In-capillary enrichment, proteolysis and separation using capillary electrophoresis with discontinuous buffers: application on proteins with moderately acidic and basic isoelectric points
Chandra A.
Nesbitt
and
Ken K.-C.
Yeung
*
Department of Chemistry and Department of Biochemistry, The University of Western Ontario, London, Ontario, Canada. E-mail: kyeung@uwo.ca; Fax: +1 519 661 3022; Tel: +1 661 2111 86439
First published on 18th October 2008
Abstract
Advances in mass spectrometry and capillary-format separation continue to improve the sensitivity of protein analysis. Of equal importance is the miniaturization of sample pretreatment such as enrichment and proteolysis. In a previous report (Nesbitt et al., Electrophoresis, 2008, 29, 466–474), nanoliter-volume protein enrichment, tryptic digestion, and partial separation was demonstrated in capillary electrophoresis followed by MALDI mass spectral analysis. A discontinuous buffer system, consisting of ammonium (pH 10) and acetate (pH 4), was used to create a pH junction inside the capillary, trapping a protein with a neutral isoelectric point, myoglobin (pI 7.2). Moreover, co-enrichment of myoglobin with trypsin led to an in-capillary digestion. In this paper, the ability of this discontinuous buffer system to perform similar in-capillary sample pretreatment on proteins with moderately acidic and basic pI was studied and reported. Lentil lectin (pI 8.6) and a multi-phosphorylated protein, β-casein (pI 5.1), were selected as model proteins. In addition to the previously shown tryptic digestion, proteolysis with endoproteinase Asp-N was also performed. Digestion of these acidic and basic pI proteins produced a few peptides with extreme pI values lying outside the trapping range of the discontinuous buffer. An alteration in the peptide trapping procedure was made to accommodate these analytes. Offline MALDI mass spectral analysis confirmed the presence of the expected peptides. The presented miniaturized sample pretreatment methodology was proven to be applicable on proteins with a moderately wide range of pI. Flexibility in the choice of protease was also evident.
Introduction
Protein identification by tandem mass spectrometry (MS/MS) generally requires samples to be pretreated with sequence-specific proteolytic digestion.1,2 Conventional proteolysis is performed in-solution in the presence of solubilized enzymes. The enzyme is generally prepared at low concentrations to minimize autodigestion. The autolyzed peptides from the enzyme can significantly suppress the analyte signals, especially for analytes at trace levels. As a result, the in-solution approach suffers from low digestion efficiency and longer reaction times. The use of immobilized enzymes on solid phases has become a popular alternative.3,4 Immobilization of enzymes minimizes autolysis,5,6 allows for the use of higher enzyme concentrations, and thus yields shorter digestion times. In addition, immobilized enzyme solid phases can be adapted as microreactors for integration with flow systems.7 For example, an immobilized trypsin reactor has been coupled to a chromatographic column and online mass spectrometry via a switching valve.4,8 Likewise, the use of monolithic on-capillary pepsin microreactors has been reported in capillary electrophoresis.9 In addition, integrated digestion reactors have also been developed on microfluidic devices for various protein sample pretreatment.10–12 The implementation of solid phases in microfluidic channels is however, not straightforward and can significantly complicate the manufacturing and operational processes, making the device less suitable for disposable or multiplexed applications. Some of the technical challenges associated with trypsin immobilized particles are the requirement of frits within the micro-channels. This often leads to clogging of the chip.13 Due to the size of the micro-channels, packing of the stationary phase can also lead to non-homogeneous particle density at the channel walls resulting in less efficient digestions.14 Alternatively, monolithic material can be synthesized in situ within the micro-channels. This approach can in theory provide a more homogeneous solid phase without the requirement of retaining frits, but they are currently not commercially available.15 Hence, devices based on open channel format remains to be more straightforward and more economically advantageous.Our research group recently developed an integrated methodology for the enrichment, digestion, and partial separation of protein samples at sub-microliter volumes in open capillaries.16 The experiment was performed on a commercial CE instrument, in which the capillary was filled with a discontinuous buffer system, consisting of an acidic buffer (pH 4.25 acetate) and a basic buffer (pH 9.75 ammonium). Upon voltage application, with the cathode placed at ammonium, and the anode placed at acetate, a sharp pH junction was formed at the boundary of the discontinuous buffer inside the capillary.17,18 Proteins and/or peptides with pI falling within the pH range between the two buffers were mobilized towards, and became trapped as zwitterions, at the pH junction. Hence, this technique was referred to as capillary isoelectric trapping (cIET). A protein standard of neutral pI, myoglobin, was chosen for the demonstration of in-capillary enrichment.18 Co-enrichment of proteins was also observed when a sample of protein mixture was introduced. This led to the co-focusing of myoglobin and trypsin, and in turn the in-capillary proteolysis. Most of the resulting peptides from myoglobin had near-neutral pI and thus also remained focused by the pH junction.
In contrast to the near-neutral pI proteins and peptides, strongly acidic (pI < 4.25) and basic (pI > 9.75) species should not be captured by the discontinuous buffer. This selective enrichment was previously demonstrated with an acidic protein, amyloglucosidase (pI 3.6), and a basic protein, cytochrome c (pI 10.6). They migrated away from the pH junction and eventually exited the capillary without enrichment.18 Nevertheless, mildly acidic and basic proteins, with pI between 4.25 and 9.75, are still expected to be enriched by cIET. In this work, the enrichment of these mildly acidic and basic proteins was studied, using lentil lectin (pI 8.6) as the model of weakly basic pI protein and β-casein (pI 5.1) as the weakly acidic pI protein.
β-Casein, a multi-phosphorylated protein, was a particularly interesting analyte. The tryptic digestion of β-casein generates two highly acidic phosphopeptides (pI 3.29 and 1.37). Compared to the other non-phosphorylated peptides, these phosphopeptides exhibit a much lower degree of ionization in mass spectrometry (MS), and thus routinely require pre-MS isolation for highly sensitive detection. The in-capillary enrichment and digestion of β-casein by cIET was performed in this work. Following the in-capillary digestion, isolation of the phosphopeptides was attempted by selectively focusing the remaining non-phosphorylated peptides based on their near-neutral pI. Offline MALDI MS was again used in this work to detect and identify the peptides which eluted from the capillary.
Finally, in addition to using trypsin, in-capillary digestion with endoproteinase Asp-N (abbreviated as ‘AspN’ in this work) was also attempted to illustrate the versatility of our methodology. While the majority of proteolysis in this field is performed using trypsin, the use of alternative enzymes is necessary for proteins lacking arginine and lysine residues.
Experimental
Reagents and samples
All solutions were prepared using 18.2 MΩ deionized water from Millipore Water Purification System (Bedford, MA, USA). Reagent grade acetic acid, ammonium hydroxide, phosphoric acid, and trifluoroacetic acid were purchased from EM Science (Gibbstown, NJ). Calcium chloride, ammonium bicarbonate and citric acid were purchased from Sigma Aldrich (Markham, ON, Canada). The zwitterionic phospholipid surfactant, 1,2-dilauroyl-sn-phosphotidylcholine (DLPC), was purchased from Avanti Polar Lipids, Inc., (Alabaster, AL) and prepared as previously described in the literature.19 DLPC forms a semi-permanent bilayer coating on the silica capillary surface upon rinsing. This coating is effective in suppressing the electroosmotic flow and minimizing protein adsorption onto the capillary wall.19,20 After the coating is formed, experiments were performed without addition of DLPC in the discontinuous buffer. Desorption of DLPC from the capillary wall coating was found to be minimal. Detrimental effects on the peptide signals were not observed in MALDI MS.21–23 Lentil lectin (lens culinaris) (MW 49,000, pI 8.6), β-casein (bovine milk) (MW 23583.3 Da, pI 5.1) and myoglobin (horse heart) (MW 16952.5 Da, pI 7.2), were used as received from Sigma-Aldrich. 2,5-Dihydroxybenzoic acid (DHB) was purchased from Sigma-Aldrich (St. Louis, MO, USA). HPLC grade acetone, methanol, and ethanol were purchased from Fisher Scientific Ltd. (Nepean, ON, Canada). L-(tosylamido-2-phenyl) ethyl chloromethyl ketone (TPCK) treated trypsin (from bovine pancreas) (MW 23,800 Da, pI 10.0) and endoproteinase Asp-N (from mutant strain of Pseudomonas fragi) (MW 24,500 Da, pI 8.65) were purchased from Sigma Aldrich. The estimated isoelectric points of the peptides from β-casein, lentil lectin, and myoglobin were computed using the online pI calculator provided by ExPASy.In-vial protein digestion
All in-vial digestions were performed in low retention vials from Axygen Scientific (Union City, CA, USA) in the presence of 50 mM ammonium bicarbonate (measured pH 8.0) and 2 mM calcium chloride (CaCl2). To generate the β-casein and lentil lectin peptide solutions, digestions were performed individually overnight at 37 °C with 1.0 mL of 1.0 µg/µL β-casein and lentil lectin. The trypsin-to-β-casein ratio was 1:20 w/w and the trypsin-to-lentil lectin ratio was 1:100 w/w. The dried samples were re-dissolved in 1.0 mL of water and stored at −20 °C prior to use.In-capillary protein enrichment
All cIET enrichment of proteins were performed on an Agilent 3D-CE Capillary Electrophoresis Instrument (Palo Alto, CA, USA) with direct UV absorbance detection at 200 nm. Data acquisition was obtained through the ChemStation software by Agilent. Fused silica capillaries were purchased from Polymicro Technologies (Pheonix, AZ, USA). The dimensions of the capillaries were 50 µm in inner diameter, 360 µm in outer diameter, 48.5 cm in total length, and 40 cm in length to detector. Prior to use, all new capillaries were flushed (1 bar) with sodium hydroxide (0.1 M) for 5 min, deionized water for 5 min, and finally with DLPC in 0.1 mM in Tris-HCl and CaCl2 for 20 min to form an inner wall coating. In between runs, the capillary was re-coated with DLPC for 10 min (1 bar). A discontinuous buffer system of pH 9.75 ammonium (10 mM) and pH 4.25 acetate (10 mM) was used for all in-capillary enrichment and digestion. The selected buffers and pH were previously established to be optimal for creating a very sharp pH junction.17 The pH of the acetate buffer was adjusted with ammonium hydroxide, and likewise the pH of the ammonium buffer was adjusted with acetic acid. The protein or peptide samples were prepared by dissolving the sample in the ammonium buffer (pH 9.75) and then allowed to fill the capillary with an internal volume of 0.95 µL. The acetate buffer was placed in the inlet reservoir (anodic electrolyte) and the ammonium buffer was placed in the outlet reservoir (cathodic electrolyte) during voltage application. A constant voltage of 30 kV was used for all experiments.In-capillary protein digestion at a pH junction
To begin in-capillary digestion, the capillary (0.95 µL) was pre-filled with protein solution prepared in the ammonium buffer (cathodic electrolyte) described above. In the experiment involving trypsin digestion of lentil lectin and β-casein, approximately 1.7 nL of trypsin solution, prepared in the acetate buffer (anodic electrolyte), was injected at the anodic end of the capillary under constant pressure of 10 mbar for 5 s. The trypsin-to-β-casein ratio was 1:20 w/w, and the trypsin-to-lentil lectin ratio was 1:15 w/w. The enzyme-to-substrate ratios for all in-capillary digestion experiments were determined based on the quantities (weights) of the enzyme and substrate injected into the capillary. The concentration of the trypsin was adjusted to obtain the desired enzyme-to-substrate ratio; e.g., 1.5 µg/µL trypsin was used with 0.05 µg/µL of β-casein, and 2.0 µg/µL trypsin was used with 0.05 µg/µL of lentil lectin.To perform AspN digestion of myoglobin, approximately 7.2 nL of AspN solution, prepared in the acetate buffer, was injected at the anodic end of the capillary under constant pressure of 20 mbar for 10 s. Since the digestion was performed at two different concentrations of myoglobin, the concentration of AspN in 7.2 nL was varied to maintain a constant ratio of 1:70 (w/w) relative to the amount of myoglobin injected into the capillary; e.g., 0.1 µg/µL AspN for 0.05 µg/µL myoglobin, and 0.02 µg/µL AspN for 0.01 µg/µL myoglobin. The cIET co-enrichment of protein and enzyme was allowed to proceed for 10 minutes, then voltage application was suspended. The CE capillary cartridge remained in the instrument, at which point the cartridge temperature was raised to 37 °C for 2 h to promote digestion. Following the incubation period, re-application of voltage was carried out to re-focus the newly formed peptides.
Offline MALDI MS analysis
Toward the end of the enrichment process, the residual EOF would bring the focused peptide band past the on-capillary detection window, which was located 8.5 cm away from the capillary outlet. The arrival time of the sample band, with incubation time subtracted, was used to determine the average EOF in that run. Based on this calculated EOF, additional voltage was applied (typically 5 to 10 minutes) to mobilize the sample band from the detection point to the capillary outlet (cathode).To transfer the sample band from the capillary onto the MALDI sample plate, the cathodic end of the capillary was re-positioned outside the instrument, while the anodic end remained inside the instrument. Elution of the capillary content was performed by applying a constant pressure (50 mbar) at the capillary inlet in 20-second intervals, yielding 35 nL fractions which were manually deposited onto the sample plate pre-spotted with matrix. Several matrices and matrix additives or co-matrices have been reported in literature to enhance the phosphopeptide response in MALDI MS. These matrices include α-cyano-4-hydroxycinnamic acid (CHCA), 2′,4′,6′-trihydroxy-acetophenone, and DHB. Examples of additives include trifluoroacetic acid (TFA), ammonium salt, phosphoric acid, hydrochloric acid, and nitrocellulose. In this work, a combination of DHB and ammonium citrate with 1% phosphoric acid was used for all samples. Sample deposition on the MALDI target was based on the three layer method previously described in literature.21,23 Briefly, the first layer consisted of 0.5 µL of 20 mg/mL DHB in acetone/methanol (1:1 by volume) and the second layer consisted of 0.04 µL of 20 mg/mL DHB in ammonium citrate/ethanol/phosphoric acid (79:20:1 by volume). After drying, the sample from the capillary was spotted manually as the third layer. Between depositions of consecutive spots, the capillary tip was cleaned by wiping to minimize carry over from spot-to-spot.
MALDI mass spectra of peptides and proteins were obtained using the following two instruments. A 4700 Proteomic Discovery System MALDI TOF/TOF MS (Applied Biosystems, Farmingham, MA) was used to perform MS analyses for the experiments on β-casein digestion by trypsin. The instrument is equipped with a 355-nm Nd:Yag laser. Both linear and reflectron modes were used, in positive ion mode, for detecting β-casein and its peptides respectively. All voltage settings were left at the default values preset by Applied Biosystems. Mass spectra were recorded as sums of 1000 shots under video monitoring in all experiments.
A Bruker Reflex IV MALDI time-of-flight MS (Bremen/Leipzig, Germany) was used to perform MS analyses on the digestion of lentil lectin and myoglobin. The instrument is equipped with a 337-nm nitrogen laser. Both linear and reflectron modes were used, in positive ion mode, for detecting the proteins and their peptides respectively. All voltage settings were left at the default values preset by Bruker. Mass spectra were recorded as sums of 100 shots under video monitoring in all experiments. In our work, considerable differences in performance between these two instrument were not found.
Results and discussion
Even though our cIET enrichment methodology had been applied on a number of proteins and peptides (including myoglobin, BSA, and tryptic peptides of myoglobin),16–18 the digestion step had only been reported with myoglobin to date. It is therefore important in this work to demonstrate its application on other proteins, particularly those with non-neutral pI.Enrichment and tryptic digestion of lentil lectin
The first study was conducted with lentil lectin, which was chosen for its weakly basic pI of 8.6. The experimental setup was similar to that used for myoglobin. Briefly, 0.05 µg/µL lentil lectin was prepared in the pH 9.75 ammonium buffer and was injected to fill the capillary. Trypsin was dissolved in the pH 4.25 acetate buffer and was injected as a small plug at the anode (Fig. 1A). Upon voltage application, a pH junction was formed, bringing the lentil lectin and trypsin together. The suppressed EOF, resulting from the DLPC coated capillary, slowly carried the pH junction and samples toward the cathode (Fig. 1B). Voltage was suspended for 2 h and the temperature of the capillary in the cartridge was raised to 37 °C for the digestion (Fig. 1C). Next, the voltage was resumed for approximately 10 min to re-focus the peptides (Fig. 1D). Finally, an applied pressure was used to elute the sample as droplets onto the MALDI target for MS analysis (Fig. 1E). When the analyte band at the pH junction passed the on-capillary UV absorption detector, a single, sharp, intense signal similar to Fig. 6 in ref. 18 was observed (not shown), confirming the successful enrichment of the basic pI protein.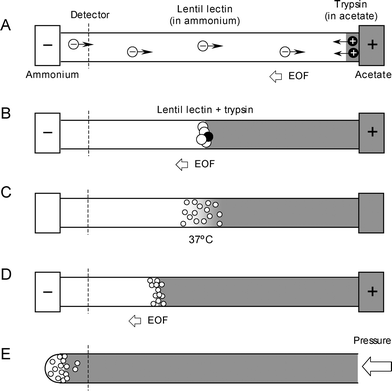 | ||
| Fig. 1 Schematic of integrated in-capillary sample preparation: injection of 0.05 µg/µl lentil lectin and trypsin in discontinuous buffer (A), co-enrichment of protein and enzyme at the pH junction (B), digestion at elevated temperature (C), re-application of voltage to refocus the peptides (D), elution of peptides as droplets for offline MALDI MS analysis (E). | ||
A list of peptides and their corresponding MS signal intensities identified from the collected fractions are shown in Table 1. Peptides were detected in 9 fractions, which was comparable to the 7–10 fractions previously observed for myoglobin under identical conditions.16 Another similarity was the distribution of the peptides; that is, the higher pI peptides were observed in the earlier fractions and the lower pI peptides were observed in later fractions. This partial separation based on pI was previously attributed to an isoelectric focusing effect of the discontinuous buffer on the peptides. The 6 identified peptides of lentil lectin corresponded to a relatively low sequence coverage of 29%.
| Peptide sequence | Theo. MM [M + H]+ | Expt. MM [M + H]+ | Calc. pI | Spot 1 | Spot 2 | Spot 3 | Spot 4 | Spot 5 | Spot 6 | Spot 7 | Spot 8 | Spot 9 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ALYSTPIHIWDR | 1471.77 | 1471.8 | 6.79 | 2726 | 129 | |||||||
| HIGIDVNSIK | 1095.62 | 1095.6 | 6.74 | 220 | 78 | |||||||
| FSPDQKNLIFQGDGYTTK | 2059.01 | 2059.0 | 5.96 | 85 | 215 | 131 | 343 | 522 | 940 | 587 | ||
| SWNLQNGER | 1103.52 | 1103.5 | 5.72 | 5054 | 507 | 175 | 231 | 143 | 78 | 113 | 201 | |
| DVVPEWVR | 999.53 | 999.5 | 4.37 | 173 | 324 | 238 | 680 | |||||
| TSQTVAVEFDTFYNAAWDPSNK | 2491.14 | 2491.1 | 4.03 | 40 | 57 |
Upon examination of the amino acid sequence of lentil lectin, the non-detected portion of the protein was from 4 large peptides with MW ranging from 3200 to 4500 Da. It was therefore logical to presume that the non-detection was a result of MALDI MS being less sensitive in detecting the larger peptides (MW > 3000). To test this hypothesis, an in-vial digestion was performed overnight on a 20-times concentrated lentil lectin solution (1.0 µg/µL). The solution was dried under vacuum to remove the volatile salt, re-solvated in the same volume of water, and was spotted (0.25 µL) for MALDI MS analysis. Intense signals from the 6 peptides listed in Table 1, as well as weak signals from 2 of these peptides with a missed cleavage were observed. As predicted, the 4 large peptides with MW > 3000 Da were not detected (data not shown). Since this control experiment eliminated the use of any capillary electrophoresis, we therefore concluded the low sequence coverage obtained from lentil lectin was not related to the effectiveness of our in-capillary digestion.
Enrichment and tryptic digestion of β-casein
Phosphorylation lowers the pI of a protein due to the addition of the highly acidic phosphate group; e.g., the pKa1 and pKa2 of the phosphate of phosphoserine are 0.9 and 6.1.24 Five phosphoserine residues are present in β-casein (bovine milk): S15, S17–19, and S35, resulting in a calculated pI of 5.1. β-Casein was selected as a model protein to test the in-capillary enrichment and digestion of weakly acidic pI proteins using discontinuous buffers. Since pI 5.1 falls within the discontinuous buffer range, β-casein is expected to be enriched at the pH junction.The cIET enrichment of β-casein was first confirmed by the UV absorption signal, which revealed the expected characteristic single-peak (data not shown). Next, in-capillary enrichment and digestion of 0.05 µg/µL β-casein was performed by incorporating trypsin as illustrated in Fig. 1. Following sample deposition and MALDI MS analysis, 6 peptide peaks were identified (20% sequence coverage) however, none of them contained any phosphoserines (data not shown).
When digested with trypsin, the phosphate groups in β-casein were distributed into two peptides: β1 with a single phosphate, and β2 with four phosphate groups. The calculated pI values of β1 and β2 were 3.29 and 1.37 respectively. On the other hand, the six non-phosphorylated peptides identified from the mass spectra had pI values ranging from 4.4 to 8.8. Given the boundary pH of our discontinuous buffer was 4.25 (acetate) and 9.75 (ammonium), the pH junction indeed selectively enriched the six observed non-phosphorylated peptides, while allowing the two highly acidic phosphopeptides to exit the capillary. Similar selective enrichment behaviour was previously demonstrated to remove amyloglucosidase (pI 3.6).18
Readers should realize that the voltage application in the current methodology occurred in two stages. The first voltage application was for the enrichment of the intact β-casein protein molecules. In the presence of the co-enriched trypsin, digestion proceeded without the applied voltage. Next, voltage was applied for the second time to refocus the resulting peptides at the pH junction. This was the step which separated the highly acidic peptides (pI < 4.25) from the remaining peptides with pI between 4.25 and 9.75. Typically, the duration of the second voltage application step was 5 to 10 minutes, adjusted depending on the magnitude of the residual EOF. Evidently, this period was adequate to completely mobilize the phosphopeptides out of the capillary.
To prevent the complete removal of phosphopeptides, the post-digestion voltage application step was shortened to 2 minutes. The intensities and pI of the identified peptides resulting from the in-capillary enrichment and digestion of β-casein are shown in Table 2. A total of ten peptides, including β1 and β2, were observed. Under the adjusted (2-minute) post-digestion focusing, the phosphopeptides were clearly separated from the non-phosphorylated peptides based on pI, while remaining inside the capillary for subsequent MALDI MS analysis.
| Peptide sequence | Theo. MM [M + H]+ | Expt. MM [M + H]+ | Calc. pI | Spot 1 | Spot 2 | Spot 3 | Spot 4 | Spot 5 | Spot 6 | Spot 7 | Spot 8 | Spot 9 | Spot 10 | Spot 11 | Spot 12 | Spot 13 | Spot 14 | Spot 15 | Spot 16 | Spot 17 | Spot 18 | Spot 19 | Spot 20 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AVPYPQR | 830.45 | 830.4 | 8.79 | 641 | 1294 | 3397 | 10236 | 6002 | 2528 | 291 | |||||||||||||
| VLPVPQK | 780.50 | 780.4 | 8.72 | 1042 | 454 | 2904 | 756 | 3018 | 7941 | 2803 | 449 | 107 | |||||||||||
| HKEMPFPK | 1013.52 | 1013.4 | 8.60 | 1444 | 840 | 3324 | 7356 | 3791 | 1023 | ||||||||||||||
| EMPFPK | 748.37 | 748.4 | 6.10 | 225 | 942 | 2332 | 2460 | 2062 | 249 | ||||||||||||||
| LLYQEPVLGPVR | 1383.65 | 1383.6 | 6.00 | 266 | 387 | 574 | 667 | 282 | |||||||||||||||
| GPFPIIV | 742.45 | 742.4 | 5.52 | 494 | 560 | 1135 | 1038 | 633 | 116 | ||||||||||||||
| YPVEPFTER | 1137.60 | 1137.5 | 4.48 | 1170 | 1908 | 492 | 2487 | 2333 | |||||||||||||||
| DMPIQAFLLYQEPVLGPVR | 2186.17 | 2186.1 | 4.37 | 134 | 58 | 449 | |||||||||||||||||
| FQpSEEQQQTEDELQDK | 2061.82 | 2061.7 | 3.29 | 90 | 135 | 118 | 169 | 110 | 126 | 80 | 106 | 99 | 56 | ||||||||||
| RELEELNVPGEIVEpSLpSpSpSEESITR | 3122.26 | 3122.3 | 1.37 | 43 | 136 | 393 | 277 | 126 | 51 | 121 | 52 | 192 | 68 |
In comparison, MALDI MS analysis was performed on a 0.1 µg/µL β-casein peptide sample pre-digested in-vial overnight. Despite the higher sample concentration, only β1 and 6 non-phosphorylated peptides (pI 3.7 to 8.8) were identified. The tetra-phosphorylated peptide, β2, was not detected, mostly likely due to ionization suppression in the presence of the non-phosphorylated peptides. This clearly demonstrated that cIET not only provided an effective means of in-capillary protein enrichment and digestion at small sample volumes, it also offered a separation of the resulting peptides based on pI. This separation was particularly useful in isolating the peptides with highly acidic pI (and in theory, peptides with highly basic pI). In this case, it was shown to be ideal for the analysis of protein phosphorylation at very small sample quantity.
AspN as alternative proteases for in-capillary digestion
Trypsin is the most commonly used enzyme in protein analysis. It cleaves at the C-terminal to lysine and arginine residues, typically generating peptides within the mass range of 500–5000 Da. For proteins low in lysine and arginine residues, alternative proteases are required; such as endoproteinases AspN and GluC.25In this section, the in-capillary digestion of myoglobin by cIET was attempted using AspN as the protease. AspN primarily cleaves on the N-terminal side of aspartic acid residues.26 Like trypsin, AspN is most active at neutral pH (between pH 6.0 and 8.5) and at an elevated temperature of 37 °C. The calculated pI of AspN is 8.65, and therefore was expected to become co-enriched with the substrate myoglobin at the pH junction for digestion. Conventional in-vial proteolysis by AspN is typically performed at 37 °C for 2 to 18 hours depending on the enzyme-to-substrate ratio. In the next experiment, the optimal digestion time was determined by monitoring the progression of in-capillary AspN proteolysis of myoglobin. The capillary was filled with a freshly mixed solution of AspN and 0.5 µg/µL myoglobin at an enzyme-to-substrate ratio of 1:70 w/w. A higher concentration of myoglobin was used since the protein enrichment step was omitted. To mimic the near-neutral pH environment at the pH junction, the enzyme and substrate were prepared in a mixture containing 10 mM pH 9.75 ammonium and 10 mM pH 4.25 acetate. The mixture was allowed to react for variable time periods (1, 2 and 4 h) in the capillary at 37 °C. Following the various reaction times, the capillary content was spotted for MALDI MS analysis. It was discovered that the same 8 peptides were found in all three cases, corresponding to a sequence coverage of 66%. The MS intensities of myoglobin and its peptides were additionally recorded, and the results suggested that the 2 h digestion time was most optimal.
The procedure of in-capillary digestion of myoglobin with AspN was similar to that with trypsin (as illustrated in Fig 1). The injection of trypsin was replaced with the injection of AspN, prepared in the same 10 mM pH 4.25 acetate buffer. Co-enrichment of enzyme and substrate took place upon voltage application. After 10 minutes, voltage application was suspended for the 2 h digestion at 37 °C, followed by re-application of voltage (5–10 min) to refocus the newly formed peptides. With an initial myoglobin concentration of 0.05 µg/µL, the MALDI MS analysis identified a total of 8 peptides (Table 3). Most of the peptides were identified in Spots 1 and 2. The pI 4.14 peptide, having a pI below the pH 4.25 of acetate, was identified in later factions. This was another example of highly acidic pI peptides being separated by the pH junction based on pI. The eight identified peptides translated to a sequence coverage of 68%. Compared to the peptides identified from the in-vial digestion, this set of peptides contained a peptide with a missed cleavage (pI 6.26) that gave rise to a slightly higher sequence coverage. Upon examination of the amino acid sequence of myoglobin, when completely digested with AspN, a total of nine peptides are produced in theory. A large peptide of 49 amino acid residues (MW 5426.46 Da, sequence coverage of 32%) was never observed by MS in this work. Hence, we concluded that similar sequence coverage was found for the in-vial and in-capillary digestions.
| Peptide sequence | Theo. MM [M + H]+ | Expt. MM [M + H]+ | Calc. pI | Spot 1 | Spot 2 | Spot 3 | Spot 4 | Spot 5 | Spot 6 | Spot 7 |
|---|---|---|---|---|---|---|---|---|---|---|
| DAIIHVLHSKHPG | 1423.78 | 1423.8 | 7.02 | 486 | ||||||
| DKFKHLKTEAEMKASE | 1891.96 | 1892.0 | 6.78 | 2805 | 1149 | |||||
| DAIIHVLHSKHPGDFGA* | 1813.93 | 1813.9 | 6.26 | 3231 | 1495 | |||||
| DAQGAMTKALELFRNDIAAKYKELGFQG* | 3085.58 | 3085.5 | 6.21 | 77 | 57 | |||||
| DIAAKYKELGFQG | 1439.75 | 1439.8 | 6.07 | 4028 | 3180 | 217 | ||||
| DAQGAMTKALELFRN | 1664.84 | 1664.8 | 6.07 | 6962 | 4762 | 119 | 169 | |||
| DIAGHGQEVLIRLFTGHPETLEKF | 2707.42 | 2707.4 | 5.36 | 1065 | 958 | |||||
| GLSDGEWQQVLNVWGKVEA | 2115.05 | 2115.1 | 4.14 | 101 | 119 | 341 | 1383 |
To illustrate the applicability of our AspN digestion on lower concentration samples, the above experiment was repeated on 0.01 µg/µL myoglobin. In this case, a total of 6 peptides were identified (pI 4.14, 5.36, 6.07, 6.07, 6.26 and 6.78 listed in Table 3). These 6 peptides were confined in two fractions: all 6 peptides were detected in Spot 1, and 4 of the 6 peptides were found in Spot 2. The observed MS signal intensities ranged from 200 to 2000, which expectedly were lower than those obtained at the higher sample concentration (Table 3). However, the sequence coverage from these 6 peptides was 66%, which was essentially unchanged.
Conclusion
The integrated approach of protein enrichment, digestion, and partial separation by cIET was applied to moderately acidic and basic proteins, including the phosphorylated protein β-casein. Compared to in-solution digestion, this integrated technique was generally found to provide higher sequence coverage and number of identified peptides. By facilitating a co-enrichment of enzymes and substrates in a confined volume at the pH junction, cIET significantly improved the effectiveness of digestion, and allowed for a shorter incubation period (2 h) when compared to the traditional in-solution digestion (overnight). For the in-capillary digestion of β-casein, very acidic phosphopeptides with high mobilities were generated. The selective enrichment nature of the pH junction was applied to the controlled isolation of these acidic peptides from the remaining peptides based on pI. This is highly beneficial for subsequent MALDI MS analysis as dispersing the peptides into different fractions minimizes ionization suppression thereby facilitating more sensitive detection of the phosphopeptides, especially the multi-phosphorylated peptides. Most importantly, this technique is capable of analysing samples at very low quantities (ng-level), and intact protein samples can be directly injected without pre-digestion. These features are ideal for sequence verification analysis of scarce recombinant proteins.In addition to trypsin, digestion with endoproteinase AspN was demonstrated. AspN was found to behave very similarly to trypsin for in-capillary digestion. Essentially the identical procedure for trypsin was applicable to AspN. This presented an opportunity to conveniently perform miniaturized digestion for studying custom designed enzymes. From a broader perspective, the use of an alternate enzyme represented another step towards performing multiple, in-capillary, small-volume sample preparation reactions for proteomics. Other ongoing development based on cIET includes in-capillary disulfide bonding reduction and sample desalting. The presented methodology bears tremendous potential in future development of nanoliter-range sample preparation.
All current and previous in-capillary digestions by cIET were conducted in conjugation with MALDI MS analysis. Future work will explore the integration with electrospray ionization (ESI) MS for online operation. This will require the use of alternate inner capillary coatings and the incorporation of organic solvents in the discontinuous buffers for better ESI compatibility.27 Research in both areas is underway.
Acknowledgements
This work was supported primarily by the Discovery Grants Program of the Natural Sciences and Engineering Research Council of Canada (NSERC), and in part by the University of Western Ontario (UWO). C.A.N thanks NSERC for financial support during her studies through the Postgraduate Scholarships program. The Bruker Reflex IV mass spectrometer was funded by the Canada Foundation for Innovation and the Ontario Innovation Trust. The Applied Biosystems 4700 Proteomics Analyzer and the Agilent capillary electrophoresis instrument were funded by the Academic Development Fund Program of UWO. Mass spectrometry service provided by the UWO MALDI MS Facility of the Schulich School of Medicine & Dentistry was also acknowledged.References
- S. Akashi, K. Suzuki, A. Akihiro, N. Yamada, E.-I. Suzuki, K. Hirayama, S. Nakamura and Y. Nishimura, Rapid Commun. Mass Spectrom., 2006, 20, 1932–1938 CrossRef CAS.
- M. Mann, P. Hojrup and P. Roepstorff, Biol. Mass Spectrom., 1993, 22, 338–345 CAS.
- J. Ji, Y. Zhang, X. Zhou, J. Kong, Y. Tang and B. Liu, Anal. Chem., 2008, 80, 2457–2463 CrossRef CAS.
- G. Massolini and E. Calleri, J. Sep. Sci., 2005, 28, 7–21 CrossRef CAS.
- L. N. Amankwa and W. G. Kuhr, Anal. Chem., 1992, 64, 1610 CrossRef CAS.
- H. Wu, J. Zhai, Y. Tian, H. Lu, X. Wang, W. Jia, B. Liu, P. Yang, Y. Xu and H. Wang, Lab Chip, 2004, 4, 588–597 RSC.
- T. Laurell and G. Marko-Varga, Proteomics, 2002, 22, 345 CrossRef.
- F. Svec, Electrophoresis, 2006, 27, 947–961 CrossRef CAS.
- J. Ma, L. Zhang, Z. Liang, W. Zhang and Y. Zhang, J. Sep. Sci., 2007, 30, 3050–3059 CrossRef CAS.
- Y. Liu, H. Lu, W. Zhong, P. Y. Song, J. Kong, P. Yang, H. H. Girault and B. Liu, Anal. Chem., 2006, 78, 801–808 CrossRef CAS.
- P. L. Urban, D. M. Goodall and N. C. Bruce, Biotechnol. Adv., 2006, 24, 42–57 CrossRef CAS.
- H. Wang, R. Oleschuk, F. Ouchen, F. Li, P. Thibault and D. J. Harrison, Rapid Commun. Mass Spectrom., 2000, 14, 1377–1383 CrossRef CAS.
- J. P. Kutter, Trends Anal. Chem., 2000, 19, 352–363 CrossRef CAS.
- A. Rios, A. Escarpa, M. C. Gonzalez and A. G. Crevillen, Trends Anal. Chem., 2006, 25, 467–479 CrossRef CAS.
- D. Josic and J. G. Clifton, J. Chromatogr., A, 2007, 1144, 2–13 CrossRef CAS.
- C. A. Nesbitt, K. Jurcic and K. K.-C. Yeung, Electrophoresis, 2008, 29, 466–474 CrossRef CAS.
- K. Jurcic, C. A. Nesbitt and K. K.-C. Yeung, J. Chromatogr., A, 2006, 1134, 317–325 CrossRef CAS.
- C. A. Nesbitt, J. T.-M. Lo and K. K.-C. Yeung, J. Chromatogr., A, 2005, 1073, 175–180 CrossRef CAS.
- J. M. Cunliffe, M. E. Baryla and C. A. Lucy, Anal. Chem., 2002, 74, 776–783 CrossRef CAS.
- C. A. Lucy, N. E. Baryla and K. K.-C. Yeung, in: M. A. Strege, A. L. Lagu (Eds.), Methods in Molecular Biology: Capillary Electrophoresis of Proteins and Peptides, Humana Press, Totowa, NJ, 2004 Search PubMed.
- H. X. Zhang, G. K. Hunter, H. A. Goldberg, G. A. Lajoie and K. K.-C. Yeung, Anal. Chim. Acta, 2007, 581, 268–280 CrossRef CAS.
- H. X. Zhang and K. K.-C. Yeung, Anal. Chem., 2004, 76, 6814–6818 CrossRef CAS.
- H. X. Zhang, C. J. Zhang, G. A. Lajoie and K. K.-C. Yeung, Anal. Chem., 2005, 77, 6078–6084 CrossRef CAS.
- J. Kyte, Structure in Protein Chemistry, Garland Publishing, New York, 1995 Search PubMed.
- F. Hillenkamp and J. Peter-Katalinic, MALDI MS: A practical guide to Instrumentation, Methods and Applications, Wiley-VCH Munster, Germany, 2007 Search PubMed.
- G. R. Drapeau, J. Biol. Chem., 1980, 255, 839–840 CAS.
- J. N. M. Ballard, G. A. Lajoie and K. K. C. Yeung, J. Chromatogr., A, 2007, 1156, 101–110 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |