SECM for imaging and detection of latent fingerprints
Meiqin
Zhang
a and
Hubert H.
Girault
*b
aDepartment of Chemistry, University of Warwick, Coventry, UK CV4 7AL
bLaboratoire d'Electrochimie Physique et Analytique, Ecole Polytechnique Fédérale de Lausanne, CH-1015 Lausanne, Switzerland. E-mail: hubert.girault@epfl.ch; Fax: +41-21-693 3667; Tel: +41-21-693 3145
First published on 14th November 2008
Abstract
Fingerprint analysis is one of the most important methods used for personal identification of evidence found at a crime scene for forensic purposes. Using scanning electrochemical microscopy (SECM) imaging, researchers can visualize the ultrastructure of human fingerprints on wet porous and non-porous surfaces by combining with silver-staining or multi-metal-deposition (MMD) technology. SECM allows investigators to image chemical activities of fingerprint surfaces with an impressively high resolution, such as the third level valuable information for confirming an identification. This methodology takes a significant advantage of the high sensitivity of SECM towards the small variation of electrochemical reaction rates at the substrate surface. In this review, we highlight the recent breakthroughs in ultrasensitive imaging and detection of latent fingerprints with a special focus on a novel application of SECM. We will also discuss our perspectives on future research directions.
 Meiqin Zhang | Meiqin Zhang obtained her PhD from Peking University under the supervision of Professor Yuanhua Shao in 2006. She then moved to a postdoctoral position with Professor Hubert H. Girault at the Ecole Polytechnique Fédérale de Lausanne (Switzerland) in 2006. Now she is awarded ‘Marie Curie Incoming International Fellowship’, working with Professor Patrick R. Unwin at the University of Warwick (UK). Her main interests are in creating new methods of bioanalysis using SECM and in particular the development of nanoparticle biosensors. |
 Hubert Girault | Hubert H. Girault is currently director of the Laboratory of Analytical and Physical Electrochemistry at the Ecole Polytechnique Fédérale de Lausanne, Switzerland. After a PhD at Southampton University (UK), he was lecturer at Edinburgh University, Scotland (1985–1992). His research activities include spectro-electrochemistry at liquid–liquid interfaces, electrochemical micro-analytical systems for immunoassay, electrophoresis for proteomic applications and electrochemical ionisation methods for mass spectrometry. In 2006, he was the recipient of the Faraday Medal from the Royal Society of Chemistry. |
1. Introduction
When a finger touches a surface, pores on the papillary ridges deposit a residue of perspiration, consisting of 99% water with the remainder made up of inorganic salts such as NaCl and organic substances such as urea. It leaves an impression of the finger's ridge pattern, referred to as a latent fingerprint, which requires physical or chemical treatments to enable visualization. Over a century after fingerprint evidence was first used to obtain a criminal conviction, the detection and identification of fingerprints are today still the cornerstone of modern crime investigation, as it is among the few biometric signatures that can be truly unique and invariable for an individual.1–3 More recently, increasing identity fraud has created a growing need for biometric technology for positive person identification in a number of non-forensic applications, such as a driver's license, welfare disbursement, access control, and credit card identification. Because biometric identifiers cannot be easily misplaced, forged or shared, they are considered more reliable for personal identification than traditional token- or knowledge-based methods.For years, forensic scientists have been seeking new methods or trying to improve existing techniques for the visualization of latent fingerprints. A wide range of physical and chemical techniques is nowadays available.4 For a given set of circumstances, the choice of the best detection techniques, or sequence of techniques, will depend on several factors that include the nature of the surface (e.g. rough, smooth, porous or non-porous), the presence of any particular contaminants, and environmental factors (e.g. whether the surface is or has been wet or not). The structured combination of optical methods (diffused reflection, luminescence, UV absorption, mass spectrometry (MS) and reflection), physical methods [powdering, vacuum metal deposition (VMD), small particle reagent], physico-chemical methods [physical developer, multi-metal-deposition (MMD), iodine, cyanoacrylate] and chemical methods (ninhydrin and its analogues, DFO, etc.), permits a rational and highly efficient processing of the secretions deposited by the fingers on a great variety of substrates.5–10 To visualize these developed fingerprints, alternative light sources (such as Crimescope® or Polilight®) or laser-based detection methods are commonly used. These conventional techniques are quite effective in the recovery of latent fingerprints under many ordinary circumstances. However, like all other existing techniques, these optical methods do not work in all possible cases and certain types of latent fingerprints or object surfaces with unique characteristics: surfaces with multicolored backgrounds, surfaces contaminated with blood or other body fluids, and other porous or non-absorbent surfaces may be problematic. The major challenges in fingerprint detection with laser techniques stem from both low emission levels from the fingerprint secretions and interferences from the background signal, for example the background fluorescence when imaging fingerprints on the surface of banknotes. In addition, the fluorescence-based readout of latent fingerprints involves expensive instrumentation and sophisticated numerical algorithms to interpret the data. In this short review, we highlight the application of an electrochemical method, scanning electrochemical microscopy (SECM), as it is a relatively simple, accurate and inexpensive technique for latent fingerprint analysis.
2. Applications and features of SECM
SECM,11,12 a scanning probe technique, has been developed into a powerful analytical tool used to measure the kinetics of heterogeneous and homogeneous reactions,13–16 to visualize and quantify heterogeneously distributed chemical activity on various sample surfaces and biological systems,17–24 and to micro- and nano-fabricate on substrate surfaces.25,26 Instrumentally, SECM consists of a disk-shaped ultramicroelectrode (UME) as the scanning probe (typically 25 µm disc diameter or smaller) scanned over a sample using a high-precision position controller. The Faradaic steady-state current established at the electrode varies according to the electrochemical properties of the sample measured using a potentiostat. In SECM experiments, the UME is brought into the vicinity of the substrate surface, where the electrochemical response of the probe is recorded as a function of the lateral probe position (x, y) for imaging sample.Compared with other scanning probe microscopic techniques, SECM offers some unique features. For example, it is capable of imaging chemical activities present at a substrate surface to provide chemical information that is complementary to the information obtained from other techniques displaying mainly the topography and morphology of the sample surface such as scanning electron microscopy (SEM) and atomic force microscopy (AFM). Moreover, SECM can image samples with relative large surface areas (centimeters square) under physiological conditions instead of non-physiological conditions such as high vacuum or surface conductivity,27 which makes SECM particularly advantageous over other scanning probe techniques for the application to imaging of latent fingerprints. Furthermore, in contrast to well-established microscopic techniques, which are based on fluorescence detection and hence need the biomolecules to be labeled with suitable fluorescence dyes, SECM measurements usually can be performed without fluorescent labeling. However, the biological materials left by the latent image on the substrate are not redox active, and cannot be imaged directly. To circumvent this difficulty, the latent image is silver stained either directly with a silver salt or indirectly on gold nanoparticles adsorbed on the image. The major advantage of the silver-staining procedure is to provide an amplification step, as the SECM is used to detect colloidal silver.
3. Silver-staining and multi-metal-deposition detection of biomolecules
Silver-staining detection of biomolecules
In proteomics, it is essential to be able to analyze and identify proteins separated by gel electrophoresis at a high sensitivity. Ideally, the detection method should have a detection limit as low as possible, a wide dynamic range and a linear relationship between the biomolecule quantity and the staining intensity.28 The detection procedure should be simple, fast to perform, non-toxic, MS-compatible, and not too expensive from an application point of view. So far, various staining methods have been developed for visualizing proteins following gel electrophoresis, such as visible organic dyes,29copper staining,30silver staining,31–33fluorescent labeling,34 and radioactive labeling.35 Of these, Coomassie Brilliant Blue (CBB) dyes have been very popular due to their low cost and relative ease of use. However, the principal limitation associated with this method is a poor detection sensitivity, which is about 8–50 ng proteins per spot. Fluorescence-based detection approaches are highly sensitive but are generally used less frequently than the CBB-based staining protocols since they suffer from the drawback of high cost due to the complex and expensive optical detection instrumentation. Although labeling of proteins using radioactive isotopes still remains the most sensitive method available, it needs special equipment and extremely complex handling procedures. On the contrary, silver staining is a relatively rapid, inexpensive, and highly sensitive alternative to these techniques mentioned above for developing a routine analytical technique.Silver staining, originally introduced in 1979 by Switzer et al.,31 has provided a breakthrough for sensitive protein detection. Later this method was applied to nucleic acid research.36 Currently, silver staining is considered as sensitive as autoradiography and fluorescence labeling techniques that are widely used to detect proteins or DNA fragments in various experiments. It offers a record of the two-dimensional electrophoresis result that can be visualized without any special equipment. There are numerous variations of the silver-staining protocols published, each with different advantages regarding convenience, safety, sensitivity, cost, reproducibility, speed and compatibility with other analytical techniques, notably MS. The basic mechanisms underlying silver staining of proteins in gels are relatively well understood. In principle, protein detection mainly relies on the binding of silver ions (Ag+) to the sulfhydryl and carboxyl groups of proteins, followed by reduction to free metallic silver (Ag0) which is insoluble and visible, allowing bands containing protein to be seen. The initial deposition of metallic silver promotes further deposition in an autocatalytic process, resulting in exceptionally high sensitivity. The protein or nucleic acid bands are visualized as spots where the reduction occurs and the image of biomolecule distribution within the gel, therefore, is based on the difference in oxidation–reduction potential between the area with and without biomolecules. Silver-staining procedures can be divided into two general categories: (1) ammoniacal silver stains, and (2) silver nitrate stains.37 The ammoniacal silver stains usually have lower background and are more sensitive but require longer procedures. Silver nitrate stains, on the other hand, are faster but slightly less sensitive.
In our work, the fingerprints were formed on porous poly(vinylidene difluoride) (PVDF) membranes and treated by silver-staining protocol with silver nitrate as described in the proteomic literature,38 and then used for SECM imaging.
Multi-metal-deposition detection of biomolecules
Nanoparticles represent an excellent biocompatibility with biomolecules and show unique structural, electronic, magnetic, optical and catalytic properties which have made them a very attractive material as a signal amplifier in the detection of antibodies, antigen proteins, DNA and RNA using optical methods or various electrochemical techniques.39,40 Recently, metallic colloid nanoparticles (e.g.gold and silver colloids) have been successfully applied to the progress of both chemical and biological sensing because of their easily controllable size distribution and long-term stability.41Silver enhancement treatment, i.e.silver deposition on gold nanoparticles, is generally used to visualize protein- or DNA-conjugated particles both in electron microscopy studies and electrochemical detection.23,42,43The application of gold colloid to the field of forensic sciences was introduced by Saunders,44 as a method for the detection of latent fingerprints. The gold nanoparticles stabilized by citrate ions adhere to the fingerprint residue and catalyze the precipitation of the Ag+ ions to metallic silver (Ag0) from the aqueous solution on the ridges, thus the autometallographic silver deposition procedure significantly enhances the developed fingerprints as a result of enlarging the size and darkening the color of the particles. It is a process known as ‘Multi-Metal-Deposition’ (MMD). The gold adherence to the fingerprint material is explained by an electrostatic interaction between the negatively charged gold colloids and the positively charged components of the fingerprint residue. Currently, MMD is widely used as a very sensitive technique that is efficient on a wide range of surfaces, both porous and non-porous.8 In particular, it is a detection method that can be used on surfaces that are problematic with other conventional techniques, e.g. polystyrene packaging, thermal paper and surfaces that are or have been wet.
In our work, the MMD samples for SECM imaging were prepared by following a procedure previously reported,7 which is based on the use of small particles of metallic gold (diameter ≈14 nm) suspended in water firstly binding to the fingerprint residues. Owing to the small particle size, it is often difficult to optically observe the fingerprints at this stage. Consequently, a second step is required to enhance the contrast, i.e. the specific chemical deposition of metallic silver onto the gold particles. As a result, the finger deposits distinctly appear as black ridges on a lighter background.
4. SECM for imaging of latent fingerprints
Imaging of protein-modified latent fingerprints on the PVDF membrane
In a series of papers,45–47 our group has described the use of SECM in the feedback mode to visualize silver- or copper-stained proteins on PVDF membranes or in poly(ethylene terephthalate) (PET) channels by generating an oxidizer at the probe to oxidize these metals. Recently, this methodology has been further extended to the forensic field of protein-modified latent fingerprint detection.48A three-electrode setup for imaging fingerprints was employed with a disk-shaped Pt UME as the amperometric SECM probe as schematically presented in Fig. 1. The counter and quasi-reference electrodes were a Pt wire and a silver wire, respectively. The protein-fingerprinted membrane was fixed on a microscope glass slide. This cell assembly was secured onto a platform which includes three screws for leveling the substrate surface. In the amperometric feedback mode of SECM measurements, the probe signal used for imaging is a Faradaic current, i, originating from the oxidation of the mediator [IrCl6]3− added to the electrolyte solution. With the probe kept at a constant potential (0.8 V vs. Ag QRE) high enough to oxidize [IrCl6]3− and placed in the bulk solution far above the surface of the fingerprinted substrate, the anodic current is completely controlled by the hemispherical diffusion of [IrCl6]3− toward the active disk of the UME and a diffusion-limited steady-state current (i∞) is observed. When the probe is brought close to the silver-stained fingerprint sample surface, electrochemical recycling of the [IrCl6]3− becomes possible by the following heterogeneous bimolecular electron transfer reaction between [IrCl6]2− and silver nanoparticles:45,47,48
 | (1) |
![Schematic (not to scale) representation of the operating principle for SECM imaging of silver-stained fingerprints on the PVDF membrane using [IrCl6]3−oxidation (Eprobe = 0.8 V vs. Ag QRE) at the probe.](/image/article/2009/AN/b815336a/b815336a-f1.gif) | ||
| Fig. 1 Schematic (not to scale) representation of the operating principle for SECM imaging of silver-stained fingerprints on the PVDF membrane using [IrCl6]3−oxidation (Eprobe = 0.8 V vs. Ag QRE) at the probe. | ||
This process leads to an increase in probe current (positive feedback effect). On the other hand, the probe current decreases monotonically with the probe approaching the blank substrate because of [IrCl6]3− diffusion hindrance (negative feedback effect). For imaging of silver-stained fingerprints, probe approach curves (i/i∞vs.probe-to-sample distance d) have been used to position the probe at an appropriate working distance over the sample. Changes in the probe current during lateral scans used for ‘chemical imaging’ reflect the topology of the silver-stained fingerprint on the membrane surface.
The feedback mode of SECM can be observed when the probe is close to the surface, usually at a distance less than twice the probe radius. Theoretically, the spatial resolution of SECM is mainly governed by the probe radius, r, i.e. a smaller probe offers a higher spatial resolution, as the probe–substrate distance is reduced. This short distance, however, is difficult to maintain with the commonly used constant-height mode of operation when imaging large areas that may be tilted with respect to the traveling probe and probe crash can occur. With the objective of imaging with a high spatial resolution a relative large area (5 × 3 mm) of fingerprinted surface, a 25 µm-diameter probe was chosen and the sample surface was leveled manually according to the information from line scan curve experiments in the x and y directions performed on a fingerprinted surface. Details of this operating process have been described elsewhere.48
An optical laser-scanner image of an inked fingerprint on the PVDF membrane using a classical writing ink is illustrated in Fig. 2a. The blue and white lines correspond to the ridges and furrows, respectively. According to a classification system known as the Henry–Galton system, loops, whorls, and arches are the basic classes of fingerprint patterns.49 The one shown in Fig. 2a belongs to the whorls as we can see that some of the ridges make a turn through at least one circuit. For comparison, a high-resolution SECM image (5 × 3 mm) of the protein-modified fingerprint with silver staining on the PVDF membrane is shown in Fig. 2b, which needed about 6 h and 15 min with a fast scanning speed of 100 µm s−1 to ensure the compatibility of the system with large surface scanning. The image area corresponds approximately to the rectangle region marked (inside the black line) in Fig. 2a. The changes observed in SECM-generated data are mainly due to variations in the electrochemical reactivity between regions that are covered with silver-stained proteins and regions that are not except that a higher background current is presented in the region of 0 ≤ y/mm ≤ 1. In Fig. 2b, a brown color has been used to emphasize the regions of significant positive feedback. The brown regions correspond to the ridges because the proteins were transferred from the ridges of the finger to the PVDF membrane substrate and then stained with silver nanoparticles. The positive feedback is due to the regeneration of [IrCl6]3− by the heterogeneous bimolecular electron transfer described in reaction (1). To the contrary, the furrows of the finger cannot transfer proteins onto the PVDF membrane surface, so [IrCl6]3− cannot be recycled. The insulating nature of the PVDF membrane blocks some of the diffusion of [IrCl6]3− to the probe surface, which leads to a lower probe current as shown in the light regions of Fig. 2b.
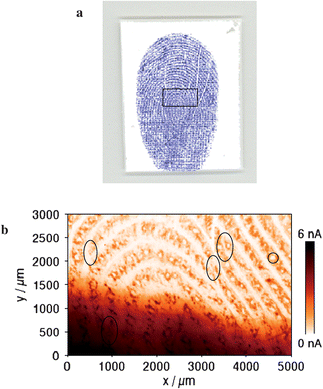 | ||
| Fig. 2 (a) An optical laser-scanner image of the classical writing ink fingerprint on the PVDF membrane. The blue and white lines correspond to the ridges and furrows, respectively. (b) An SECM image (5 × 3 mm) of the silver-stained fingerprint on the PVDF membrane. The image area corresponds approximately to the rectangle region marked (inside black line) in Fig. 2a. Pore shape shown in the circle. The ovals represent the characteristic points. The brown and white lines correspond to the ridges and furrows, respectively. Measuring conditions: Pt UME of 25 µm-diameter as the SECM probe, 2 mM K3IrCl6 in 0.1 M KNO3, Eprobe = 0.8 V vs. Ag QRE, a probe–substrate separation of approximately 15 µm and a lateral scan rate of 100 µm s−1. | ||
A significant feature of this proposed technique is the ability of obtaining a high-resolution image for the pore shape (marked with a circle in Fig. 2b). Champod et al. describe three general levels of information that can be distinguished in fingerprints:3 the first level involves the overall pattern of the papillary ridges, yet there exist many variations and sub-patterns. The second level information refers to the major deviations of the ridge paths from general patterns (marked with ovals in Fig. 2b). The third level of information refers to the shape and relative position of the pores of each part of a ridge. For the moment, the first and second levels are very important for the verification of a person's identity. Comparatively little research, however, has been done on the third level information because the image resolution of the fingerprints is often not good enough for determining a match based on pore or ridge shape. As can be seen from Fig. 2b, pores (40–120 µm in diameter) and relative locations of ridges were clearly imaged. If the size of the SECM probe was decreased (e.g. <2 µm diameter), the quality of images will be further improved. Therefore, the SECM image of fingerprints is a potential tool for obtaining the third level information to support and confirm a human identification.
Imaging of MMD-enhanced latent fingerprints on glass
Very recently, the fingerprints developed by the MMD technique have been imaged electrochemically in the feedback mode, where the silver layer on the gold nanoparticles is re-oxidized upon the passage of the scanning probe.50 The operating principle for SECM imaging of MMD-enhanced latent fingerprints on glass is the same as described above. Changes in the probe current during lateral scans used for ‘electrochemical imaging’ reflect the topology of the MMD-enhanced fingerprint on the glass surface. Here, in order to image a relatively large area (3 × 2 mm) of the fingerprint with a high resolution, we chose a 20 µm-diameter probe and the probe–substrate distance was kept to about 4 µm.An optical laser-scanner image of an MMD-enhanced fingerprint on glass before SECM imaging is shown in Fig. 3a. For comparison, the rectangle region (3 × 2 mm) marked (inside the black line) was imaged by SECM, shown in Fig. 3b, which need about 5 h with a scan rate of 300 µm s−1 to ensure the compatibility of the system with large surface scanning. The brown and white lines represent the ridges and furrows, respectively. The changes observed in SECM-generated data are mainly due to variations in the electrochemical reactivity between ridges that are covered with silver deposits and furrows that are not. The white band is present in the region of 1.78 ≤ y/mm ≤ 1.84 in Fig. 3b. This is due to the repeated line scans performed in the x direction for leveling the substrate before SECM imaging, where metallic silver is completely dissolved in the solution. As can be observed in Fig. 3b, SECM of the MMD-enhanced fingerprint provides an image with a good contrast and a high resolution.
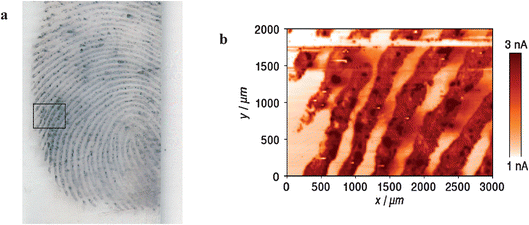 | ||
| Fig. 3 (a) An optical laser-scanner image of the MMD-enhanced latent fingerprint on glass. The black and white lines correspond to the ridges and furrows, respectively. (b) A high-resolution SECM image (3 × 2 mm) of the sample with a scan rate of 300 µm s−1. The SECM image area corresponds approximately to the rectangle region marked (inside black line) in Fig. 3a. The brown and white lines correspond to the ridges and furrows, respectively. Measuring conditions: Pt UME of 20 µm-diameter as the SECM probe, 1 mM K3IrCl6 in 0.1 M KNO3, Eprobe = 0.8 V vs. Ag QRE, a probe–substrate distance of approximately 4 µm. | ||
Conclusions and outlook
A novel electrochemical strategy for imaging latent fingerprints on both porous and non-porous surfaces under wet conditions is presented: fingerprints were first developed by silver staining for protein-modified fingerprints on the PVDF membrane and the silver on gold multi-metal-deposition method for latent fingerprints on glass, and then revealed by SECM with [IrCl6]3− acting as redox mediator to detect the silver dissolution in the feedback mode. High-resolution SECM images of fingerprints in both cases were achieved and their quality is already comparable with state-of-the-art optical detection techniques.Work is in progress to extend this approach to image benzoquinone-tagged latent fingerprints. This method has the potential to develop fingerprint detection on a range of difficult surfaces that are known to provide poor results with current techniques, such as paper, plastic, banknotes and wet surfaces. Moreover, it is also compatible with the previously used detecting techniques of bloody latent fingerprints and other metal-stained samples (VMD sample)51 where a small amount of gold is deposited under high vacuum onto the exhibit and then followed by the deposition, onto the gold layer, of a much thicker layer of zinc. However, before such an imaging technique could be readily applied on items submitted for examination in routine casework, it would need to be improved by employing a multi-electrode probe with high speed for parallel imaging so that larger scan images can be carried out within a much shorter timescale.52 A more practical constant-distance mode of SECM should be employed to solve technological problems associated with high-resolution imaging for very rough or curved surfaces by applying shear-force, alternating-current-based distance control systems or combining with AFM.53–56 In addition, new probes such as soft tips could also be used to avoid tip crash during imaging for the fingerprints on rough or tilted surfaces.
Acknowledgements
This work was supported by Ecole Polytechnique Fédérale de Lausanne. M. Z. thanks Marie Curie Incoming International Fellowships (040126: ‘SECM-CRDS’) from the 6th Framework Programme on Research, Technological Development and Demonstration of European Commission, and acknowledges Professor Patrick R. Unwin for the fellowship application.References
- R. D. Olsen, Scott's Fingerprint Mechanics, Charles C. Thomas, Springfield, IL, 1978 Search PubMed.
- Advances in Fingerprint Technology, ed. H. C. Lee and R. E. Gaensslen, CRC Press, Boca Raton, 2nd edn, 2001 Search PubMed.
- C. Champod, C. Lennard, P. Margot and M. Stoilovic, Fingerprints and other Ridge Skin Impressions, CRC Press, Boca Raton, 2004 Search PubMed.
- Fingerprint Development Handbook, Home Office Scientific Development Branch, Heanor Gate Printing Limited, Heanor, Derbyshire, UK, 2nd edn, 2005 Search PubMed.
- G. Payne, B. J. Reedy, C. Lennard, B. Comber, D. Exline and C. Roux, Forensic Sci. Int., 2005, 150, 33–51 CrossRef CAS.
- D. R. Ifa, N. E. Manicke, A. L. Dill and R. G. Cooks, Science, 2008, 321, 805 CrossRef CAS.
- G. S. Sodhi and J. Kaur, Forensic Sci. Int., 2001, 120, 172–176 CrossRef CAS.
- B. Schnetz and P. Margot, Forensic Sci. Int., 2001, 118, 21–28 CrossRef CAS.
- M. Sametband, I. Shweky, U. Banin, D. Mandler and J. Almog, Chem. Commun., 2007, 11, 1142–144 RSC.
- U. Ramminger, U. Nickel and B. Geide, J. Forensic Sci., 2001, 46, 288–293 CAS.
- A. J. Bard, F.-R. F. Fan and M. V. Mirkin, Scanning Electrochemical Microscopy, ed. A. J. Bard, Marcel Dekker, New York, 1994, vol. 18, pp. 244–391 Search PubMed.
- Scanning Electrochemical Microscopy, ed. A. J. Bard and M. V. Mirkin, Marcel Dekker, New York, 2001 Search PubMed.
- M. V. Mirkin, T. C. Richards and A. J. Bard, J. Phys. Chem., 1993, 97, 7672–7677 CrossRef CAS.
- A. L. Barker, M. Gonsalves, J. V. Macphercon, C. J. Slevin and P. R. Unwin, Anal. Chim. Acta, 1999, 385, 223–240 CrossRef CAS.
- Z. Zhang, Y. Yuan, P. Sun, B. Su, J. Guo, Y. Shao and H. H. Girault, J. Phys. Chem. B, 2002, 106, 6713–6717 CrossRef CAS.
- P. Sun, F. Li, Y. Chen, M. Zhang, Z. Zhang, Z. Gao and Y. Shao, J. Am. Chem. Soc., 2003, 125, 9600–9601 CrossRef CAS.
- F.-R. F. Fan and A. J. Bard, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 14222–14227 CrossRef CAS.
- B. R. Horrocks, D. Schmidtke, A. Heller and A. J. Bard, Anal. Chem., 1993, 65, 3605–3614 CrossRef CAS.
- H. Shiku, T. Akeda, H. Yamada, T. Matsue and I. Uchida, Anal. Chem., 1995, 67, 312–317 CrossRef CAS.
- G. Wittstock, K.-J. Yu, H. B. Halsall, T. H. Ridgeway and W. R. Heineman, Anal. Chem., 1995, 67, 3578–3582 CrossRef CAS.
- G. Wittstock and W. Schuhmann, Anal. Chem., 1997, 69, 5059–5066 CrossRef CAS.
- J. V. Macpherson and P. R. Unwin, Anal. Chem., 2001, 73, 550–557 CrossRef CAS.
- J. Wang, F. Y. Song and F. M. Zhou, Langmuir, 2002, 18, 6653–6658 CrossRef CAS.
- A. Amemiya, J. D. Guo, H. Xiong and D. A. Gross, Anal. Bioanal. Chem., 2006, 386, 458–471 CrossRef CAS.
- Y. Yatziv, I. Turyan and D. Mandler, J. Am. Chem. Soc., 2002, 124, 5618–5619 CrossRef CAS.
- I. Turyan, M. Etienne, D. Mandler and W. Schuhmann, Electroanalysis, 2005, 17, 5–6.
- M. Zhang, B. Su, F. Cortes-Salazar, M. Hojeij and H. H. Girault, Electrochem. Commun., 2008, 10, 714–718 CrossRef CAS.
- R. Westermeier and R. Marouga, Biosci. Rep., 2005, 25, 19–32 CrossRef CAS.
- V. Neuhoff, N. Arold, D. Taube and W. Ehrhardt, Electrophoresis, 1988, 9, 255–262 CAS.
- C. Lee, A. Levin and D. Branton, Anal. Biochem., 1987, 166, 308–312 CrossRef CAS.
- R. C. Switzer, C. R. Merril and S. Shifrin, Anal. Biochem., 1979, 98, 231–237 CrossRef.
- H. Blum, H. Beier and H. J. Gross, Electrophoresis, 1987, 8, 93–99 CAS.
- T. Rabilloud, L. Vuillard, C. Gilly and J. Lawrence, J. Cell. Mol. Biol., 1994, 40, 57–75 Search PubMed.
- A. Bermudez, J.-R. Daban, J. R. Garcia and E. Mendez, Biotechniques, 1994, 16, 621–624 CAS.
- P. H. O'Farrell, J. Biol. Chem., 1975, 250, 4007–4021.
- M. Christensen, L. Sunde, L. Bolund and T. F. Orntoft, Scand. J. Clin. Lab. Invest., 1999, 59, 167–168 CrossRef CAS.
- C. R. Merril, Nature, 1990, 343, 779–780 CrossRef CAS.
- B. K. Sørensen, P. Højrup, E. Østergård, C. S. Jørgensen, J. Enghild, L. R. Ryder and G. Houen, Anal. Biochem., 2002, 304, 33–41 CrossRef CAS.
- S. Schultz, D. R. Smith, J. J. Mock and D. A. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 996–1001 CrossRef CAS.
- J. J. Gooding, Electroanalysis, 2002, 14, 1149–1156 CrossRef CAS.
- L. He, M. D. Musick, S. R. Nicewarner, F. G. Salinas, S. J. Benkovic, M. J. Natan and C. D. Keating, J. Am. Chem. Soc., 2000, 122, 9071–9077 CrossRef CAS.
- T. A. Taton, C. A. Mirkin and R. L. Letsinger, Science, 2000, 289, 1757–1760 CrossRef CAS.
- J. J. Storhoff, R. Elghanian, R. C. Mucic, C. A. Mirkin and R. L. Letsinger, J. Am. Chem. Soc., 1998, 120, 1959–1964 CrossRef CAS.
- G. Saunders, International Association for Identification, 74th Annual Educational Conference, June, 1989, Pensacola, USA Search PubMed.
- M. Carano, N. Lion, J.-P. Abid and H. H. Girault, Electrochem. Commun., 2004, 6, 1217–1221 CrossRef CAS.
- M. Carano, N. Lion and H. H. Girault, J. Electroanal. Chem., 2007, 599, 349–355 CrossRef CAS.
- M. Zhang, G. Wittstock, Y. Shao and H. H. Girault, Anal. Chem., 2007, 79, 4833–4839 CrossRef CAS.
- M. Zhang and H. H. Girault, Electrochem. Commun., 2007, 9, 1778–1782 CrossRef CAS.
- E. R. Henry, Classification and Uses of Finger Prints, Georges Routedge, London, 4th edn, 1900 Search PubMed.
- M. Zhang, A. Becue, M. Prudent, C. Champod and H. H. Girault, Chem. Commun., 2007, 38, 3948–3950 Search PubMed.
- P. Theys, Y. Turgis, A. Lepareux, G. Chevet and P. F. Ceccaldi, Int. Criminal Police Rev., 1968, 217, 106–108 Search PubMed.
- A. L. Barker, P. R. Unwin, J. W. Gardner and H. Rieley, Electrochem. Commun., 2004, 6, 91–97 CrossRef CAS.
- A. Hengstenberg, C. Kranz and W. Schuhmann, Chem.–Eur. J., 2000, 6, 1547–1554 CrossRef CAS.
- M. A. Alpuche-Alviles and D. O. Wipf, Anal. Chem., 2001, 73, 4873–4881 CrossRef CAS.
- P. M. Diakowski and Z. Ding, Phys. Chem. Chem. Phys., 2007, 9, 5966–5974 RSC.
- J. V. Macpherson and P. R. Unwin, Anal. Chem., 2000, 72, 276–285 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |