Consensus multivariate methods in gas chromatography mass spectrometry and denaturing gradient gel electrophoresis: MHC-congenic and other strains of mice can be classified according to the profiles of volatiles and microflora in their scent-marks
Simeone
Zomer
a,
Sarah J.
Dixon
a,
Yun
Xu
a,
Susanne P.
Jensen
b,
Huitu
Wang
c,
Clare V.
Lanyon
d,
Anthony G.
O'Donnell
d,
Anthony S.
Clare
c,
L. Morris
Gosling
b,
Dustin J.
Penn
e and
Richard G.
Brereton
*a
aCentre for Chemometrics, School of Chemistry, University of Bristol, Cantocks Close, Bristol, UK BS8 1TS
bEvolutionary Biology Group, University of Newcastle upon Tyne, Newcastle upon Tyne, UK NE2 4HH
cSchool of Marine Science and Technology, Ridley Building, Newcastle University, Newcastle upon Tyne, UK NE1 7RU
dSchool of Biology and Psychology, Division of Biology, IRES, University of Newcastle upon Tyne, Newcastle upon Tyne, UK NE1 7RU
eKonrad Lorenz Institute for Ethology, Austrian Academy of Sciences, Savoyenstr. 1a, A-1160 Vienna, Austria
First published on 19th November 2008
Abstract
House mice (Mus domesticus) communicate using scent-marks, and the chemical and microbial composition of these ‘extended phenotypes’ are both influenced by genetics. This study examined how the genes of the major histocompatibility complex (MHC) and background genes influence the volatile compounds (analysed with Gas Chromatography Mass Spectrometry or GC/MS) and microbial communities (analysed using Denaturating Gradient Gel Electrophoresis or DGGE) in scent-marks produced by congenic strains of mice. The use of Consensus Principal Components Analysis is described and shows relationships between the two types of fingerprints (GC/MS and DGGE profiles). Classification methods including Support Vector Machines and Discriminant Partial Least Squares suggest that mice can be classified according to both background strain and MHC-haplotype. As expected, the differences among the mice were much greater between strains that vary at both MHC and background loci than the congenics, which differ only at the MHC. These results indicate that the volatiles in scent-marks provide information about genetic similarity of the mice, and support the idea that the production of these genetically determined volatiles is influenced by commensal microflora. This paper describes the application of consensus methods to relate two blocks of analytical data.
1 Introduction
The major histocompatibility complex (MHC) is a multi-gene family found in vertebrates, and these highly polymorphic loci encode cell-surface antigen-presenting proteins.1 Because of their important role in the immune system, MHC genes have been investigated in great depth by immunologists, and increasingly by evolutionary biologists aiming to understand how selection maintains high levels of allelic diversity at MHC loci. MHC diversity is thought to be driven by co-evolving pathogens and parasites; however, another non-exclusive possibility is that sexual selection plays a role.2–6 Much evidence indicates that MHC genes influence odour and mating preferences in mice,5–10 which may adaptively function to increase offspring resistance to pathogens and parasites;5,6 however, it is unclear how MHC genes influence odour. Several mechanisms have been proposed,9,10 including the ‘microflora hypothesis’, which suggests that MHC genes influence the growth of commensal microbiota that control variation in individual odour.11,12 This idea is supported by evidence that MHC genes influence commensal microflora in the intestine13 and scent-marks14 of congenic laboratory mice; however, studies examining the odour of animals reared in germ-free environments have produced mixed results.9 Some recent studies support the idea that MHC genes influence odour by altering the pool of peptides in the urine of mice (‘the peptide hypothesis’).15 These mechanisms are not mutually exclusive, and it has been suggested that MHC-bound peptides provide the precursors that are metabolised and made volatile by commensal microflora (i.e. the ‘peptide-microflora hypothesis’).9 One of the unsolved questions has been determining whether MHC-dependent microflora might influence urinary volatiles. In this study, we examined how MHC genes influence both microflora communities and volatile compounds in mouse scent-marks, and we applied new methods to analyse these two large and complex datasets. More specifically, we applied a chemometric procedure to compare and examine the microbiological and chemical signals of different strains of mice that vary in their genetic background and MHC-haplotype to answer the following questions:(1) Can we distinguish (and predict) the genetic strain of a mouse, including strains that differ genetically only at the MHC locus (‘MHC congenics’), using the volatile compounds in scent-marks measured by Gas Chromatography Mass Spectrometry or GC/MS?
(2) Can we also distinguish mouse strains using the microbial communities in scent-marks, using molecular genetic profiles (Denaturating Gradient Gel Electrophoresis or DGGE)?
(3) How does the MHC compare to the rest of the genome (background genes) in its influence on urinary volatiles (GC/MS) and microflora (DGGE)?
(4) Is there a relationship between the pattern of volatiles (GC/MS) and pattern of microflora (DGGE)?
Mouse scent-marks are likely to be multivariate in nature, in that no one single compound is uniquely responsible for the signal – rather like recognising a human face – it is a combination of features that allow for unique recognition. Chemometrics approaches work on multivariate models, that is models taking into account the correlation between the variables, and as such have an important role to play in biology where there are likely to be interactions between variables.
In this study, samples of scent-marks were obtained by inserting glass slides into mouse cages containing a male mouse, which are usually scent-marked by individual mice. This is a form of olfactory communication that provides information about signaller quality to competitors and mates.16 From these samples of scent-marks, DNA was extracted and the microbial community was genetically fingerprinted using DGGE (16S rRNA gene) to obtain a profile of the commensal microflora,14 and the chemical profile was analysed using GC/MS after sampling with solid phase microextraction (SPME).
The data analysis procedure began with a feature extraction process where raw microbiological and chemical data were summarized in two distinct data matrices.
Each matrix is composed of sample vectors whose elements identify a set of variables common to all samples in the population, which correspond to the relative intensity of gel bands (DGGE) and the areas of common chromatographic peaks (GC/MS). Following feature extraction, a series of processing steps including scaling, normalization and standardisation are applied in order to maximize the interpretability of the information embedded in the matrices. The last part of the procedure involves a variety of multivariate pattern recognition techniques to extract the information of interest from the processed data with which to answer the relevant questions. The main trends emerging from the two blocks are investigated using Consensus Principal Components Analysis (C-PCA)17–19 that identifies the common directions of variation and can be posed in relation to factors of interest such as background genetics and MHC-haplotypes of mice. The exploratory analysis is then completed with an estimation of the degree of overlap between the microbiological and chemical profile to verify the microflora hypothesis. Afterwards, other techniques such as Partial Least Squares Discriminant Analysis (PLS-DA)20–24 and Support Vector Machines (SVM)25,26 were applied to determine whether the fingerprint recorded can be used for prediction purposes. Results indicate that the mouse strains emerge clearly in both profiles recorded and that this information can be used to produce predictive models. The strains of the mice contribute to shape both the GC/MS and DGGE profiles, with the whole strain differences having a stronger effect than MHC. Finally the profiles show a certain degree of overlap.
2 Experimental
2.1 Animals
Twenty-eight male congenic mice from four strains (two MHC-congenic strains) were obtained from Harlan UK: BALB.B/OlaHsd (strain BALB/c, MHC-haplotype b), BALB/cOlaHsd (strain BALB/c, haplotype d), C57BL/10ScSnOlaHsd (strain B10, haplotype b) and C57BL10.D2/nOlaHsd (strain B10, haplotype d). Mice were single-housed in MB2 (20 × 10 × 13 cm) cages in mixed strain racks and maintained under controlled temperature (17–28 °C) and light conditions (reversed 12 h : 12 h light : dark cycle with lights on at 22:00 hours), with ad libitum access to water and food (SDS rat and mouse breeding diet RM3 (P), Special Diet Services, Witham, Essex, UK), shredded paper nesting material and cardboard tubes and softwood chewing sticks for enrichment. Scent-marks were collected when the mice were aged 14–23 weeks.2.2 Scent-mark collection
Glass microscope slides (76 × 26 × 1.5 mm thick) were frosted on one side using an abrasive, silicon carbide (Carborundum), and cut into two strips each measuring 76 × 13 mm. They were cleaned with a specialist glass cleaner (Dri-Decon), rinsed in tap water and oven-dried overnight at 110 °C. For scent-mark collection, three strips of frosted glass slide were placed frosted side up on a clean glass slide (76 × 38 × 1.5 mm thick) resting on a foam pad (76 × 38 × 5 mm thick) on an upside-down aluminium mini bread tin (10 × 5.4 × 3.2 cm deep). A second mini bread tin with a square hole (65 × 30 mm) cut out was placed on top to hold the slides in place. This device raised the area to be scent-marked 4.4 cm above the substrate, reducing the contamination by materials from the cage bottom. The device was placed in a mouse cage for scent-mark collection for 1.5 h at the beginning of the dark phase (i.e. the animal's active period). The slides were then removed from the metal holders, placed in a silanised 40 ml EPA glass vial with a polypropylene (PP) screw cap and a Silicone/PTFE septum and stored in a freezer at −80 °C. The vials were further stored in 32 oz glass jars with a solid PP Teflon-lined cap to limit contaminants such as CO2 penetrating the septum. Vials were silanised in order to reduce the adhesiveness of the glass surface with a 2% solution of dichlorodimethylsilane in chloroform, left in a fume cupboard overnight, rinsed in tap water and dried for a minimum of 3 h at 110 °C. After scent-mark collection, the metal tins and clear glass slides were hand-washed and the foam pads machine-washed at 60 °C with Dri-Decon and left to dry overnight on top of a 110 °C oven.2.3 SPME fibre
Volatile compounds were extracted from scent-marks with a Solid Phase Micro Extraction (SPME) procedure. In order to assess the recovery and sensitivity of different fibre types with our sample of interest – mouse scent-marks – five commercially-available types were compared by exposure to identical samples. All SPME fibres were supplied by Supelco (Bellefonte, PA) and were pre-conditioned by inserting them into the GC injector according to the manufacturer's instructions. As the amount of analytes in real scent-marks was not constant and not controllable over time, simulated scent-marks were used. These were prepared by injecting 10 µl mouse urine onto clean frosted glass strips which had previously been sealed in an EPA glass vial. Ten µl urine gave a comparable GC/MS signal to that of authentic scent-marks. Sampling was done at 60 °C for 30 min in order to maximize recovery and the number of peaks. A total of 107 volatile compounds was detected, although no fibre detected all compounds. The SPME fibre that recovered most volatiles (95 peaks) was a 50/30 divinylbenzene/Carboxen on PDMS and was employed in further analysis.2.4 Chemical analysis
Immediately before SPME sampling, 2 µl methanol solution containing the internal standards fluorobenzene (8 ng), toluene-d8 (8 ng), 4-bromofluorobenzene (8 ng), 1,2-dichlorobenzene-d4 (8 ng) and hexanoic acid-d11 (40 ng) was injected onto the scent-marked collecting materials. The air-tight glass vial was mounted onto a magnetic stirrer and submerged in a 60 ± 1 °C water bath. It was imperative that the scent-marked glass strips remained separated in the vial to avoid impeding the release of volatiles into the headspace. The headspace of the vial was sampled for 60 min (unless indicated otherwise) with the SPME fibre exposed from the protective needle by pushing the plunger forward of the manual holder. After sampling, the fibre was retracted and the needle was removed from the vial. The SPME needle was then inserted into the GC injector and the fibre was exposed to the carrier gas by pushing the plunger of the manual holder. The analytes were thermally desorbed for 3 min in the hot GC injector. The analysis of all extracts was performed on an Agilent 6890N Gas Chromatograph coupled to a 5973 inert Mass Selective Detector (Agilent Technologies, Stockport, UK). The technical specifications of the GC capillary column was: Zebron, ZB-Wax, ∅ 0.25 mm × 30 m, film thickness 0.25 µm, 100% poly(ethylene glycol) (Phenomenex UK Ltd, Macclesfield, UK). The GC injection port liner used was an SPME injection sleeve of 0.75 mm ID (Supelco, Bellefonte, PA). The carrier gas used was helium. The injector temperature was 230 °C in a pulsed splitless mode at a pulse pressure of 25 psi for 2 min. After 2 min, carrier gas was introduced at a constant flow at 1.00 ml min−1. The 69 min oven temperature ramp program was: 40 °C (hold for 5 min), then to 100 °C at 3 °C min−1 (5 min) followed by to 150 °C at 5 °C min−1 (5 min), and finally to 230 °C at 5 °C min−1 (8 min). The spectrometer was run with the following parameters: positive electron ionization mode (EI+), source energy 70 eV, full scan in the mass range of m/z 35–400 amu, no solvent delay, interface transfer line 250 °C. The detector signals were collected, integrated and recorded using an MSD Chemstation (Agilent Technologies).2.5 Microbiological analysis
DNA was extracted from the glass slides using the Fast Prep DNA SPIN kit (Anachem) with modifications. Glass slides were rotated at room temperature with the cell suspension and cell lysing buffer for 30 min after which the nucleic acids were extracted as described by the manufacturer. The low amounts of DNA recovered from the samples necessitated that all samples were amplified twice with polymerase chain reaction (PCR).14 Mouse-specific, 16S rRNA gene sequence profiles were produced by separation of the heterogeneous PCR products using DGGE, which was performed using the DCode system (Bio-Rad, Hercules, CA, USA) as described previously.27 A reference marker, constructed using clones of marker bacteria, was included on each gel to verify the continuity of the gradient and to facilitate comparison between patterns. Gels were stained for 1 h in 1X TAE buffer containing Sybr-Green (molecular probes) prior to digitization.2.6 Computation
All computations were performed on a PC desktop Dell Dimension 2400, 2.20 GHz, 1 GB Ram, with Matlab 6.5 (Mathworks) using in-house software. SVM was applied using the OSU-SVM ver. 3.00 Toolbox by Maet al.283 Chemometric methods
Experiments are setup in a way to compare odours from mice that vary systematically in their genetic background and MHC-haplotype. The dataset is composed of four distinct groups of samples as follows: (i) strain BALB, MHC-haplotype H-2d; (ii) strain BALB, MHC-haplotype H-2b; (iii) strain C57BL10, MHC-haplotype H-2d; and (iv) strain C57BL10, MHC-haplotype H-2b. Hereafter, these groups will be referred as to S1H1, S1H2, S2H1 and S2H2 respectively. This design allows comparing mice with same genetic background but different MHC-haplotype (S1H1vs. S1H2; S2H1vs. S2H2) as well as mice with different genetic background but with same MHC-haplotype (S1H1vs. S2H1; S1H2vs. S2H2).Chemical and microbiological data are both acquired for 34 mice (dataset α) divided in the four groups indicated above (9 S1H1, 9 S1H2, 8 S2H1 and 8 S2H2). In addition, further GC/MS analysis is carried out on another 58 mice (dataset β, 13 S1H1, 11 S1H2, 15 S2H1 and 17 S2H2). These two datasets are used for two different purposes. Dataset α allows an initial exploration of the data, where main trends can be searched for after merging the microbiological and chemical information and can be subsequently posed in relation to the genetics of mice, i.e. their strain as well as MHC-haplotypes. A consensus analysis enables to verify which measure in each block contributes to the patterns in the data and most importantly whether and to what extent, similar trends are reproduced in the microbiological and chemical profile. This may provide clues to verify the possible shaping of the commensal microflora by the MHC-haplotypes.
Dataset β, which consists of more instances, can be used to build up a statistical model to verify whether the chemical profile can be used to predict the genetics of mice. Dataset β will train a classifier that will be tested on the instances of dataset α.
3.1 Feature extraction
The purpose of this operation is to extract from the raw data a defined number of variables for each sample to arrange into two distinct data matrices that will summarize the information obtained by the chemical and microbiological analyses. Each sample must be described by the same number of variables and each variable must refer to the same entity across all samples. This preparation phase is necessary for most analytical techniques and can be more or less laborious depending on the characteristics of the data.29In DGGE, the variables correspond to the intensities of the bands identified along the gel. Feature extraction is straightforward as it requires a digitization of the image of the DGGE gel (Bionumerics gel analysis software, Applied Maths, Sint-Martens-Latem, Belgium) where lanes are aligned relative to internal markers and the intensity of the bands is measured and quantified.30,31
For GC/MS, variables correspond to the areas of the peaks having a common chemical origin in different chromatograms of all samples32,33 to form peak tables. Although faster alternatives are available for data screening (e.g. using Total Ion Currents or Single Ion Chromatograms), this method is preferred because it directly relates to the chemical components in the mixture, hence making feasible the identification of potential marker compounds in a subsequent phase of the analysis. However, the procedure is laborious, particularly because chromatograms may contain several hundreds of compounds per sample, the signal-to-noise ratio may be low, and peaks may partially overlap, so peaks must be compared on the basis of their spectral and retention characteristics. The procedure involves four main stages and the basis of the first three steps is described in detail elsewhere34 and takes advantage of the MS information to align peaks in different chromatograms and form a peak table. The main steps are summarized below.
(1) Smoothing of single ion chromatograms. Noise is smoothed with a DB5 (Daubechies wavelet) wavelet filter and three levels of decomposition, chosen based on the data under investigation: because the data contain a significant amount of high frequency noise which is caused by the column (high bleeding) such a wavelet appeared to be the most effective one with very little distortion.
(2) Determining the position and intensity of peaks. Next the first derivative is calculated using a Savitzky–Golay five-point quadratic filter.35 The profile is then processed using a switch-position loop. A peak start is taken as whenever the first derivative becomes positive. A peak maximum is taken when two sequential first derivative points are negative and either the peak is at least seven datapoints wide or the peak is at least four times higher than the lowest non-zero value in the chromatogram. The second criterion is necessary to detect partially overlapped peaks found on the shoulder of other peaks that are not sufficiently large. A peak end is taken as when two sequential first derivative points are positive. This step is performed on all the mass ions and peaks with same retention time for different ions are then merged together. The combination of their intensity is used to estimate their area while the combination of ions is assigned as the spectrum of the compound. The procedure is then applied on all samples and the dataset matrix is deduced by means of a pair-wise comparison of peaks across samples on the basis of their spectral profile. The intensity of peaks is determined by our previously published method.34
(3) Matching peaks from a common origin. The procedure starts from the first peak of the first sample, looking for peaks within a defined retention time window across the second sample. If a peak with a similar MS is found, the areas are allocated in the same column of the matrix (same variable), otherwise the unmatched peak is regarded as originating from a new compound and is placed in a new column and assigned as a new variable. The procedure is repeated for all peaks detected in all samples. Various further failsafe mechanisms are incorporated into our method.34
(4) Removing rare peaks. Because a number of unique peaks are detected in only a small portion of samples and have low diagnostic power for a class, only those detected in at least four samples are retained for further analysis.
3.2 Pattern recognition
After the signal processing step described above, the microbiological and chemical information acquired with DGGE and GC/MS is summarized in two distinct matrices (XM and XC). Both are composed of 34 row vectors (samples of dataset α) with 30 and 780 variables respectively, corresponding to different gel bands and chromatographic peaks.A series of additional processing steps must be applied to make the application of the following pattern recognition techniques for exploration and classification effective. It is desirable to scale the data using a logarithmic transformation in order to reduce the influence of large peaks, and to reflect the fact that intensities are often log-normally distributed. However, an additional problem then relates to the sparseness of XM and XC, as both matrices contain a relatively high proportion of zeros because some bands and peaks are found only in a portion of the samples: we find in many areas of metabolomic profiling that marker peaks are present only in a portion of samples. In order to compute logarithms of the peaks that are either absent or below the detection we replace the zeros with half of the lowest intensity peak detected in the dataset. Next, each sample is normalized along rows so that the sum of variables equals one: this allows maximizing the comparison between samples, removing unwanted effects such as the quantity of material sampled and analysed. Finally data are standardised along columns to attribute to each variable an equal weight.
In the last few years, modifications of the PCA have been introduced to break the descriptors into several blocks to improve the interpretation of the models, one of which is consensus PCA (C-PCA).18,19,38 In our case it is initially of interest to verify what is the ‘consensual’ view obtained from merging the information available from XM and XC and to determine what is the contribution of each of the two profiles. In C-PCA, the data matrix X is broken down in different blocks (XC and XM in our case) and a consensus direction among all the blocks is sought. The method starts by selecting a super-score t that is multiplied on all blocks to give the block loadings. The block loadings are normalized to length one and then multiplied through the respective blocks to give the block scores. The block scores are combined in the super-block T. Then a PCA iteration on T is performed to give the super-weight wT that is normalized to length one and multiplied onto the super-block T to obtain an improved estimate of the super-score t. This is repeated until convergence of t. The super-score T summarizes the main trends between all blocks and block scores TM and TC represent the individual contribution of each block that are related to the former by means of the weight vector w (Fig. 1). The algorithm adopted here is the one originally proposed that incorporates a block-scaling to balance the contribution of XC and XM relative to the number of variables. In addition, to stabilise the algorithm, the super-scores are initialised as suggested18 with the first score vector calculated from the super-matrix determined as follows:
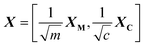
where tr indicates the trace of the matrix. The coefficient is standardised, i.e. it spans the interval [0,1] and returns 1 in case of perfect correlation. In addition its value is invariant to scaling and rotation, therefore the coefficient can be effectively used also on the super-space extracted with PCA, to measure to what extent the configurations derived give the same view of the samples. This solution may be preferred to comparing directly XM and XC because PCA reduces the data-space of the blocks to their essential variability, smoothing out the noise present in XM and XC.
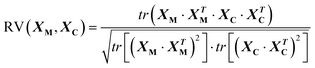
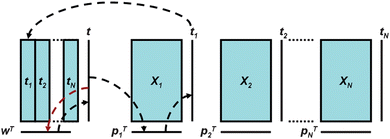 | ||
| Fig. 1 Principles of C-PCA. The scores obtained from each data-block t1…tN, are joined into a matrix from which a super-score is extracted to describe the overall trend in the data. | ||
The first technique used is PLS-DA. SVM is used as an alternative in its basic form (LIN-SVM) and with a radial basis kernel (RBF). Further details about SVMs are discussed elsewhere.25,26,40
The optimal parameters of the model (number of components for PLS-DA, penalty error C for LIN-SVM, C and radial width σ for RBF-SVM) are determined by a leave-one-out procedure: a model with a certain parameter setting (i.e. a given number of PLS components for PLS-DA or kernel parameters, i.e.C and σ, for SVM) is built using 57 of the 58 samples of dataset β and its prediction ability tested on the sample left out, with the procedure repeated for each sample. For PLS-DA, the number of PLS components varied from one to ten and for SVM, C and σ were varied according to a grid search procedure. The parameters of the best model, i.e. the one with the lowest error rate, is then used to build the predictive model on the full dataset β and this model was tested on the 34 samples that compose dataset α.
4 Results and discussion
The feature extraction procedure summarizes the information recorded by DGGE and GC/MS into two data matrices XM (34 × 30) and XC (34 × 780). The DGGE matrix is illustrated in Fig. 2. The relatively low number of variables in XM allows a direct visual inspection where it appears clearly the presence of bands related to the strains, e.g. band nos. 1 and 27 for S1 as well as band nos. 18, 19 and 21 for S2. To a minor extent, bands characteristic of the MHC are also observable, e.g. band no. 14 for H2 and band no. 15 for H1. The higher dimensionality of XC does not allow drawing such direct conclusions on the second block of data, hence requiring multivariate modelling to summarize the main trends.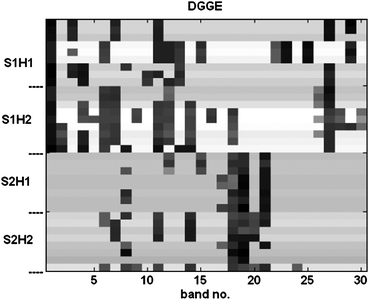 | ||
| Fig. 2 DGGE (XM) matrices obtained by feature extraction. | ||
4.1 Exploratory analysis
Consensus PCA is applied on both blocks XM and XC to extract the consensual view on the samples of dataset α (Fig. 3). The first two super-scores (on the top) indicate a main trend that directly relates to the strain membership. S1 samples are located towards the left of the plot (filled-in symbols), while the S2 samples (open symbols) distribute in a rather compact cluster in the top right quadrant. A less pronounced distinction can also be detected in relation to MHC, where H1 samples (circles) and H2 samples (triangles) tend to separate along the top right–bottom left diagonal. The related block scores plots for XM and XC exhibit a similar distribution, although XC is characterised by a higher dispersion. Fig. 4 shows the norm of the super-scores (top), indicating that the first two are in fact predominant and describe a large portion of the data-blocks. The weights (bottom) indicate that XM is explained well by the first two super-scores, while XC contributes more to the following components. In addition, with some overlap, the third and fourth super-scores (Fig. 3, bottom) further indicate a trend relating to MHC, with samples partially separated again along the top left–bottom right diagonal. In addition, S2 samples seem to be somewhat less dispersed as they distribute closer to the centre of the plot, hence explaining less variation. Also, the blocks scores in these two components appear more markedly different from each other.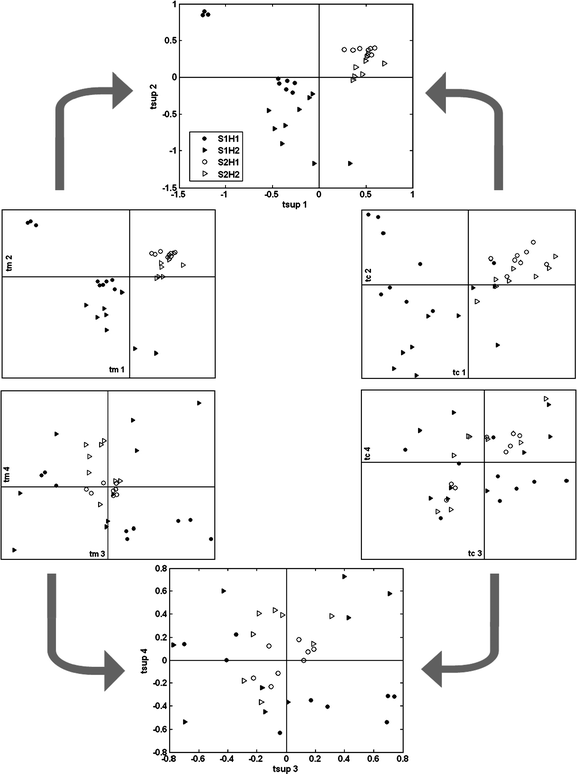 | ||
| Fig. 3 C-PCA. The scatter plot of super-scores tsup (middle) are derived from the scores tm of block XM (on the left) and the scores tc of the block XC (on the right). Symbols for the four groups SnHn where n = 1 or 2 are defined in the top graph. | ||
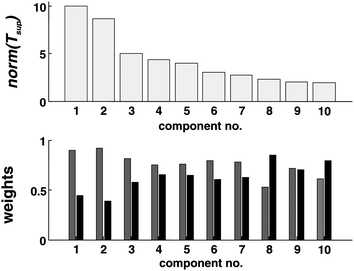 | ||
| Fig. 4 Size of the super-scores tsup (top) and weights of the block scores (bottom) tm (grey) and tc (black). | ||
Whereas supervised feature selection could be performed to obtain better apparent graphical separation between the groups, such supervised methods can lead to overfitting of the data and are only legitimate if one is sure in advance that there will be a separation. It can be shown41 that using a completely random dataset, by choosing the variables that appear most diagnostic, apparently good separation can be obtained. In hypothesis-based biology, it is often dangerous to perform supervised feature selection prior to graphical visualisation of results, although legitimate to simplify the data using criteria unrelated to the hypothesis being studied; for example, we remove rare variables from the dataset that occur in less than four samples.
Summarizing, the results indicate that the genetic strain of the mice influences the composition of the commensal microbiota from scent-marks as well as the volatile compounds, with a stronger trend associating to the strain membership and a weaker but clearly visible trend associating with MHC-haplotype. Nevertheless, the weights (Fig. 4, bottom) for the chemical data suggest that a substantial amount of variation is not explained by these factors. This is further confirmed when calculating the amount of overlap between XM and XC, with the RV coefficient having a value of 0.47. The correlation value substantially increases when the blocks are reduced to their essential variability using the scores of the principal component space. Fig. 5 shows where the RV coefficient exceeds a value of 0.65 for each possible combination of the PCs. Each subset combination of PCs is considered because factors of higher order may relate to trends in the data that are unique for a block (e.g. instrumental drift in GC/MS), hence affecting the correlation negatively. It is possible to observe that the first two components are well correlated and PC2 from XM resembles PC2, PC3 and PC4 from XC. The correlation rises to a maximum of 0.84 for the configurations given by the first two PCs of both XM and XC. The matrices XM and XC show a degree of overlap, but also have a substantial variation that is specific to each block. This is expected, particularly for XC, where the matrix is composed by several hundred variables and may result in a richer and more complex pattern. Systematic variations that effect solely either XM or XC may decrease the amount of correlation. Examples may be the occasional presence of contaminants during the sampling phase or instrumental drift. In addition, it can be noticed that some genetically related factors may contribute to the distribution of only one of the two blocks, for example, volatiles that that do not interact with the metabolism of the microflora, or in turn, MHC-induced chemicals that interact with the microflora but that the sampling technique combined with GC/MS is unable to determine.
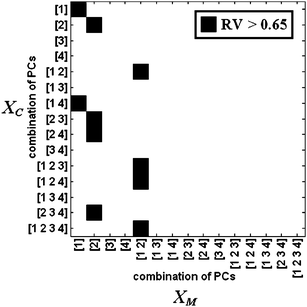 | ||
| Fig. 5 Map of the RV coefficients. | ||
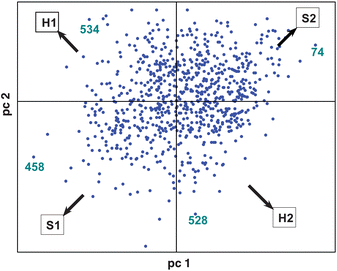
The identification of potential chemical markers in XC relating to genetics can be readily carried out looking at the block loadings (Fig. 6). In PCA, the loadings represent the weights of the original variables in determining the block scores as illustrated in Fig. 3. Relevant variables for a given group observed in the plot of the scores locate in an analogous axis position in the loadings plot. It was observed that the block scores of the first two components allow a separation between S1 and S2 along the top left–bottom right diagonal and a (milder) separation between H1 and H2 along the bottom left–top right diagonal. Some potential markers, located at the extremes of such directions in the loadings plots are taken and their quantitative distribution examined in Fig. 7. Peak nos. 458 and 74 display a distribution consistent with S1 and S2 respectively, while peak nos. 534 and 528 are related to H1 and H2. The mass spectra of these four compounds are presented in Fig. 8.
 | ||
| Fig. 6 Block loadings of XC. | ||
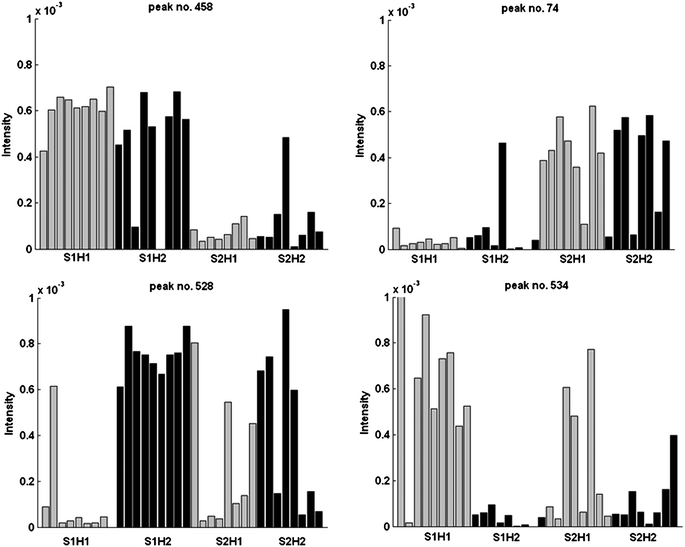 | ||
| Fig. 7 Potential markers for MHC and strains in the GC/MS data. | ||
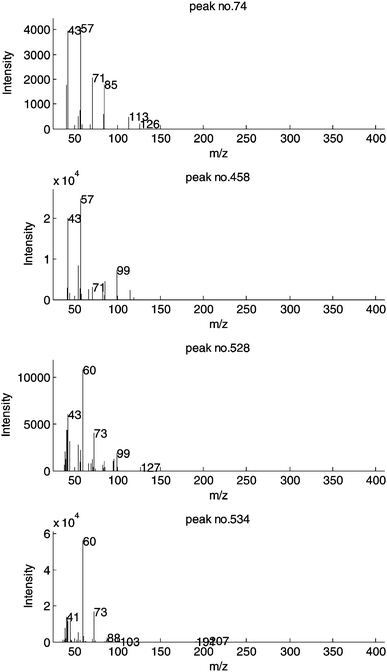 | ||
| Fig. 8 Mass spectra of four potential marker peaks. | ||
4.2 Classification
The use of the chemical information as a fingerprint to predict genetics is summarized in Table 1. Optimisation with a leave-one-out procedure leads to the selection of a PLS-DA model with two components for strain classification and three components for haplotypes. The optimal value of the penalty error for LIN-SVM is C = 0.01 both for strains and haplotypes. For RBF-SVM, optimisation returns C = 50, σ = 0.01 for strains, and C = 100, σ = 0.0005 for haplotypes as models.| Correct classification (%) | ID misclassified samples | |||||
|---|---|---|---|---|---|---|
| Training set | Test set | |||||
| Strains | Haplotypes | Strains | Haplotypes | Strains | Haplotypes | |
| LIN-SVM | 100 | 100 | 97.06 | 88.24 | No. 23 | Nos. 2, 3, 12, 34 |
| RBF-SVM | 100 | 100 | 100.00 | 85.29 | N/a | Nos. 2, 3, 4, 12, 34 |
| PLS-DA | 92.86 | 100 | 97.06 | 85.29 | No. 34 | Nos. 2, 14, 18, 28, 34 |
All three classifiers perform very similarly, with no substantial differences. Auto-prediction on the samples of dataset β, which were used to build the model, returns a perfect classification. Most notably, testing on a separate dataset α indicates that strain membership (genetic background) can be better predicted than haplotype membership (MHC-haplotype). However, a rate of 85–88% for classifying by MHC-haplotype further indicates that this feature is consistently present in the data and can be implemented to generate a predictive model that performs satisfactorily.
5 Conclusions
Our goals were to examine whether the volatile compounds (analysed by GC/MS) or microbial communities (analysed with DGGE fingerprinting) in scent-marks differ among strains of mice, and if so, whether there are any relational features between the two extended phenotypes. Further, we examined the effects influenced by MHC-genes as well as the whole genetic background (i.e. the entire genome excluding the MHC region). The experimental design allowed us to compare the effects of MHC genes with those of the whole genetic background (i.e. the entire genome excluding the MHC region), while controlling environmental sources of variation (e.g. diet, social conditions, etc.). To our knowledge, this is one of the first studies to provide evidence that MHC and other genes influence the composition of volatile compounds in urine (and the first analytical study on scent-marks),7,8 and it is the first study to simultaneously compare the patterns of volatiles with microflora communities.The raw data analysed with DGGE and GC/MS are organised by means of a feature extraction process in two distinct matrices XM (DGGE) and XC (GC/MS) that can be multivariately modelled. Consensus PCA shows that the genetic background (S1 and S2, Fig. 3) has a strong influence in shaping both XM and XC but a milder trend is clearly observable also in reference to the MHC-haplotype. Matrix correlation analysis indicates that XM and XC are partially correlated, but also bring consistent complementary information. These findings are associations, and further experimental manipulations are necessary to determine whether genetics influences individual microflora in scent-marks, and whether these influence profiles of volatile compounds.
Classification by means of PLS-DA and SVM demonstrates that the chemical signatures in scent-marks provide a surprisingly effective fingerprint for predicting mouse strain, as we found that the strains are perfectly classified, and MHC-haplotypes were also predicted with high accuracy.
Acknowledgements
Alexandra Katzer, Dr Penn's administrative assistant, is thanked for her superb organisational skills. This work was sponsored by ARO Contract DAAD19-03-1-0215. Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the United States Government. This work is approved for public release, distribution unlimited.References
- D. J. Penn and P. Ilmonen, Encyclopedia of Life Sciences, Macmillan, London, 2005 Search PubMed.
- D. J. Penn and S. Fischer, The Biologist, 2004, 51, 207–211 Search PubMed.
- K. Yamazaki, E. A. Boyse, V. Mike, H. T. Thaler, B. J. Mathieson, J. Abbott, J. Boyse, Z. A. Zayas and L. Thomas, J. Exp. Med., 1976, 144, 1324 CrossRef CAS.
- C. Wedekind, T. Seeback, F. Bettens and A. J. Paepke, Proceedings of the Royal Society of London Series B-Biological Sciences, 1995, 260, 245 Search PubMed.
- D. J. Penn, Ethology, 2002, 108, 1 CrossRef.
- D. J. Penn and W. K. Potts, Am. Nat., 1999, 153, 145 CrossRef.
- M. V. Novotny, H. A. Soini, S. Koyama, D. Wiesler, K. E. Bruce and D. J. Penn, J. Chem. Ecol., 2007, 33, 417–434 CrossRef CAS.
- A. Willse, A. M. Belcher, G. Preti, J. H. Wahl, M. Thresher, P. Yang, K. Yamazaki and G. K. Beauchamp, Anal. Chem., 2005, 77, 2348 CrossRef CAS.
- D. J. Penn and D. W. Potts, Adv. Immunol., 1998, 69, 411 Search PubMed.
- K. Yamazaki, K. A. Singer and G. K. Beauchamp, Genetica, 1998, 104, 235 CrossRef.
- K. Yamazaki, G. K. Beauchamp, Y. Imai, J. Bard, S. P. Phelan, L. Thomas and E. A. Boyse, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 8413 CrossRef CAS.
- H. M. Schellinck, E. Rooney and R. E. Brown, Phys. Behav., 1995, 57, 1005 Search PubMed.
- P. Toivanen, J. Vaahtovuo and E. Eerola, Infect. Immun., 2001, 69, 2372 CrossRef CAS.
- C. V. Lanyon, S. P. Rushton, A. G. O'Donnell, M. Goodfellow, A. C. Ward, M. Petrie, S. P. Jensen, L. M. Gosling and D. J. Penn, FEMS Microbiol. Ecol., 2007, 59, 576 CrossRef CAS.
- T. Leinders-Zufall, P. Brennan, P. Widmayer, P. Chandramani, A. Maul-Pavicic, M. Jager, X. H. Li, H. Breer, F. Zufall and T. Boehm, Science, 2004, 306, 1033 CrossRef CAS.
- L. M. Gosling and S. C. Roberts, Advances in the Study of Behaviour, 2001, 30, 169 Search PubMed.
- S. Wold, S. Hellberg, T. Lundstedt, M. Sjöström and H. Wold, Proc. Symp. on PLS Model Building: Theory and Application, Frankfurt am Main, 1987 Search PubMed.
- J. A. Westerhuis, T. Kourti and J. F. MacGregor, J. Chemom., 1998, 12, 301 CrossRef CAS.
- A. K. Smilde, J. A. Westerhuis and S. de Jong, J. Chemom., 2003, 17, 323 CrossRef CAS.
- S. J. Dixon, Y. Xu, R. G. Brereton, A. Soini, M. V. Novotny, E. Oberzaucher, K. Grammer and D. J. Penn, Chemom. Intell. Lab. Syst., 2007, 87, 161 CrossRef.
- B. K. Alsberg, D. B. Kell and R. Goodacre, Anal. Chem., 1998, 70, 4126 CrossRef CAS.
- R. G. Brereton, Applied Chemometrics for Scientists, Wiley, Chichester, 2007, ch. 10 Search PubMed.
- M. Sjöström, S. Wold and B. Söderström, in Proceedings of PARC in Practice, Elsevier Science Publishers, Amsterdam, 1985 Search PubMed.
- L. Ståhle and S. Wold, J. Chemom., 1987, 1, 185.
- Y. Xu, S. Zomer and R. G. Brereton, Crit. Rev. Anal. Chem., 2006, 36, 177 CrossRef CAS.
- V. N. Vapnik, The Nature of Statistical Learning Theory, Springer, New York, 2nd edn, 2000 Search PubMed.
- G. Muyzer, E. C. de Waal and A. G. Uitterlinden, Appl. Environ. Microbiol., 1993, 59, 695 CAS.
- J. Ma, Y. Zhao and S. Ahalt, Ohio State University, OSU SVM Classifier Matlab Toolbox ver 3.00, http://www.ece.osu.edu/%7Emaj/osu_svm.
- R. O. Duda, P. E. Hart, D. G. Stork, Pattern Classification, Wiley Interscience, New York, 2nd edn, 2001 Search PubMed.
- J. Postma, B. P. J. Geraats, R. Pastoor and J. D. van Elsas, Phytopathology, 2005, 95, 808 Search PubMed.
- P. J. Mouser, D. M. Rizzo, W. F. M. Roling and B. M. Van Breukelen, Environ. Sci. Technol., 2005, 39, 7551 CrossRef CAS.
- H. Idborg-Bjorkman, P. O. Edlund, O. M. Kvalheim, I. Schuppe-Koistinen and S. P. Jacobosson, Anal. Chem., 2003, 75, 4784 CrossRef.
- H. Shen, J. C. Carter, R. G. Brereton and C. Eckers, Analyst, 2003, 128, 287 RSC.
- S. J. Dixon, R. G. Brereton, H. A. Soini, M. V. Novotny and D. J. Penn, J. Chemom., 2006, 20, 325–340 CrossRef.
- A. Savitzky and M. J. E. Golay, Anal. Chem., 1964, 36, 1627 CrossRef CAS.
- R. G. Brereton, Chemometrics: Data Analysis for the Laboratory and Chemical Plant, Wiley, Chichester, 2003 Search PubMed.
- S. Wold, K. Esbensen and P. Geladi, Chemom. Intell. Lab. Syst., 1987, 2, 37 CrossRef CAS.
- S. J. Qin, S. Valle and M. J. Piovoso, J. Chemom., 2001, 15, 715 CrossRef CAS.
- R. P. Escoufier, Appl. Stat., 1976, 25, 257 CrossRef.
- S. Zomer, R. G. Brereton, J. F. Carter and C. Eckers, Analyst, 2004, 129, 175 RSC.
- R. G. Brereton, TrAC, Trends Anal. Chem., 2006, 25, 1103 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |