Ultrasensitive detection of coliforms by means of direct asymmetric PCR combined with disposable magnetic amperometric genosensors
Óscar A.
Loaiza†
a,
Susana
Campuzano†
b,
María
Pedrero
a,
Pedro
García
b and
José M.
Pingarrón
*a
aDepartamento de Química Analítica, Facultad de CC. Químicas, Universidad Complutense de Madrid, Madrid, E-28040, Spain. E-mail: pingarro@quim.ucm.es; Fax: +34913944329; Tel: +34913944315
bDepartamento de Microbiología Molecular, Centro de Investigaciones Biológicas, CSIC, Ramiro de Maeztu 9, Madrid, E-28040, Spain
First published on 4th November 2008
Abstract
An extremely sensitive procedure for the isolation and detection of DNA from bacterial cell cultures is described. Direct asymmetric PCR amplified products from E. coli cultures are specifically detected, at a concentration level as low as 0.01 cfu mL−1 (cfu, colony forming unit), using disposable magnetic DNA hybridization amperometric sensors with no need for culture preconcentration steps.
Introduction
Traditional pathogen microorganism detection methods are time consuming due to the need of cell culturing. Nowadays, there is a great interest in biotechnological developments delivering highly specific, reproducible and low-cost screening methods for the detection of pathogen microorganisms. Thus, the use of biosensors has been proposed as a mean of obtaining rapid positive or negative results from a sample,1 as well as a way to accomplish purification and concentration steps by elimination of sample matrix inhibition via collection and capture of target analytes from the sample matrices. In this context, immobilization of DNA probes on magnetic beads (MBs) has demonstrated its usefulness for purification and/or preconcentration purposes. The use of MBs in the development of DNA hybridization sensors has been reviewed recently.2 Moreover, the use of DNA probes immobilized on magnetic nanoparticles has been proposed recently as a preliminary step for polymerase chain reaction (PCR) amplification, thus simplifying the concentration and purification process of the target DNA.3 Furthermore, combination of magnetic isolation with appropriate electrochemistry leads to highly sensitive detection of DNA hybridization.4,5Although DNA biosensors based on the integration of a sequence-specific probe and an electrochemical signal transducer are considered as really attractive due to their simplicity, low instrumentation cost and the possibility of obtaining real time accurate detection with very low detection limits,6 PCR amplification of the DNA target is still necessary due to the low abundance and extreme complexity of the non-amplified targets. PCR has been used since the middle of the 1980s in forensic, medical, environmental and food sciences, the amplified nucleic acids being detected usually by gel electrophoresis with very low specificity, given that no sequence information of amplified DNA is attained.7 More recently, different protocols to optimize detection of PCR amplified DNA were proposed, including asymmetric PCR, which predominantly amplifies one DNA strand, and direct PCR (DPCR), which allows amplification and detection of specific target nucleic acid sequences inside individual cells with no need for DNA extraction. Asymmetric PCR can result in higher sensitivity than symmetric PCR due to the presence in the PCR product of the single-stranded fragment in a high proportion, which can hybridize non-competitively with the probe.8 This implies also a faster hybridization of amplicons at the developed genosensor. Due to its speed, simplicity and minimal sample manipulation, DPCR has been demonstrated to be useful to detect and quantify bacteria in environmental samples,9 even from a sample containing one single cell.10 In this approach, the factor determining the amount of DNA available is cell lysis efficiency, which can be enhanced by employing methods to permeabilize the bacteria cell membranes to allow entry of reagents for amplification, and retard the diffusion of PCR products away from the cells, also not destroying their morphology or the microscale structure of the microbial community.11
In this communication, direct asymmetric PCR has been used to improve the sensitivity of the specific detection of a gene related to the Enterobacteriaceae bacterial family. Direct asymmetric PCR has been combined with disposable magnetic DNA sensors based on the coupling of streptavidin-peroxidase to biotinylated lacZ gene target sequences.12 Enzyme amplification of amperometric DNA assay detection was nicely reported previously by Heller's group.13
Experimental
Disposable magnetic amperometric genosensors were constructed as described previously.12 The biotinylated capture probe sequence 5′-biotin-CAG GAT ATG TGG CGG ATG AGC GGC A-3′, synthesized by Sigma Genosys, was used in the detection of amplified lacZ, taking advantage of its hybridization to target sequences located within the respective regions of amplified DNA chains. Because conventional coliform monitoring is based on the detection of the activity of the gene product (β-galactosidase) of lacZ produced by coliform bacteria,14 a 326-bp region closer to the amino terminal group of the E. coli lacZ gene15,16 was selected as suitable target for PCR amplification. This was carried out using asymmetric DPCR with the following oligonucleotides (Sigma Genosys):• lacZ forward primer: 5′-ATG AAA GCT GGC TAC AGG AAG GCC.
• lacZ reverse primer: 5′-biotin-GGT TTA TGC AGC AAC GAG ACG TCA.
DNA stock solutions (nominally 100 µM) were prepared in a buffer solution containing 10 mM Tris-HCl (Sigma) and 1 mM EDTA (Sigma) (TE, pH 8.0). More dilute solutions of the oligomers were prepared in a 50 mM Tris-HCl (Scharlau) and 20 mM NaCl buffer solution (Tris-HCl, pH 7.2). Biotinylated probe solutions and amplicons final concentrations were determined by UV-Vis molecular absorption spectrometry using a Thermo Scientific NanoDrop™ 1000 Spectrophotometer. All oligonucleotide stock solutions were stored frozen at −20 °C. On the other hand, a 1% Triton X-100 (Probus) solution was used to lyse bacteria cells. All chemicals used were of analytical-reagent grade, and deionised water was obtained from a Millipore Milli-Q purification system.
The employed procedures to carry out the pre-treatment and modification of the gold screen-printed electrodes (AuSPEs), the magnetic beads (MBs) modification, the deposition of MBs onto tetrathiafulvalene (TTF)-modified AuSPEs, and the amperometric measurements, were similar to those described previously.12 Electrochemical instrumentation consisted of an ECO Chemie Autolab PSTAT 10 potentiostat with the software package GPES 4.9. Gold screen-printed electrodes (220AT, 4-mm ∅) were purchased from Dropsens (Oviedo, Spain), and included a silver pseudo-reference electrode and a gold counter electrode. An overnight pure culture of E. coli JM109 was grown at 37 °C for 24 h. Previous calibration studies with this microorganism17 allowed correlation of optical density at 600 nm with the number of viable bacteria per millilitre of culture. The culture was then heated in a 100 °C water bath for 15 min to kill all the bacteria. After this process, the genetic material of E. coli cells is liberated in solution. Serial dilutions from this solution in double distilled water allowed different solutions of genetic material corresponding to different E. coli cells concentrations to be obtained. A 1 mL aliquot of each culture concentration was centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 5 min to pellet the bacteria cells. Once the pellet was re-suspended in 50 µL of 1% Triton X-100 to lyse the bacterial cells, the samples were boiled for 10 min, and then rapidly cooled in an ice-water mixture for 3 min to precipitate the debris. A 10 µL aliquot of the supernatant obtained in each case was used as template for the asymmetric DPCR. A 100 µL PCR reaction mixture containing the above mentioned 10 µL aliquot, 1.6 µL of the 5 µM forward primer solution, 16 µL of the 5 µM reverse biotinylated primer solution, and other components following the protocol for PCR with Taq DNA polymerase, was employed. PCR experiments were carried out using an Applied Biosystems 2720 thermal cycler, with the following conditions: denaturation of E. coli, genomic DNA at 94 °C for 3 min followed by 35 cycles for 1 min at 94 °C; annealing for 1 min at 60 °C; extension for 1 min at 72 °C, and 3 min final extension. This process produced biotinylated single-stranded lacZ gene fragments as predominant PCR product. Then, these products were purified by using a High Pure PCR Product Purification Kit (Roche), screened by gel electrophoresis in 1.5% agarose gel, and visualized by ethidium bromide (EtBr) staining.
000 rpm for 5 min to pellet the bacteria cells. Once the pellet was re-suspended in 50 µL of 1% Triton X-100 to lyse the bacterial cells, the samples were boiled for 10 min, and then rapidly cooled in an ice-water mixture for 3 min to precipitate the debris. A 10 µL aliquot of the supernatant obtained in each case was used as template for the asymmetric DPCR. A 100 µL PCR reaction mixture containing the above mentioned 10 µL aliquot, 1.6 µL of the 5 µM forward primer solution, 16 µL of the 5 µM reverse biotinylated primer solution, and other components following the protocol for PCR with Taq DNA polymerase, was employed. PCR experiments were carried out using an Applied Biosystems 2720 thermal cycler, with the following conditions: denaturation of E. coli, genomic DNA at 94 °C for 3 min followed by 35 cycles for 1 min at 94 °C; annealing for 1 min at 60 °C; extension for 1 min at 72 °C, and 3 min final extension. This process produced biotinylated single-stranded lacZ gene fragments as predominant PCR product. Then, these products were purified by using a High Pure PCR Product Purification Kit (Roche), screened by gel electrophoresis in 1.5% agarose gel, and visualized by ethidium bromide (EtBr) staining.
Results and discussion
Disposable magnetic DNA sensors, using screen-printed gold electrodes (4 mm diameter) as drop-on sensor, were employed as amperometric transducers.12Fig. 1 shows schematically the fundamentals of the magnetic genosensor. A biotinylated 25-mer capture probe was attached to streptavidin-modified magnetic beads and hybridization with the biotinylated target was allowed to proceed. Then, a streptavidin-peroxidase polymer was attached to the biotinylated target, and the resulting modified magnetic beads were captured by a neodymium magnet on the surface of TTF-AuSPEs. Hybridization was monitored amperometrically at −0.15 V after the addition of 0.35 mM hydrogen peroxide. Confined-TTF on the electrode surface mediated the catalytic reduction of H2O2 by HRP, the electrochemical reduction of the generated TTF+ being measured at the above mentioned potential. No amperometric response was observed when H2O2 was added to the TTF-AuSPE modified with the magnetic beads-labelled ss-DNA before the hybridization process. Application of asymmetric PCR using genomic DNA extracted from an E. coli culture to amplify a 326 bp DNA fragment containing the target sequence allowed a detection limit for the amplicons of 2.5 aM to be obtained.12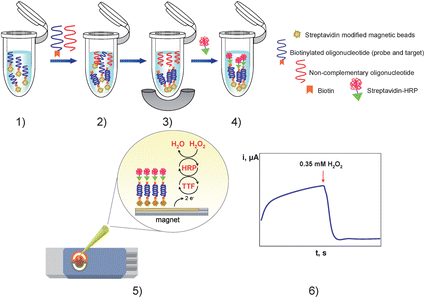 | ||
| Fig. 1 Schematic representation of the enzyme amplification protocol: (1) probe-modified magnetic beads washing step; (2) hybridization with the target lacZ gene probe; (3) hybrid-modified magnetic beads separation and non-complementary oligonucleotide extraction; (4) enzymatic labelling with streptavidin-HRP; (5) hybrid-modified magnetic beads deposition on the TTF-Au/SPEs; (6) amperometric detection of the mediated reduction of H2O2 with TTF. | ||
In this communication, we report a method to improve even more the sensitivity of this PCR-product assay as well as to reduce the assay time, which will allow the methodology to be used as a screening tool for the analysis of real samples. These advantages can be achieved by using asymmetric direct PCR, i.e., without previous genomic DNA isolation. Following the procedure described in the Experimental section, we obtained 326-bp biotinylated amplicons from the E. coli culture. Gel electrophoresis was used to confirm DPCR amplification. Fig. 2 shows, from left to right, the DNA size marker, the asymmetric DPCR products using supernatant obtained from different E. coli cultures (see Experimental section), and the PCR blank control (purified water as DNA template). The electrophoretic results confirmed the successful amplification of PCR products with the right size (326 bp) in spite of the lower EtBr staining efficiency for ssDNA. Fig. 2 also shows that a 326-bp DPCR product can be obtained with only 1 colony forming unit (cfu) per 100 mL.
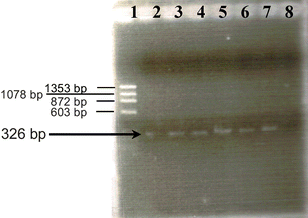 | ||
| Fig. 2 Gel electrophoresis detection of E. coli lacZ gene asymmetric DPCR products. Lane 1: Molecular weight marker (ΦX174-HaeIII digest). Lanes 2–7: asymmetric DPCR products for different cell concentrations (0.01 (2), 0.1 (3), 1 (4), 10 (5) 100 (6) and 1.0 × 104 cfu mL−1 (7)). No band was obtained with purified water used as negative PCR control (lane 8). | ||
Asymmetric DPCR samples were obtained from 0.01, 0.1, 1, 10, 100, 1 × 103 and 1.0 × 104 cfu mL−1 bacteria cultures. All these samples were 4 × 106-times diluted with PBS of pH 7.4, and 11.4 µL aliquots of the diluted preparations were tested following the procedure described previously for synthetic oligonucleotides.12Fig. 3 shows the amperograms recorded for a DPCR blank control (omitting the addition of the culture supernatant during the amplification process) and for 0.01, 100 and 1 × 104 cfu mL−1E. coli cultures. As it can be seen, the PCR blank control suggested a negligible non-specific adsorption. Moreover, amperograms obtained for asymmetric DPCR amplicons from 0.01, 100 and 1 × 104 cfu mL−1E. coli cultures exhibited similar responses, as well as those recorded from 0.1, 1, 10 and 1 × 103 cfu mL−1 (data not shown). This was expected since the final amplicon concentration was levelled off by PCR itself (see Fig. 2). These results were confirmed by repeating the measurements three times. Therefore, despite the high sensitivity achieved, it does not seem possible to quantify coliform density using asymmetric DPCR. However, taking into account that European Regulation N° 1441/2007 establishes that non-contaminated drinkable water should exhibit absence of E. coli in 100 mL of sample, and that the developed method allows an easy differentiation between 1 cfu E. coli/100 mL culture and the asymmetric DPCR blank control, the obtained results clearly demonstrate the usefulness of the combined asymmetric DPCR-magnetic disposable genosensor approach to specifically assess the presence or absence of E. coli contamination in real samples. Although the achieved goal is related to detection more than to quantification, a possibility to properly quantify coliforms at so low concentration levels would be the use of real-time-PCR systems, where samples can be taken in the exponential phase of amplification.18
![Amperometric signals obtained for the asymmetric DPCR blank control (1) and for asymmetric DPCR samples obtained from 0.01 (2), 100 (3) and 1 × 104 (4) cfu mL−1E. coli bacteria cultures. Supporting electrolyte, 0.1 M PBS buffer (pH 7.4) solution. [H2O2] = 0.35 mM, TTF loading, 2.5 µmol. Magnetic beads mass, 150 µg. Incubation conditions: biotinylated probe loading, 100 pmol, tinc = 30 min; thyb = 30 min. Enzymatic labelling: enzyme polymer loading, 1 µg; tinc = 30 min. Eapp = −0.15 V vs. Ag/AgCl.](/image/article/2009/AN/b815307h/b815307h-f3.gif) | ||
| Fig. 3 Amperometric signals obtained for the asymmetric DPCR blank control (1) and for asymmetric DPCR samples obtained from 0.01 (2), 100 (3) and 1 × 104 (4) cfu mL−1E. coli bacteria cultures. Supporting electrolyte, 0.1 M PBS buffer (pH 7.4) solution. [H2O2] = 0.35 mM, TTF loading, 2.5 µmol. Magnetic beads mass, 150 µg. Incubation conditions: biotinylated probe loading, 100 pmol, tinc = 30 min; thyb = 30 min. Enzymatic labelling: enzyme polymer loading, 1 µg; tinc = 30 min. Eapp = −0.15 V vs. Ag/AgCl. | ||
It is important to remark that the developed approach exhibits relevant analytical advantages with respect to other E. coli detection methods reported in the literature. As few as 1–5 E. coli cells in 100 mL of water were claimed to be detected by PCR amplification and electrophoresis using radiolabelled gene probes.16 However, application of this methodology to routine monitoring is difficult since visualization of the product by hybridization with a radioactive probe may require 2 to 3 days of exposure.19 Comparatively, the combined asymmetric DPCR-magnetic disposable genosensor approach allows a faster detection (only 5.5 hours are required) with a similar sensitivity. In fact, genomic sensors show higher detection limits. Thus, Mo et al.14 developed a quartz crystal microbalance (QCM) sensor based on a lacZ specific thiolated ssDNA probe able to detect 1 µg mL−1 target DNA and several viable E. coli cells in 100 mL of water. Another QCM DNA sensor based on nanoparticle amplification for the detection of E. coli O157:H7 eaeA gene, and using asymmetric PCR with biotin-labelled primers,20 was able to detect 2.67 × 102 cfu mL−1.
Concerning the comparison with genosensors using electrochemical transduction, a label-free electrochemical assay for the detection of RNA from coliform bacteria using DNA probe-coated magnetic beads and monitoring of changes in the oxidation state of guanine nucleotides, quantitatively detected 107E. coli cells in 4 h.21 Farabullini et al, described the simultaneous detection of four different food pathogenic bacteria (Salmonella enterica, Lysteria monocytogenes, Staphylococcus aureus and E. coli O157:H7) by means of a disposable electrochemical genosensor array.22 Unmodified PCR products obtained from the corresponding bacteria genomic DNAs were captured at the electrode surface via sandwich hybridization with thiol-tethered probes immobilized on the screen-printed array and biotinylated signalling probes. Electrochemical detection was carried out coupling the resulting biotinylated hybrids with alkaline phosphatase, allowing the detection of mixtures of DNA samples from different bacteria at the nanomolar level. Finally, Lermo et al. reported recently an electrochemical assay based on PCR specific amplification of the E. coli O157:H7 eaeA gene and detection of amplicons using HRP as enzyme marker.23 Using a bulk-modified avidin biocomposite and a magneto sensor, 4.5 and 0.45 ng µL−1 of the original bacteria genome were detected, respectively, after 10 PCR cycles.
Conclusions
The use of disposable magnetic DNA hybridization amperometric sensors, which combine the use of MBs for DNA isolation and hybridization and of TTF-mediated HRP-amplified detection, has demonstrated to constitute an efficient strategy for rapid, specific and sensitive detection of asymmetric DPCR amplified products obtained directly from E. coli bacterial cultures, and thus simplifying sample treatment procedures. The use of hybridization sensors avoids one of the major drawbacks of PCR analysis, false-positive results, while coupling with asymmetric DPCR allows the detection of as few as one E. coli cfu per 100 mL, and therefore demonstrating its usefulness for the assessment of drinkable water supplies safety. Moreover, the use of disposable mass-produced SPEs allows the preparation and handling of up to 30 sensors per day, one single assay taking only hours to be completed, as opposed to days for culture-based techniques. All these features are important for the application of the developed genosensors to the detection of Enterobacteriaceae in real samples. However, currently, the genosensor can only act as an alarm or screening device (yes or no system), and work is currently in progress to adapt it for the quantitative detection of coliforms. An added value of the proposed strategy is the possibility to be easily adapted to detect other significant pathogenic microorganisms, since it requires only a single DNA probe for such purpose.Acknowledgements
The financial support of Santander/Complutense Research Project PR 27/05-13953, and of the Spanish Ministerio de Educación y Ciencia Research Project CTQ2006-02743BQU is gratefully acknowledged. Oscar A. Loaiza acknowledges a pre-PhD fellowship of the Universidad Complutense de Madrid. Susana Campuzano acknowledges a “Juan de la Cierva” contract to the Spanish Ministerio de Educación y Ciencia.Notes and references
- J. M. Simpson and D. V. Lim, Biosens. Bioelectron., 2005, 21, 881–887 CrossRef CAS.
- E. Paleček and M. Fojta, Talanta, 2007, 74, 276–290 CrossRef CAS.
- M. Fuentes, C. Mateo, A. Rodríguez, M. Casqueiro, J. C. Tercero, H. H. Riese, R. Fernández-Lafuente and J. M. Guisán, Biosens. Bioelectron., 2006, 21, 1574–1580 CrossRef CAS.
- J. Wang, Electroanalysis, 2007, 19, 769–776 CrossRef CAS.
- J. Wang, G. U. Flechsig, A. Erdem, O. Korbut and P. Grundler, Electroanalysis, 2004, 16, 928–931 CrossRef CAS.
- F. Lucarelli, S. Tombelli, M. Minunni, G. Marrazza and M. Mascini, Anal. Chim. Acta, 2008, 609, 139–159 CrossRef CAS.
- E. Giakoumaki, M. Minunni, S. Tombelli, I. E. Tothill, M. Mascini, P. Bogani and M. Buiatti, Biosens. Bioelectron., 2003, 19, 337–344 CrossRef CAS.
- S. K. Poddar, Molecular Cellular Probes, 2000, 14, 25–32 Search PubMed.
- K. A. Fode-Vaughan, C. F. Wimpee, C. C. Remsen and M. L. P. Collins, BioTechnol., 2001, 31, 598–607 Search PubMed.
- K. A. Fode-Vaughan, J. S. Maki, J. A. Benson and M. L. P. Collins, Lett. Appl. Microbiol., 2003, 37, 239–243 CrossRef CAS.
- R. E. Hodson, W. A. Dustman, R. P. Garg and M. A. Moran, Appl. Environ. Microbiol., 1995, 61, 4074–4082 CAS.
- O. A. Loaiza, S. Campuzano, M. Pedrero, M. I. Pividori, P. García and J. M. Pingarrón, Anal. Chem., 2008 DOI:10.1021/ac801319b.
- Y. Zhang, A. Pothukuchy, W. Shin, Y. Kim and A. Heller, Anal. Chem., 2004, 76, 4093–4097 CrossRef CAS.
- X. T. Mo, Y. P. Zhou, H. Lei and L. Deng, Enzyme Microb. Technol., 2002, 30, 583–589 CrossRef CAS.
- A. Kalnins, K. Otto, U. Rüther and B. Müller-Hill, EMBO J., 1983, 2, 593–597 CAS.
- A. K. Bej, R. J. Steffan, J. DrCesare, L. Haff and R. M. Atlas, Appl. Environ. Microbiol., 1990, 56, 307–314 CAS.
- J. Sambrook, E. F. Fritsch, T. Maniatis, in Molecular cloning: a laboratory manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y., 1989 Search PubMed.
- F. Lucarelli, G. Marrazza and M. Mascini, Biosens. Bioelectron., 2005, 20, 2001–2009 CrossRef.
- A. Rompré, P. Servais, J. Baudart, M. R. de-Roubin and P. Laurent, J. Microbiol. Methods, 2002, 49, 31–54 CrossRef.
- X. Mao, L. Yang, X. L. Su and Y. Li, Biosens. Bioelectron., 2006, 21, 1178–1185 CrossRef CAS.
- M. J. LaGier, C. A. Scholin, J. W. Fell, J. Wang and K. D. Goodwin, Mar. Pollut. Bull., 2005, 50, 1251–1261 CrossRef CAS.
- F. Farabullini, F. Lucarelli, I. Palchetti, G. Marrazza and M. Mascini, Biosens. Bioelectron., 2007, 22, 1544–1549 CrossRef CAS.
- A. Lermo, E. Zacco, J. Barak, M. Delwiche, S. Campoy, J. Barbé, S. Alegret and M. I. Pividori, Biosens. Bioelectron., 2008, 23, 1805–1811 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2009 |