Electrochemical characterization of enzymatic activity of yeast cells entrapped in a poly(dimethylsiloxane) microwell on the basis of limited diffusion system
Hitoshi
Shiku
*a,
Shun
Goto
a,
Sungbong
Jung
a,
Kuniaki
Nagamine
a,
Masahiro
Koide
b,
Tomosato
Itayama
b,
Tomoyuki
Yasukawa
c and
Tomokazu
Matsue
*a
aGraduate School of Environmental Studies, Tohoku University, 6-6-11, Aramaki-Aoba, Sendai 980-8579, Japan. E-mail: shiku@bioinfo.che.tohoku.ac.jp; matsue@bioinfo.che.tohoku.ac.jp
bEnvironmental Chemistry Division, National Institute for Environmental Studies, 16-2 Onogawa, Tsukuba 305-8506, Japan
cGraduate School of Material Science, University of Hyogo, Hyogo, Japan
First published on 20th October 2008
Abstract
A highly sensitive and quantitative analysis was performed using a poly(dimethylsiloxane) (PDMS) microwell array in a scanning electrochemical microscopy setup. A microelectrode with a relatively large seal radius was used to cover the top of the cylindrical PDMS microwell (96 pL). The voltammogram for 4 mM ferrocyanide resulted in a charge value of 38 nC, suggesting that almost 100% of the reductant in the microwell was converted to the oxidation current. When genetically modified yeast cells were entrapped in the microwell, the accumulation of p-aminophenol (PAP) produced by expressing β-galactosidase (βGAL) was successfully observed.
1. Introduction
Analytical chemistry within small volumes has shown significant impacts to explore new research fields including single-cell analysis1–6 and single molecule dynamics.7,8 Various micro- and nanostructures were constructed and combined with highly sensitive detection systems based on optical and electric amplifiers for the purpose of simplifying sample preparation, shortening the time per assay, and elevating the throughput of the analysis. Among these micro- and nanostructures, poly(dimethylsiloxane) (PDMS) microchamber arrays7,9 or microchannels8,10–12 are probably the most simple and flexible devices that can be used as lab-on-a-chip devices.The electrochemical measurement system is also possible to realize parallel and rapid assays with a small-volume microwell array.13 However, electrochemical studies of small-volume samples require at least two electrodes to maintain the entire electric circuit connected during the measurements.14–23 Therefore, the electrode must be designed inside the microchamber14–19 or inserted into the small space under a micromanipulator operation.20–23 In the experimental setup based on scanning electrochemical microscopy (SECM),24,25 however, it is relatively easy to form a confined ultra-small volume.26–28 The electrochemical behavior of the microelectrode observed in the microchamber is drastically different from that observed when the electrode is placed in the bulk solution. In the present study, we combined the SECM technology with the PDMS cylindrical microwell array to quantitatively evaluate the enzymatic activity of recombinant yeast cells expressing β-galactosidase (βGAL). As a model system, a yeast-two-hybrid strain was selected because it is widely used not only for screening protein–protein interactions but also for detecting environmental pollutants.5,6,29–36 Schwartz–Mittelman et al. demonstrated the electrochemical evaluation of the estradiol activities of various compounds by using human estrogen receptor-alpha.31–32
The accumulated product from the yeast cells in the microwell is converted to electric charge. The enzymatic activity of the yeast cells in the confined system is quantitatively analyzed and compared with that in the open system based on the spherical diffusion theory. We find that the cellular activity is strongly affected by the designs of the microchamber and detection system. The confined PDMS microwell system allows the accumulation of the product, and therefore, is advantageous for highly sensitive analyses. However, we must be careful to discuss the enzymatic activity and mass transfer rate in small volume analysis because these parameters might change depending on the environmental conditions.
2. Materials and methods
2.1 Reagents
The (100) silicon monocrystal wafer (thickness: 230 µm, optically polished on both sides) was purchased from SUMO Co., Tokyo, Japan. The negative photoresist, SU-8-3050, was purchased from Microchem Co. USA. PDMS (Sylgard 184) was purchased from Dow Corning, Co. USA. 17β-Estradiol was purchased from Sigma, USA. p-Aminophenyl-β-D-galactopyranoside (PAPG) was purchased from Tokyo Chemical Industry Co., Ltd., Japan. Triton X-100 was purchased from Polysciences, Inc., USA. Dimethyl sulfoxide (DMSO) and p-aminophenol (PAP) were purchased from Wako Pure Chemicals, Japan. All the solutions were prepared using distilled and deionized water purchased from Direct-Q (Millipore, USA).2.2 Fabrication of the PDMS microwell array
The PDMS microwell array was fabricated by curing the prepolymer on the (100) Si substrate with a master. The master was photolithographically patterned using the negative photoresist (SU-8 3050). A 10 : 1 mixture of the silicon elastomer and the curing agent was poured on the master and left at 80 °C for 1 h for curing the prepolymer. The PDMS replica was then peeled from the substrate. The diameter and depth of the PDMS microwell were both 50 µm.2.3 Yeast strain and growth conditions
The yeast strain used in the present study was Saccharomyces cerevisiae Y190, donated by Dr Fujio Shiraishi from the National Institute for Environmental Studies. The expression plasmid contains the human estrogen receptor α (hERα),31–35 instead of the medaka estrogen receptor α (medERα: Oryzia latipes),29 fused with GAL4 DBD (binding domain). The plasmid containing coactivator TIF2 fused with the GAL4 activation domain (GAL4 AD) was also introduced into the yeast cells carrying the β-galactosidase reporter gene.33,34 The cells were preincubated for 24 h at 30 °C with shaking at 100 rpm in a modified Sabouraud dextrose (SD) medium (without tryptophan and leucine).The cell suspension (60 µL) was then mixed with the medium (60 µL) containing 10 nM 17β-estradiol and 2.0% (v/v) DMSO, and incubated for 4 h at 30 °C with shaking at 100 rpm to induce the β-galactosidase expression. The medium was exchanged to a 80 µL of Z-buffer (60.0 mM Na2HPO4·12H2O, 39.7 mM NaH2PO4·2H2O, 10.0 mM KCl, 10.0 mM MgSO4·7H2O; pH 7.0) including 0.3% (v/v) Triton X-100, a nonionic surfactant to incubate for 1 h at 30 °C with shaking at 100 rpm (final concentration of the yeast cells was 1 × 107cells mL−1).
2.4 Electrochemical detection of β-galactosidase activity in yeast cells
The PDMS microwell array was irradiated with O2 plasma at 100 W for 1 min in order to make the surface of the well hydrophilic. Generally, the O2 plasma treated PDMS surface maintains the sufficient hydrophilic nature for 1 h to smoothly introduce aqueous solutions within the microwell array. Under aqueous solution, however, the hydrophilic nature of the O2 treated PDMS surface is maintained for at least 12 h.The yeast cell suspension (100 µL) was first dispensed on the PDMS microwell array and stabilized for 10 min. Then, the suspension was withdrawn using a filter paper to remove the excess yeast cells present on the outer surface of the microwell. The liquid and yeast cells remained the inside the PDMS wells only. Secondary, the measuring solution was further poured on the PDMS microwell array gently. For the electrochemical measurements, the PDMS microwell array containing the yeast cells was carefully soaked in the Z-buffer solution containing 7.4 mM PAPG and 0.3% Triton X-100. The concentration of Triton X-100 in the measuring solution was determined to optimize the activity of the yeast cells. The mass-transfer of PAPG and PAP through the cellular membranes was sufficiently promoted, but βGAL still remained in the yeast cells. In this study, the number of cells in the PDMS microwell was controlled to be less than 150 so that the yeast cells remain at the bottom (50 µm ∅) of the cylindrical PDMS microwell at monolayer level. The exact number of cells in the well was manually counted from the photograph recorded for each microwell in which the electrochemical measurement was performed.The electrochemical measurement was carried out using an SECM system including a potentiostat (HA1010mM8; Hokuto Denko Corp., Tokyo, Japan), an inverted microscope (Nikon diaphot T200), and a motor-driven XYZ stage (Chuo-Seiki M9103). An Ag/AgCl-saturated KCl electrode was used as the reference/counter electrode. A Pt-microelectrode (radius: 10 µm; radius of the tip including the insulator part: 85 µm) was used as the working electrode. The β-galactosidase (βGAL) activity expressed in the yeast cells was monitored by detecting the oxidation current for PAP, a product of the enzyme-catalyzed hydrolysis of PAPG inside the trapped cells.29–32,37–40 The electrochemical experiment was performed on a 4–6 cm2 -piece of the PDMS microwell array sheet set in a disposable 60 mm-diameter culture dish. The Ag/AgCl reference electrode was set on the PDMS microwell array sheet. The distance between the working and the reference electrode was about 10–20 mm. In the case that the glass seal part of the working electrode completely covers the PDMS microwell, electrochemical measurement is not available because current does not flow. However, we have recognized that there is an electric connection due to leakage of the ionic flow between the working and the reference electrodes even when reactant within the microwell was consumed almost 100%.
3. Results and discussion
Fig. 1(a) shows the schematic illustration of the experimental setup. The top opening of the PDMS cylindrical microwell is covered with the disk plane of a working microelectrode tip to accumulate PAP produced by βGAL in the yeast cells. The time period between the covering of the PDMS microwell with the microelectrode tip and the application of the potential was defined as the accumulation time, taccu. As taccu increases, the concentration of PAP in the microwell also increases. After maintaining the tip potential at 0.0 V for taccu, the potential was stepped to +0.3 V to oxidize PAP accumulated in the microwell. The product of the PAP oxidation is quinone imine (QI). Fig. 1(b) shows an optical micrograph of the PDMS microwell array. The height of the SU-8 mold of the PDMS microwell was 50 ± 1 µm measured with a surface profiler. The diameter of the microwell was 50 ± 1.3 µm under the optical microscopic observation.Prior to the measurements using the yeast cells, the electrochemical behavior in the confined microwell was characterized by cyclic voltammetry. Fig. 2 shows the cyclic voltammograms (CVs) of 4.0 mM K4Fe(CN)6/0.1 M KCl in the PDMS microwell. CVs shown in the present study were performed at scan rate of 20 mV s−1. The microelectrode was positioned at various heights (z) from the upper surface of the PDMS microwell (+300 to −100 µm). The point at z = 0 defines the position where the tip touches the upper surface of the PDMS microwell. The negative z value does not reflect the actual z-position of the tip, but indicates that the microelectrode pushes down the top of the PDMS microwell. According to the spherical diffusion theory, when the microelectrode tip is sufficiently far from the upper surface of the PDMS microwell (z = +300 µm), the CV has a typical sigmoidal shape, and the oxidation current for Fe(CN)64− reaches the steady state in the positive potential region. The shape of the voltammogram drastically changed at z = 0, showing an oxidation peak at +0.4 V due to the limited diffusion. For z values less than −10 µm, the oxidation current in the positive potential region (more positive than +0.7 V) was found to become almost zero, indicating that Fe(CN)64− in the microwell was almost completely consumed. When the potential was scanned in the negative direction, a reduction peak was observed at +0.15 V. The reduction current observed in less than −0.05 V originates from the oxygen dissolved in the solution.12,42 The relatively large peak separation of 0.35 V is mainly due to the solution resistance, which depends on several parameters of the experimental setup, including the seal size of the tip electrode and the position of the tip (z). The electric charges estimated from the areas under the oxidation and reduction peaks were 38 and 32 nC, respectively.
 | ||
| Fig. 1 (a) Schematic illustration of the experimental setup. The top of the PDMS cylindrical microwell (diameter: 50 µm; depth: 50 µm) is covered with the disk plane of a working microelectrode tip to accumulate the PAP produced by the βGAL in the yeast cells. The accumulation time was defined as taccu. After maintaining the tip potential at 0 V for taccu, the potential was stepped to +0.3 V to oxidize the PAP accumulated in the microwell. Schemes of the enzymatic and electrochemical reaction were also showen. (b) Photograph of the PDMS micriowell array. Bar, 50 µm. | ||
 | ||
| Fig. 2 (a) Cyclic voltammograms in 4 mM K4Fe(CN)6/0.1 M KCl for the PDMS microwell. Scan rate: 20 mV s−1. The microelectrode was located at various z-positions (+300 to −20 µm). The point at z = 0 was defined as that where the tip touched the top of the PDMS microwell. (b) Plot of the electric charge versus z (+300 to −100 µm). Tip radius: 10.5 µm; Insulator radius: 85 µm. | ||
Fig. 2(b) shows the plot of the electric charge of the oxidation peak as a function of z. The charge was almost constant (38.22 ± 0.143 nC) when the z value was less than −15 µm. This result suggests that the confined volume (96 pL) in the present experimental setup is preserved, regardless the position of the microelectrode tip in the relatively wide range, z = −15 to −100 µm. Therefore, we can precisely analyze the electrochemical behavior in the small volume of the confined cavity formed with the PDMS microwell and microelectrode cap. In this article, the electrochemical measurements for the yeast cells entrapped in the PDMS microwell were performed at z = −30 µm. The experimental setup we introduce here requires a probe positioner but need not control the vertical position with less than 10 µm preciseness. Since the microwell is made of a PDMS elastomer, the tip and microwell are less likely to damage, and therefore, both can be used repeatedly. There exist many other methods for carrying out small-volume electrochemistry using paraffin oil,21,23mercury,28 oil/water droplets,19,22 or various microfabricated 3D structures.14–18 The present method utilizing the PDMS microwell has great advantages from the viewpoints of cost, simplicity, reproducibility, and throughput. The CV of 0.82 mM PAP oxidation within the microwell was also performed and shown in Fig. 3. At +0.3 V, the PAP oxidation current could be obtained whereas PAPG not oxidized.29,41 The charge for the PAP oxidation 14.9 ± 0.95 nC (n = 8) was in good agreement with the expected theoretical value 15.3 nC in the confined microwell of 96 pL. The CV behaviours of the PAP for near (z = −30 µm) and far from the PDMS (z = + 100 µm) are basically the same tendencies as those of the K4Fe(CN)6 system, however, when the potential was scanned back in the negative direction, a reduction peak was small at z = −30 µm probably because the product from the PAP oxidation, QI was not chemically stable.
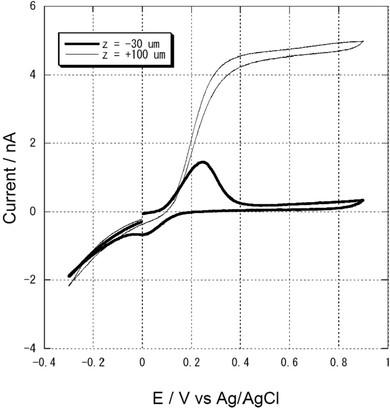 | ||
| Fig. 3 Cyclic voltammograms in 0.82 mM PAP/Z-buffer for the PDMS microwell. Scan rate: 20 mV s−1. The microelectrode was located attaching (z = −30 µm) or far from the PDMS microwell (z = +100 µm). Tip radius: 12 µm. | ||
Next, we quantitatively determined the amount of PAP produced by βGAL in the yeast cells. Fig. 4 shows the results of the chronoamperometry on the PDMS microwell entrapping 144 yeast cells. Before the potential was stepped from 0 V to +0.3 V to detect PAP, the microelectrode tip was positioned to cover the top of the PDMS microwell for different accumulation times (taccu = 1–31 min), and the current responses obtained without the yeast cells (taccu = 1, 11 min) were recorded. The current responses obtained with the yeast cells were considerably larger than those obtained without the yeast cells. We calculated the electric charge and time integration of the current to quantitatively evaluate the βGAL activity. The charges obtained for taccu = 1 min and 11 min are approximately 5 nC and 11 nC, respectively. The 6 nC increase in the charge during the accumulation time clearly indicates that PAP is produced by the βGAL activity in the yeast cells, and is accumulated in the PDMS microwell within 10 min. When yeast cells do not exist, the background charge was independent of taccu. The baseline current at 0 V fluctuated between −95 and −145 pA (n = 12, data not shown), probably due to the reduction in the concentrations of the dissolved oxygen and PAPG;40 however, the background charges recorded at the +0.3 V potential step was almost constant (∼3 nC). This low background charge is probably due the oxidation of PAP produced from PAPGvia chemical (nonenzymatic) hydrolysis in solution.
 | ||
| Fig. 4 Chronoamperometric results for the PDMS microwell entrapping 144 yeast cells. Before the potential was stepped from 0 V to +0.3 V, the microelectrode tip was positioned to cover the top of the PDMS microwell for various taccu, from 1 to 31 min (taccu = 1, 6, 11, 21, 31 min). The same procedures were also performed for the PDMS well without the yeast cells. Measuring solution: 7.4 mM PAPG, 0.3% Triton-X100T, Z-buffer. Tip radius: 12 µm. Inset: photograph of the PDMS microwells measured with (well A) and without (well B) yeast cells. | ||
Fig. 5 and 6 show the plots of the electric charge and enzymatic activity, respectively, as a function of taccu. The measurements were performed three times, and the standard deviation (SD) was 2.2 to 7.8% of the each average value. The electric charge was converted to the βGAL activity (PAP product/mol s−1cell−1) by using the reaction electron number for PAP (nET = 2) and the Faraday constant (96500 C mol−1). Fig. 5 illustrates that the accumulation of PAP in the pL level of small volume chamber has been successfully detected. However, it should be noted that the PAP accumulation rate is not liner to taccu and that the βGAL activity deceases with taccu as shown in Fig. 6.
 | ||
| Fig. 5 Plot of the electric charge versustaccu. The measurement was performed thrice under each set of experimental conditions (n = 3). | ||
 | ||
| Fig. 6 Enzymatic activity (PAP production rate per cell) as a function of taccu. | ||
This type of confined PDMS microwell system allows another electrochemical analysis categorized in a steady-state method. In Fig. 4, the oxidation current for the microwell with 144 yeast cells is greater than that for the microwell without yeast cells by 11 pA, even 150 s after the potential step. This phenomenon implies that the rate of PAP production by yeast cells is equal to the rate of oxidation of PAP at the microelectrode, and the overall processes in the microwell reach the steady state. Under these circumstances, the βGAL activity can also be evaluated from the steady-state oxidation current; this value was found to be 3.96 × 10−19 mol s−1cell−1, which corresponded well with the value estimated from the PAP accumulation experiment shown in Fig. 6. This consistency in the values was unexpected because the steady-state method is principally different from the PAP accumulation technique, even though both can be carried out in the confined PDMS microwell system. For example at taccu = 31 min, PAP accumulated within the microwell is estimated to be 8.8 × 10−14 mol, which corresponds to 12% of the initial amount of PAPG (7.26 × 10−13 mol) in the PDMS microwell whereas the PAP concentration is almost zero when the PAP oxidation current is in the steady state (150 s after the potential step to 0.3 V was applied).
Finally, the βGAL activity of yeast in the PDMS microwell was evaluated by the open-system method based on the spherical diffusion theory. In this method, the concentration profile of PAP was measured keeping the microwell uncovered. When a biologically active sample is localized at a spot on a solid support, the reactant and/or product forms a spherical concentration profile near the sample.43,44 The mass transfer rate can be obtained by employing Fick's diffusion equation. Fig. 7 shows the oxidation current profile at the central axis of the well as a function of the height (z) from the top surface of the PDMS well entrapping 86 yeast cells. The tip scan rate and tip potential were set at 9.8 µm s−1 and +0.3 V, respectively. The tip current was converted to the PAP concentration using a calibration curve ([CPAP/mM] = 1.558 × 10−1 × [I/nA] for Fig. 7). The process for estimating the mass transfer rate from the spherical concentration profile has been described elsewhere.43 The mass transfer rate for PAP production in the open system was estimated to be 4.91 × 10−18 mol s−1cell−1 by using the diffusion coefficient of PAP, 7.1 × 10−6 cm2 s−1.45 For comparison, the PAP accumulation experiments were also performed using the same microwell accommodating 86 yeast cells, and the PAP production rate in this case was found to be 3.73 × 10−19 mol s−1cell−1 at taccu = 11 min, which corresponded to only 7.6% of the βGAL activity obtained in the open system. By comparing the two experimental systems for several PDMS microwells accommodating 39–144 yeast cells (n = 6), we found that the βGAL activity in the confined PDMS microwell system is 4.3–16.3% of that in the open system. These results claim that the cellular enzymatic and metabolic activities may be strongly affected by the microenvironmental conditions, including the size and shape of the microwell and the density of the cellular sample in the microwell. In the case of the open system, the substrate, PAPG, could be supplied very quickly. However, as the PAPG is present in excess (7.4 mM) even in the confined PDMS microwell system, the supply rate does not cause any significant difference in the PAP production activity. Quinone imine (QI), the product of the electro-oxidation of PAP, has a relatively higher chemical activity than PAPG or PAP; further, it does not diffuse in the confined PDMS microwell system. However, in the open system shown in Fig. 7, QI produced at the scanning tip could be easily diluted. As shown in Fig. 4, the βGAL activity does not change and shows a good reproducibility when the same experiment is performed for three times, even at taccu = 31 min; therefore, the βGAL inhibition by QI might not be irreversible.
 | ||
| Fig. 7 Oxidation current profile as a function of the height (z) from the top of the PDMS well entrapping 86 yeast cells recorded at the center of the well in the measuring solution of 7.4 mM PAPG, 0.3% Triton-X100T, Z-buffer. Tip scan rate: 9.8 µm s−1; Tip potential: +0.3 V vs.Ag/AgCl; Tip radius: 12 µm. | ||
4. Conclusions
A confined small volume was formed using a PDMS microwell array and a cap microelectrode with a relatively large seal radius. In the cyclic voltammetric measurements, the ferrocyanide ion entrapped in the microwell was oxidized with 100% efficiency, suggesting that a reproducible and quantitative electrochemical analysis was possible using this device. We also succeeded in evaluating the βGAL activity of recombinant yeast cells in the confined PDMS microwell. βGAL catalyzes the hydrolysis of PAPG to produce PAP, which is accumulated in the microwell. The accumulated PAP was quantitatively detected by amperometry using the abovementioned device. We have evaluated the βGAL activity by three different methods: the accumulation method, steady-state method, and open-system method. The βGAL activity estimated by the accumulation method was in good agreement with that estimated by the steady-state method. However, these βGAL activities estimated in the confined PDMS microwell were found to be considerably smaller than those estimated by the open-system method based on the spherical diffusion theory. This remarkable difference in the activities is probably because the concentrations of QI near the yeast cells are expected to be smaller for the open PDMS microwell than for the confined PDMS microwell. From the results obtained in the present work, we conclude that the cellular metabolic and enzymatic activities are considerably affected by the environmental conditions near the sample cells.Acknowledgements
This work was partly supported by Grants-in-Aid for Scientific Research (18101006 and 19750055) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) and by the R & D Project for Environmental Nanotechnology from the Ministry of Environment.References
- C. Yi, C.-W. Li, S. Ji and M. Yang, Anal. Chim. Acta, 2006, 560, 1–23 CrossRef CAS.
- C. N. LaFratta and D. R. Walt, Chem. Rev., 2008, 108, 614–637 CrossRef CAS.
- P. S. Dittrich and A. Manz, Nat. Rev. Drug Discovery, 2006, 5, 210–218 CrossRef CAS.
- S. Yamamura, H. Kishi, Y. Tokimitsu, S. Kondo, R. Honda, S. R. Rao, M. Omori, E. Tamiya and A. Muraguchi, Anal. Chem., 2005, 77, 8050–8056 CrossRef CAS.
- I. Biran and D. R. Walt, Anal. Chem., 2002, 74, 3046–3054 CrossRef CAS.
- R. D. Whitaker and D. R. Walt, Anal. Biochem., 2007, 360, 63–74 CrossRef.
- Y. Rondelez, G. Tresset, K. V. Tabata, H. Arata, H. Fujita, S. Takeuchi and H. Noji, Nat. Biotechnol., 2005, 23, 361–365 CrossRef CAS.
- L. Cai, N. Friedman and X. S. Xie, Nature, 2006, 440, 358–362 CrossRef CAS.
- A. Groisman, C. Lobo, H. Cho, J. K. Campbell, Y. S. Dufour, A. M. Stevens and A. Levchenko, Nat. Methods, 2005, 2, 685–649 CrossRef CAS.
- F. K. Balagadde, L. You, C. L. Hansen, F. H. Arnold and S. R. Quake, Science, 2005, 309, 137–140 CrossRef.
- W. DiLuzio, L. Turner, M. Mayer, P. Garstecki, D. B. Weibel, H. C. Berg and G. M. Whitesides, Nature, 2005, 435, 1271–1274 CrossRef CAS.
- T. Saito, C.-C. Wu, H. Shiku, T. Yasukawa, M. Yokoo, T. Ito-Sasaki, H. Abe and T. Matsue, Analyst, 2006, 131, 1006–1011 RSC.
- Z. Lin, Y. Takahashi, Y. Kitagawa, T. Umemura, H. Shiku and T. Matsue, Anal. Chem., 2008, 80, 6830–6833 CAS.
- W. Cheng, N. Klauke, H. Sedgwick, G. L. Smith and J. M. Cooper, Lab Chip, 2006, 6, 1424–1431 RSC.
- X. Cai, N. Klauke, A. Glidle, P. Cobbold, G. L. Smith and J. M. Cooper, Anal. Chem., 2002, 74, 908–914 CrossRef CAS.
- C. D. T. Bratten, P. H. Cobbold and J. M. Cooper, Anal. Chem., 1998, 70, 1164–117 CrossRef CAS.
- Z. P. Aguilar, W. R. Vandaveer and I. Fritsch, Anal. Chem., 2002, 74, 3321–3329 CrossRef CAS.
- J. C. Ball, D. L. Scott, S. Daunert, J. Wang and L. G. Bachas, Anal. Chem., 2000, 72, 497–501 CrossRef CAS.
- K. Nakatani, M. Sudo and N. Kitamura, J. Phys. Chem. B, 1998, 102, 2908–2913 CrossRef CAS.
- N. Gao, X. Wang, L. Li, X. Zhang and W. Jin, Analyst, 2007, 132, 1139–1146 RSC.
- T. Yasukawa, A. Glidle, J. M. Cooper and T. Matuse, Anal. Chem., 2002, 74, 5001–5008 CrossRef CAS.
- R. Kashyap and M. Gratzl, Anal. Chem., 1998, 70, 1468–1476 CrossRef CAS.
- R. A. Clark, P. B. Hietpas and A. G. Ewing, Anal. Chem., 1997, 69, 259–263 CrossRef CAS.
- G. Wittstock, M. Burchardt, S. E. Pust, Y. Shen and C. Zhao, Angew. Chem., Int. Ed., 2007, 46, 1584–1617 CrossRef CAS.
- A. Schulte and W. Schuhmann, Angew. Chem., Int. Ed., 2007, 46, 8760–8777 CrossRef CAS.
- P. Sun and M. V. Mirkin, Anal. Chem., 2007, 79, 5809–5816 CrossRef CAS.
- F.-R. F. Fan, J. Kwak and A. J. Bard, J. Am. Chem. Soc., 1996, 118, 9669–9675 CrossRef CAS.
- M. V. Mirkin, L. O. Bulhoes and A. J. Bard, J. Am. Chem. Soc., 1993, 115, 201–204 CrossRef CAS.
- T. Yasukawa, K. Nagamine, Y. Horiguchi, H. Shiku, M. Koide, T. Itayama, F. Shiraishi and T. Matsue, Anal. Chem., 2008, 80, 3722–3727 CrossRef CAS.
- M. Badihi-Mossberg, V. Buchner and J. Rishpon, Electroanalysis, 2007, 19, 2015–2028 CrossRef CAS.
- A. Schwartz-Mittelman, A. Baruch, T. Neufeld, V. Buchner and J. Rishpon, Bioelectrochemistry, 2005, 65, 149–156 CrossRef CAS.
- A. Schwartz-Mittelman, T. Neufeld, D. Biran and J. Rishpon, Anal. Biochem., 2003, 317, 34–39 CrossRef.
- J. Nishikawa, K. Saito, J. Goto, F. Dakeyama, M. Matsueo and T. Nishihara, Toxcol. Appl. Pharmacol., 1999, 154, 76–83 Search PubMed.
- F. Shiraishi, H. Shiraishi, J. Nishikawa, T. Nishihara and M. Morita, J. Environ. Chem., 2000, 10, 57–64 Search PubMed.
- S. Arulmozhiraja, F. Shiraishi, T. Okumura, M. Iida, H. Takigami, J. S. Edmonds and M. Morite, Toxicol. Sci., 2005, 84, 49–62 CrossRef CAS.
- C. K. Chow and S. P. Palecek, Biotechnol. Prog., 2004, 20, 449–456 CrossRef CAS.
- I. Biran, L. Kilmenty, R. H. Aronis, E. Z. Ron and J. Rishpon, Microbiology, 1999, 145, 2129–2133 CAS.
- T. Kaya, K. Nagamine, N. Matsui, T. Yasukawa, H. Shiku and T. Matsue, Chem. Commun., 2004, 248–249 RSC.
- K. Nagamine, S. Onodera, A. Kurihara, T. Yasukawa, H. Shiku, R. Asano, I. Kumagai and T. Matsue, Biotechnol. Bioeng., 2007, 96, 1008–1013 CrossRef CAS.
- C. Zhao, J. K. Sinha, C. A. Wijayawardhana and G. Wittstock, J. Electroanal. Chem., 2004, 561, 83–91 CrossRef CAS.
- N. Matsui, T. Kaya, K. Nagamine, T. Yasukawa, H. Shiku and T. Matsue, Biosens. Bioelectron., 2006, 21, 1202–1209 CrossRef.
- H. Shiku, T. Saito, C.-C. Wu, T. Yasukawa, M. Yokoo, H. Abe, T. Matsue and H. Yamada, Chem. Lett., 2006, 35, 234–235 CrossRef CAS.
- T. Kaya, D. Numai, K. Nagamine, S. Aoyagi, H. Shiku and T. Matsue, Analyst, 2004, 129, 529–534 RSC.
- H. Shiku, T. Shiraishi, S. Aoyagi, Y. Utsumi, M. Matsudaira, H. Abe, H. Hoshi, S. Kasai, H. Ohya and T. Matsue, Anal. Chim. Acta, 2004, 522, 51–58 CrossRef CAS.
- O. Niwa, Y. Xu, H. B. Halsall and W. R. Heineman, Anal. Chem., 1993, 65, 1559–1563 CrossRef.
| This journal is © The Royal Society of Chemistry 2009 |