be calculated from:33
| ST = SE × DE × DR × DC | (3) |
| DR = Fv × Cb | (4) |
400 molecules/s in each microchannel with a total system throughput of 4.02 × 105 molecules/s in the 30 channel network.
As noted above, scaling down the microchannel width and pitch can be used to further increase the ST of the present system. For example, reducing the channel width to 3 µm with a pitch of 3 µm and using the 10× objective to produce a reasonable collection efficiency (NA = 0.5) will increase the number of channels that can be imaged to 133 with a corresponding ST of 1.78 × 106. A further reduction in channel width to 1 µm with an inter-channel spacing of ∼400 nm (diffraction-limited resolution is ∼λ/2) will allow the system to image 571 microchannels using the 10× objective increasing ST to 7.65 × 106 molecules/s. While this represents a significant increase in ST over our previous work,33 it also indicates exceptional increase in ST over previous research reports.34,39–46 For example, Van Orden et al. reported an ST of 2000 DNA fragments s−1 using a CCD operated in a snap-shot mode,34 while Ma et al. reported an ST of ∼8500 molecules s−1 from their single molecule spectroscopy studies.43 Recently, Chansin et al. speculated a throughput of ∼1500 molecules s−1 using their synthetic nanopores to measure DNA translocation events.46
Detection of single DNA/fluorophore conjugates
For these experiments, we utilized the 40× (NA = 0.75) objective and the optimized field strength (80 V/cm) for producing a high single molecule SNR to enhance sensitivity for the detection of single fluorophore molecules that were conjugated to ssDNAs. Solutions of ssDNAs labeled with AlexaFluor 660 were prepared in 1× TTE buffer and diluted to 750 pM in the same buffer. The samples were electrokinetically pumped through the microfluidic network and an exposure time of 100 ms was selected for the CCD per image frame when operated in FTM. With the 40× microscope objective, the laser excitation volume was calculated to be 0.12 nL, which in this case is not the sample probe volume due to the fact that the irradiation area was sub-divided into smaller elements defined by the pixels comprising the EMCCD camera. The probe volume was actually calculated from the size of each individual pixel of the EMCCD (8 µm × 8 µm) and the total magnification of the optical system (40×), producing a probe volume of 0.8 fL. Based on this probe volume size and the concentration used for these experiments (750 pM), the single molecule occupancy was 0.3 and the double occupancy probability was 0.09. Fig. 4a shows a fluorescence image of individual ssDNA molecules end-labeled with AlexaFluor 660 migrating through the fluidic device. From this image, single molecule streaks were not observed as was seen in Fig. 2b. This was a result of the binning operation performed on the image (3 × 3 bin), the relatively small linear velocity used and the data image shown in Fig. 4a taken from a single frame. To enhance spot visualization of the single molecules, we acquired 10 images of the blank (1× TTE buffer) from which the standard deviation was calculated, which was found to be 42 units for an average background of 2019 CCD units per pixel; a threshold value was selected as three-times the standard deviation above the average background (2145), which was subtracted from a sample sub-frame (25 × 42 pixels) as indicated in Fig. 4a. This also reduced false positive signals resulting from statistical variations in the background. Application of this threshold condition generated the sub-frame image shown in Fig. 4a. This sub-frame represents an approximate 5 µm (wide) × 8.4 µm (length) area within a single microchannel. From this image, fluorescence from single molecules were clearly visible, which could be individually counted to extract quantitative information. This same operation was applied to an image acquired when there was only buffer in the microfluidic network with this image shown in Fig. 4b. As can be seen from this image, there was no fluorescence registered as that observed in Fig. 4a. | ||
| Fig. 4 (a) Detection of individual dye molecules (AlexaFluor 660) end-labeled to a 60 base ssDNA distributed in different microfluidic channels (five channels are shown) for a sample concentration of 750 pM. The dye/ssDNA conjugate was pumped through the fluidic channels using a field strength of 80 V/cm (linear velocity = 0.01 cm/s). To further visualize the fluorescent photon bursts in this image, a sub-frame (25 × 42 pixels) was expanded and quantitative information extracted by applying a threshold condition to this sub-frame (see text for details). (b) Image acquired when the fluidic channels were filled with buffer only, and the processed sub-frame with application of the threshold condition as described in the text. | ||
From careful inspection of the sub-frame image shown in Fig. 4a, we were able to count 64 photon bursts generated from single ssDNA-AlexaFluor 660 conjugates. From the size of the image taken (5 µm × 8.4 µm × 1.4 µm, Pv) and the concentration of the conjugates used herein (750 pM, Cb), we could calculate the number of expected events (Nev) from:
| Nev = CbPv | (5) |
Different concentrations of ssDNA-AlexaFluor 660 conjugates were next evaluated using our multi-channel single molecule detector system and processed with the same threshold value as noted above. An average of eight images per dye concentration was plotted with the calibration plot constructed from the number of photon burst events versus the conjugate concentration. This analysis was performed on a sub-frame image with a size of 25 × 42 CCD pixels. The number of spots (single molecule events) observed was linear with the sample concentration yielding a correlation factor of 0.98 (data not shown).
Fluorescence quenching assay
We were next interested in evaluating the capability of our system to monitor biochemical reactions using fluorescent probes to signal the extent of the reaction. We employed a model test case using an oligonucleotide duplex (15 bp) with one strand (sense strand) labeled with AlexaFluor 660 at its 5′-end and the complementary strand (anti-sense strand) labeled with the Black Hole Quencher-3 (BHQ-3) at its 3′-end to create sufficient proximity for resonance energy transfer quenching of the fluorescence from AlexaFluor 660 for the duplexed form of this construct (see Fig. 5a). Melting of the duplex could be affected by introducing thermal energy into the system, producing ssDNAs that would result in the generation of fluorescence from the sense strand. Therefore, a melt analysis for this oligonucleotide could be generated by plotting the number of single molecule events observed from the sense strand versus the temperature.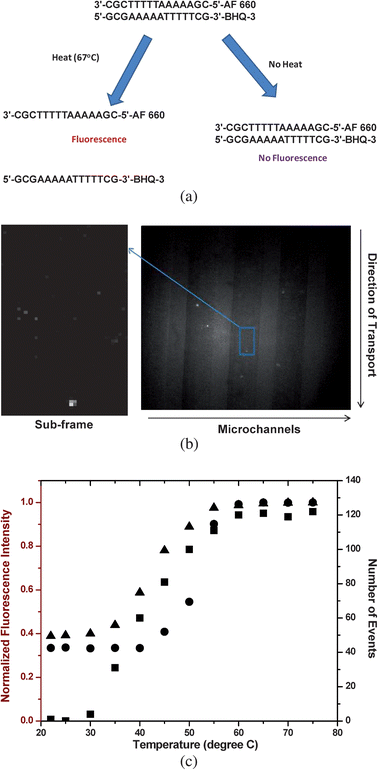 | ||
| Fig. 5 (a) Fluorescence single molecule quenching assay for determining duplex Tm using a 15-base pair oligonucleotide labeled with AlexaFluor 660 on its 5′-end and its complement labeled with the Black Hole Quencher-3 (BHQ-3) on its 3′-end. (b) Fluorescence single molecule image of the 15-bp oligonucleotide when the sample was heated to 67 °C. Fluorescence from single DNA/AlexaFluor 660 molecules were visible as shown in this image due to duplex melting, producing ssDNAs. Also shown is the sub-frame of this image (37 × 67 pixels) that was processed as described in Fig. 4. (c) Plot of number of single molecule events counted over a fixed interval of time at different sample temperatures (■, concentration = 750 pM). Also shown is a plot of the normalized fluorescence intensity of the duplexed DNA at different temperatures using a bulk measurement (dsDNA concentration = 40 nM, ●; and 1 nM, ▲) measured using a conventional fluorometer (λex = 675 nm). | ||
For these experiments, the duplexed samples (750 pM) were prepared in Tris/HCl buffer containing 20 mM Tris-HCl, 10 mM MgCl2 and 10 mM KCl at pH 7.5. The microfluidic device was equipped with a strip heater, which was placed 200 µm upstream from the imaging area. The duplexes were electrokinetically driven through the fluidic device at the desired temperature using an 80 V/cm field strength. When operated at room temperature, there was no fluorescence signal registered on the detector. The absence of fluorescence photon bursts in the EMCCD image clearly indicated that the reporter dye was sufficiently quenched by BHQ-3 and no single-stranded constructs could be seen (data not shown). This is in agreement with previous studies using this fluorophore–quencher pair with a reported 96% quenching efficiency.47
To generate ssDNA in the flowing sample, the strip heater was then turned on with the temperature set to 67 °C. Fig. 5b shows a single molecule fluorescence image of photon bursts generated from the ssDNA constructs when imaged at this temperature. To clearly visualize these spots and extract quantitative information by counting the single molecule events, the threshold condition was applied as discussed earlier. A sub-frame (37 × 67 pixels) was selected and processed (see Fig. 5b). The presence of fluorescent photon bursts in the 67 °C image versus that taken at room temperature is expected; the thermal energy introduced into the system produced a sufficient population of ssDNAs due to thermal denaturation of the duplexed DNA and thus, the energy transfer interaction between the two labels is eliminated because of proximity considerations.
A melt analysis was then conducted on the oligonucleotide duplex by taking images of single molecules traversing through the interrogation zone at different temperatures and counting the number of events (i.e., ssDNAs) at each temperature investigated. This analysis generated a sigmoidally-shaped plot (see Fig. 5c) with a sharp rise in the number of single molecule events between 30 and 50 °C with a midpoint at ∼40 °C. This midpoint represents the melting temperature (Tm) of the duplex, which is defined as the temperature in which 50% of the DNA is in its double-stranded form and 50% in its single-stranded form. This value agrees favorably with that obtained from a theoretical prediction (Tm = 41.7 °C) using oligonucleotide analysis software.48,49
The melting temperature of the duplex was also estimated by measuring fluorescence from a bulk sample of the duplex (1 nM) as a function of temperature using a standard fluorometer (λex = 675 nm). Fig. 5c shows the resulting plot of fluorescence intensity obtained at various temperatures, which generated a Tm value of 43 °C. The slight deviation of this value (43 °C) compared to the single molecule result could be attributed to the slightly higher concentration of the duplex used in the bulk solution since the inter-strand duplex Tm depends on its concentration.48 This supposition was further supported by the experimental Tm (50 °C) obtained for a 40 nM solution of this same duplex (see Fig. 5c).
As an example of the necessity for high throughput processing required to perform this measurement, we were able to reduce the processing time by a factor directly related to the number of channels imaged during the single molecule measurement, which in this case yielded a 5-fold reduction in processing time compared to a single point SMD flow-based scenario. Implementation of a smaller microchannel width and pitch producing a higher packing density of channels would significantly increase the data processing rate. For 1 µm wide channels and a pitch of 400 nm, the number of channels that could be effectively imaged using a 200 µm FoV would be 143 channels, reducing the processing time by 143-fold.
Conclusion
We have demonstrated the ability to detect single molecules using a frame transfer EMCCD in a high throughput format using a series of microchannels packed into the FoV of our imaging system. The system was configured in an epi-illumination format with beam shaping optics to generate a collimated output adequate for wide-field illumination and providing adequate irradiance and SNR to observe photon bursts from single fluorophore molecules. The present design allowed imaging 30 microchannels when λ-DNAs were stained with an intercalating dye. This represents a 12-fold increase in the sample throughput (4.02 × 105 molecules/s) over our previous work,33 which employed a CCD operated in a time-delayed integration mode. The reported optical system also provided, for the first time, single fluorophore sensitivity for a multi-channel flow-based single molecule experiment as demonstrated by the photon burst detection of ssDNAs end-labeled with AlexaFluor 660. However, in this case, a 40× objective with its high NA was required to collect sufficient numbers of fluorescent photons to produce adequate SNR for detection of the single fluorophore molecules. This resulted in a decrease in the system sample processing throughput due to the reduced FoV (200 µm vs. 1600 µm for a 5× objective), which permitted the simultaneous interrogation of 5 channels (40× objective) versus 30 channels (5× objective). However, reducing the microchannel width and pitch can significantly improve the sampling throughput for the 40× objective using optical lithography to produce a molding tool for micro-replicating high density fluidic networks.The results obtained from a fluorescence quenching assay associated with DNA duplex melting demonstrated the ability of our system to generate high throughput data processing appropriate for evaluating the fate of a biochemical reaction by monitoring population differences between reactants and/or products in a single molecule approach. The appealing aspect of the single molecule approach is the ability to use low concentrations of precious biological reactants and enzymes, the exquisite sensitivity associated with single molecule detection to record subtle changes in the reaction profile and the ability to monitor reactions efficiently in ultra-small volumes. This will be particularly attractive for high throughput screening applications, where the effects of elements from a large combinatorial library must be screened against a therapeutic target (i.e., bioenzyme). Owing to the large size of most combinatorial libraries (>106 elements), high throughput processing is critical.
Acknowledgements
The authors acknowledge support of this work through the National Institutes of Health (EB-006639), the National Science Foundation (EPS-0346411) and the Louisiana Board of Regents. The authors would also like to thank Prof. Lloyd M. Davis of the University of Tennessee Space Institute for helpful discussions on the development of the wide-field-of-view imaging system.References
- L. A. Sklar, B. S. Edwards, R. S. Larson, E. Prossnitz, B. Andrejewski, T. Bennett, T. Buranda, A. Chigaev, T. Foutz, C. Jackson, A. Key, F. Kuckuck, R. Potter, S. Ramirez, P. Simons, S. Young and G. Lopez, Clinical Cancer Research, 2001, 7, 3701S–3701S Search PubMed.
- R. Ehret, W. Baumann, M. Brischwein, M. Lehmann, T. Henning, I. Freund, S. Drechsler, U. Friedrich, M. L. Hubert, E. Motrescu, A. Kob, H. Palzer, H. Grothe and B. Wolf, Fresenius’ Journal of Analytical Chemistry, 2001, 369, 30–35 Search PubMed.
- G. W. Aherne, E. McDonald and P. Workman, Breast Cancer Research, 2002, 4, 148–154 Search PubMed.
- W. Zheng, R. H. Spencer and L. Kiss, Assay and Drug Development Technologies, 2004, 2, 543–552 Search PubMed.
- B. A. Posner, Current Opinion in Drug Discovery & Development, 2005, 8, 487–494 Search PubMed.
- H. Wesche, S. H. Xiao and S. W. Young, Combinatorial Chemistry & High Throughput Screening, 2005, 8, 181–195 Search PubMed.
- S. A. Sundberg, Curr. Opin. Biotechnol., 2000, 11, 47–53 CrossRef CAS.
- K. R. Oldenburg, J. H. Zhang, T. M. Chen, A. Maffia, K. F. Blom, A. P. Combs and T. D. Y. Chung, J. Biomol. Screening, 1998, 3, 55–62 CrossRef CAS.
- C. X. Huang, M. A. Quesada and R. A. Mathies, Anal. Chem., 1992, 64, 967–972 CrossRef CAS.
- D. J. Lockhart and W.E.A., Nature, 2000, 405, 827–836 CrossRef CAS.
- G. MacBeath, A. N. Koehler and S. L. Schreiber, J. Am. Chem. Soc., 1999, 121, 7967–7968 CrossRef CAS.
- G. MacBeath, Genome Biol., 2001, 2, 2005.
- G. MacBeath and L. S. Schreiber, Science, 2000, 289, 1760–1763 CAS.
- J. West, M. Becker, S. Tombrink and A. Manz, Anal. Chem., 2008, 80, 4403–4419 CrossRef CAS.
- S. P. Dittrich, K. Tachikawa and A. Manz, Anal. Chem., 2006, 78, 3887–3907 CrossRef CAS.
- M. G. Whitesides, Nature, 2006, 442, 368–373 CrossRef CAS.
- T. Vilkner, D. Janasek and A. Manz, Anal. Chem., 2004, 76, 3373–3386 CrossRef CAS.
- M. B. Wabuyele, H. Farquar, W. Stryjewski, R. P. Harmer, S. A. Soper, Y. W. Cheng and F. Barany, J. Am. Chem. Soc., 2003, 80, 6937–6945 CrossRef.
- E. Barkai, Y. Jung and R. Silbey, Annu. Rev. Phys. Chem., 2004, 55, 457–507 CrossRef CAS.
- E. B. Shera, N. K. Seitzinger, L. M. Davis, R. A. Keller and S. A. Soper, Chemical Physics Letters, 1990, 174, 553–557 CrossRef CAS.
- S. A. Soper, Q. L. Mattingly and P. Vegunta, Anal. Chem., 1993, 65, 740–747 CrossRef CAS.
- W. E. Moerner and D. P. Fromm, Rev. Sci. Instrum., 2003, 74, 3597–3619 CrossRef CAS.
- C. Zander, Fresenius’ J. Anal. Chem., 2000, 366, 745–751 CrossRef CAS.
- B. B. Haab and R. A. Mathies, Anal. Chem., 1999, 71, 5137–5145 CrossRef CAS.
- T. A. Byassee, W. C. Chan and S. Nie, Anal. Chem., 2000, 72, 5606–5611 CrossRef CAS.
- M. B. Wabuyele, S. M. Ford, W. Stryjewski, J. Barrow and S. A. Soper, Electrophoresis, 2001, 22, 3939–3948 CrossRef CAS.
- S. Nie, D. T. Chiu and R. N. Zare, Anal. Chem., 1995, 67, 2849–2857 CrossRef CAS.
- V. J. Sweedler, Crit. Rev. Anal. Chem., 1993, 24, 59–98 CAS.
- O. Bernard and C. Coates, Laser Focus World, 2005, 41, 133–134 Search PubMed.
- R. H. Petty, Microsc. Res. Technol., 2007, 70, 687–709 Search PubMed.
- J. D. Denvir, G. Coates and C. Collin, Proc. SPIE, 2002, 4626, 502–512.
- A. O'Grady, Proc. SPIE, 2006, 6093, 60930S.
- M. J. Emory and S. A. Soper, Anal. Chem., 2008, 80, 3897–3903 CrossRef CAS.
- A. Van Orden, R. A. Keller and P. W. Ambrose, Anal. Chem., 2000, 72, 37–41 CrossRef CAS.
- M. Christenson, Single Mol., 2000, 1, 177–179 CrossRef CAS.
- W. J. Hiller, T. A. Kowalewski and T. Tatarczyk, Proc. SPIE, 1992, 1801, 595–601 CAS.
- H.-W. Li and E. S. Yeung, J. Photochem. Photobiol., A, 2004, 172, 73–79.
- R. A. Mathies and K. Peck, Biophysical Journal, 1990, 57, A189–A189 Search PubMed.
- A. Castro, F. R. Fairfeild and E. B. Shera, Anal. Chem., 1993, 65, 849–852 CrossRef CAS.
- J. T. Petty, M. E. Johnson, P. M. Goodwin, J. C. Martin, J. H. Jett and R. A. Keller, Anal. Chem., 1995, 67, 1755–1761 CrossRef CAS.
- Z. P. Huang, J. T. Petty, B. Oquinn, J. L. Longmire, N. C. Brown, J. H. Jett and R. A. Keller, Nucleic Acids Res., 1996, 24, 4202–4209 CrossRef CAS.
- H. P. Chou, C. Spence, A. Scherer and S. Quake, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 11–13 CrossRef CAS.
- Y. F. Ma, M. R. Shortreed and E. S. Yeung, Anal. Chem., 2000, 72, 4640–4645 CrossRef CAS.
- T. Anazawa, H. Matsunaga and E. S. Yeung, Anal. Chem., 2002, 74, 5033–5038 CrossRef CAS.
- J. R. Krogmeier, I. Schaefer, G. Seward, G. R. Yantz and J. W. Larson, Lab Chip, 2007, 7, 1767–1774 RSC.
- G. A. T. Chansin, R. Mulero, J. Hong, M. J. Kim, A. J. Demello and J. B. Edel, Nano Lett., 2007, 7, 2901–2906 CrossRef CAS.
- S. A. E. Marras, R. F. Kramer and S. Tyagi, Nucleic Acids Res., 2002, 30, e122 CrossRef CAS.
- J. Santa Lucia, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 1460–1465 CrossRef CAS.
- R. Owczarzy and M. Behlke, Molecular Genetics and Biophysics, Integrated DNA Technology, 2005, 1–4 Search PubMed.
| This journal is © The Royal Society of Chemistry 2009 |