Split hybridisation probes for electrochemical typing of single-nucleotide polymorphisms†
Fausto
Lucarelli
a,
Silvia
Capponcelli
b,
Giovanna
Marrazza
*a,
Luca
Sangiorgi
b and
Marco
Mascini
a
aDepartment of Chemistry, University of Florence, via della Lastruccia 3, 50019 Sesto F.no, Florence, Italy. E-mail: giovanna.marrazza@unifi.it; Fax: +39 055 5253396
bModulo di Familiarità e Genetica, Istituti Ortopedici Rizzoli, via di Barbiano 1/10, 40136, Bologna, Italy
First published on 18th October 2008
Abstract
This paper describes the development of a highly selective single-nucleotide polymorphisms (SNPs) typing method based on the use of split hybridisation probes and demonstrates the concept through the electrochemical analysis of single-base mutations in actual patient samples. The requirement that two probes hybridised adjacent to one another to allow for stabilisation (via base-stacking) and binding of the allele-specific oligonucleotide (ASO), imparted highly stringent selectivity criteria to the assay. Simple rules for tuning the characteristics of such stacking/ASO probe pairs and achieve full mismatch discrimination at ambient conditions (with no need to strictly control the temperature) are provided. All genotyping experiments were indeed performed at room temperature, using the planar surface of disposable probe-modified gold electrodes as the genosensing platform. The ability to detect nanomolar amounts of a synthetic target even within a vast excess of single-base substituted sequences gave strong evidence of the specificity of the split probes assay. Proving the general validity of this genotyping approach, application of the analytical pathway was further demonstrated for clinical targets (amplified from the human TP53gene) whose mutational site was poorly accessible, being part of a thermodynamically stable hairpin. In combination with use of auxiliary oligonucleotides (which restored the availability of each pre-defined hybridisation site), the assay demonstrated the ability to fully discriminate single-base mutations with detection limits in the high picomolar range (total analysis time: 60 min). Our specific probe design, hybridisation and signal transduction paths make the analytical process remarkably simple, relatively low cost and, thus, well suited for low throughput analysis of clinically relevant samples.
Introduction
Sequence variations at the single nucleotide level (the so-called single nucleotide polymorphisms, SNPs) occur in our DNA roughly once every 500–1000 bases.1 While some of these base changes are “silent” and do not affect functioning of the organism, some other have been strongly correlated to different forms of cancer and the occurrence of fatal and long term diseases. The ability to map the presence of such mutations at well defined loci is, therefore, providing revolutionary opportunities in both diagnosis and treatment of diseases.The number of available strategies to recognize the presence of single-nucleotide polymorphisms within specific genes is, nowadays, continuously increasing. Large scale SNPs analyses are, for example, frequently carried out using high-throughput oligonucleotide microarrays which rely on the allele-specific hybridisation of surface-immobilised capture probes.2 Alternative and even more selective methodologies rely on the fidelity of the enzymes involved in DNA replication and repair, such as polymerases, ligases and endonucleases. Among them, single-base extension,3–5oligonucleotide ligation6,7 and invasive cleavage8 reactions offer unsurpassed levels of selectivity and sensitivity. Other hybridisation-based approaches achieve single-nucleotide resolution through complicated washing steps which demand strict temperature and buffer composition control9. SNPs are also currently detected by comparing the optical melting profiles of matched and mismatched duplexes.10,11 Sophisticated strategies of “active” hybridisation have used electric fields on a microelectronic device to regulate nucleic acids transport, hybridisation and stringency.12–14 Mismatched duplexes have also been discerned by measuring a change in the electronic coupling within the base-pair stack, rather than relying on the different thermodynamic stability of fully complementary and mismatched hybrids.15,16 Alternatively, different groups have taken advantage of the improved mismatch discrimination capabilities of (oligo)nucleotide-functionalised nanoparticles,17,18 very short allele-specific probes,19–21 structured22,23 (hairpin-like) oligonucleotides and probes based on DNA analogues such as PNA24–26 and LNA.27 Depending on the specific application, several of these methodologies are, however, excessively demanding in terms of costs, analysis time or complexity of probe design. Moreover, despite strong research efforts, a number of papers still report on the inability to effectively suppress mishybridisation events, with consequent observation of relatively strong non-specific signals.
Addressing aspects which could help promoting the analysis of point mutations even at a decentralised level, this paper describes the development of a highly specific yet low cost SNPs typing assay based on the use of split hybridisation probes.28–33 While this method cannot compete with the levels of parallelisation reached by many commercially available genotyping platforms (e.g., Affymetrix, Illumina, Corbett, Sequenom, etc.), our strategy could be suitably applied when only small numbers of SNPs have to be analysed. In a conventional allele-specific hybridisation assay, the region that surrounds the SNP is recognised by a single ASO probe. In contrast, the proposed method relies on the use of (at least) two oligonucleotides which are hybridised to the longer sample in a tandem fashion. Specifically, a very short and labelled ASO probe is hybridised to the complementary sequence in a contiguous arrangement34 with a longer and stabilising oligo, denoted as the “stacking probe” (Fig. 1A).
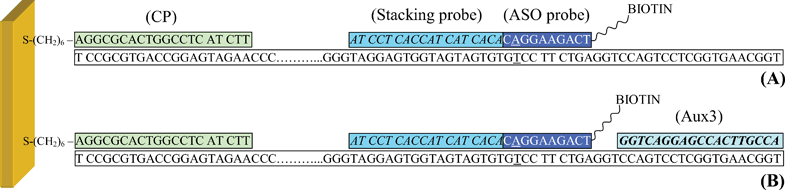 | ||
| Fig. 1 Schematic representation of TP53 samples hybridised at the surface of the gold sensor: (A) assay format involving use of the split (Stacking/ASO) probes only; (B) combined use of the split probes and the auxiliary oligonucleotide Aux3. CP: surface-tethered capture probe; stacking probe: STK; ASO probe: SP2. | ||
The reason why the proposed stacking/ASO probe pairs make the selectivity of the assay particularly favourable is twofold. On one hand, the ASO probes were designed in a way that, in the absence of stacking probe, no direct hybridisation to the target sequence was allowed. The requirement that both STK and ASO probe hybridised adjacent to one another to allow for ASO probe stabilisation via base-stacking35 and binding, thus imparted the first selectivity criterion. The method then took advantage from the fact that the short duplex generated by the ASO probe is extremely sensitive to single-nucleotide substitutions.
Owing to their inherent characteristics (which combine compatibility with the design of high-throughput microarrays36 with lower costs), electrochemical detection technologies have recently emerged as the most promising and amenable to drive the transfer of DNA detection protocols to the point-of-care setting.37–43 Meeting the need for low cost, technically and operationally simplified analytical protocols, all genotyping experiments shown in this study were performed at ambient conditions using the planar surface of disposable capture probe-modified screen-printed gold electrodes as the genosensing platform.44 Accordingly, electrochemical transduction of the hybridisation events was accomplished using an enzyme label45–50 which converted its electro-inactive substrate into an electroactive derivative.
Unlike most of the common allele-specific typing protocols (which require precise temperature control for allele discrimination), an excellent selectivity against single-base mismatches was achieved at room temperature, even using high ionic strength hybridisation and post-hybridisation washing buffers. To prove the usefulness and general validity of this genotyping approach, application of the analytical pathway was additionally demonstrated for a clinical target particularly difficult to handle because it is prone to fold into stable secondary structures. In particular, these tests involved recognition of a T>A missense mutation within a 169 bp sequence amplified from exon 7 (codon 257) of the TP53 tumour suppressor gene. This TP53 c.770T>A mutation at codon 257 was found on lymphocytes of a phenotypic Li–Fraumeni syndrome (LFS) family undergoing genetic counselling at Rizzoli Orthopaedic Institute (Bologna, Italy). The LFS (MIM #151623), an autosomal dominant disorder that predisposes individuals to multiple forms of cancer,51 is characterised in 70% of the affected families by TP53 germ line mutations.52 Patients affected by LFS develop different forms of cancer which are histologically indistinguishable from those of the general population but that usually occur at younger age.53
Experimental
Materials
NAP-10 columns of Sephadex G-25 were obtained from Amersham Pharmacia Biotech. TE buffer 20× (200 mM Tris-HCl; 20 mM EDTA; pH 7.5), Picogreen™ and λ-DNA standard solution (100 μg mL−1) were obtained from Molecular Probes. All other reagents were of analytical grade and from Sigma–Aldrich. MilliQ water was used throughout this work.All synthetic oligonucleotides (listed in Table 1) were obtained from MWG Biotech AG. Prior to use, the thiol-modified probe was treated with dithiothreitol, purified by elution through a NAP-10 column of Sephadex G-25 and then quantified by UV measurements at 260 nm. All oligonucleotide stock solutions were prepared in 0.5 M sodium phosphate buffer ([Na+] = 0.82 M, pH 7.0) and stored frozen.
| Oligo name | Sequence (5′–3′) |
|---|---|
| a CP: thiol-tethered capture probe; T1, T2, l-T2, T3, T4: oligonucleotide targets; STK: stacking probe; SP1, SP2, SP3, SP4: biotin-labelled ASO probes; SP257: biotin-labelled reporter sequence; Aux, Aux2 and Aux3: auxiliary oligonucleotides. Codon 257 is highlighted in italics with the site of mutation underlined. | |
| CP | HS–(CH2)6–AGGCGCACTGGCCTCATCTT |
| T1 | GGAGTCTTCC![[A with combining low line]](https://www.rsc.org/images/entities/i_char_0041_0332.gif) GTGTGATGATGGTGAGGATTAAGATGAGGCCAGTGCGCCT GTGTGATGATGGTGAGGATTAAGATGAGGCCAGTGCGCCT |
| T2 | GGAGTCTTCC![[T with combining low line]](https://www.rsc.org/images/entities/i_char_0054_0332.gif) GTGTGATGATGGTGAGGATTAAGATGAGGCCAGTGCGCCT GTGTGATGATGGTGAGGATTAAGATGAGGCCAGTGCGCCT |
| T3 | GGAGTCTTCC![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) GTGTGATGATGGTGAGGATTAAGATGAGGCCAGTGCGCCT GTGTGATGATGGTGAGGATTAAGATGAGGCCAGTGCGCCT |
| T4 | GGAGTCTTCC![[G with combining low line]](https://www.rsc.org/images/entities/i_char_0047_0332.gif) GTGTGATGATGGTGAGGATTAAGATGAGGCCAGTGCGCCT GTGTGATGATGGTGAGGATTAAGATGAGGCCAGTGCGCCT |
| l-T2 | TGGCAAGTGGCTCCTGACCTGGAGTCTTCCGTGTGATGATGGTGAGGATGGGCCAAGATGAGGCCAGTGCGCCT |
| STK | ATCCTCACCATCATCACA |
| SP1 |
CGGAAGACT–TEG–biot |
| SP2 |
CGGAAGACT–TEG–biot |
| SP3 |
CGGAAGAC–TEG–biot |
| SP4 |
CGGAAGAC–TEG–biot |
| SP257 |
CCATCATCACACGGAAGACTCCAG–TEG–biot |
| Aux | GGGCCTGTGTTATCTCCTAGGTTGG |
| Aux2 | CCAGGTCAGGAGCCACTTGCCA |
| Aux3 | GGTCAGGAGCCACTTGCCA |
Methods
Patients and mutational screening
The genomic DNA from 3 members of a phenotypic LFS family was extracted, under informed consent, from patients’ lymphocytes using a QIAamp DNA Mini Kit (QIAGEN). The mutational screening of TP53exons 4–11 was then performed by PCR-amplification and subsequent sequencing of the PCR products. All primer pairs and PCR conditions are listed in Table S-1 of the ESI.†Correct amplification was confirmed by agarose gel electrophoresis; the products were then purified, preconcentrated using a QIAquick PCR Purification Kit (QIAGEN) and sequenced. Exon 7 sequence analysis of the family proband (mother) revealed the presence of 2 different nucleotide sequences, one corresponding to that reported on Blast (GenBank U94788)54 and the other carrying the c.770T>A missense point mutation (Figure S-1A, ESI†). Exon 7 sequence analysis of the son showed only the presence of the sequence carrying the c.770T>A missense point mutation (Figure S-1B, ESI†). This mutation was not found on exon 7 sequence of the daughter that corresponded to that reported on Blast (Figure S-1C, ESI†). Since the mutational screening revealed the presence of the c.770T>A mutation in both the mother and the son, a 169 bp fragment comprising the mutational site (Figure S-2, ESI†) was PCR amplified starting from the DNA of all members of this family. Generation of double-stranded samples (instead of single-stranded asymmetric amplification products) was deliberately decided to challenge the functionality of the split probes. The concentration of the amplicons was determined by fluorescence measurements, using the Picogreen™ dye and a TD-700 fluorometer (Analytical Control).
Bio-modification of the sensor surface
Materials and procedures to screen-print the transducers and the protocol employed for modifying the gold surfaces with the thiol-tethered capture probe were as previously described.55 Briefly, the gold surface of the working electrodes was exposed overnight to 10 μL of the thiolated probe solution (0.5 μM in 0.5 M sodium phosphate buffer pH 7.0). The immobilisation step was then followed by treatment with 6-mercapto-1-hexanol (10 μL, 1 mM aqueous solution; 30 min). Prior to hybridisation, the modified electrodes were washed twice with 15 μL of sodium phosphate buffer.With the specific aim to keep the analytical procedure as simple as possible, any use of thermostatic devices to control hybridisation and post-hybridisation temperatures was deliberately avoided. All subsequent steps of the analytical procedure (hybridisation, labelling and detection) were thus carried out at room temperature (21–23 °C) in an air-conditioned environment.
Allele-specific analysis by means of the split probes assay
All synthetic oligonucleotide targets (T1, T2, T3 and T4) were simply diluted to the desired concentration using an 0.5 M sodium phosphate buffer which contained 100 nM of the stacking probe (STK) and 100 nM of a biotinylated ASO probe (SP1, SP2, SP3 or SP4). The PCR products amplified from the TP53gene additionally required the presence of 100 nM of specific “auxiliary” oligonucleotides (Table 1). These oligos included a sequence which hybridised the sample immediately downstream of the capture probe (Aux) and a similar one which recognised the region downstream of the ASO probes (Aux2 or Aux3). The samples were thermally denatured using a boiling water bath (5 min at 100 °C) and then quickly cooled in an ice-water bath for 1 min. A 10 μL aliquot of these solutions was finally interacted with the probe-modified electrodes for 30 min. Negative control samples included either the blank or a fully non-complementary sequence solution (both comprising split probes and auxiliary oligonucleotides). After hybridisation, the electrode surfaces were washed twice with 15 μL of DEA buffer (0.1 M diethanolamine, 0.1 M KCl, 1 mM MgCl2, pH 9.6).Labelling with alkaline phosphatase and electrochemical detection
The steps of labelling and electrochemical detection were performed as previously optimised.55 The biotinylated hybrids were labelled via interaction with 10 μL of a solution which contained 1.5 × 10−3 U μL−1 of the streptavidin-alkaline phosphatase conjugate and 10 mg mL−1 of BSA (as the blocking agent) in DEA buffer. After 10 minutes, the sensors were washed twice with 15 μL of DEA buffer. The planar electrochemical cells were then incubated with 110 μL of an α-naphthyl phosphate solution (1 mg mL−1 in DEA buffer). After 20 minutes, the oxidation signal of the enzymatically-produced α-naphthol was measured by DPV (modulation time = 0.05 s; interval time = 0.15 s; step potential = 5 mV; modulation amplitude = 70 mV; potential scan: from 0 to +0.55 V). All electrochemical measurements were performed with a μAUTOLAB type III (Eco Chemie). Each result is the mean and standard deviation of at least three measurements.Results and discussion
As an extension of our preliminary studies19 and to meet the need for reliable yet simplified and relatively low cost detection technologies, this paper describes the development of an electrochemical SNPs typing platform which relies on the use of split (stacked) hybridisation probes. Fig. 1A schematically illustrates the concept, with the region that surrounds the mutational site (the underlined base) targeted by the coaxially hybridised stacking and ASO probes. According to our experience,44 the processes of target capture and allele-specific identification were operated by physically distinct probes. No successive rounds of hybridisation were, however, required. Wild-type and mutant targets were all captured at the electrode surface by a unique capture probe (CP), designed to recognise the 3′ end of these sequences. Concurrently, the site of mutation (which was located far from the surface-hybridised termini) was identified by the stacking/ASO probe pair.Notably, several of the previously published papers describing related methodologies of stacking hybridisation, suffered from a number of drawbacks. Unsatisfactory levels of selectivity were, for example, observed despite a strict control of the temperature during the hybridisation phase and the subsequent application of stringent washing steps with warm and low ionic strength buffers.31,33 Use of radioactive labels, which are extremely sensitive but dangerous during handling and subsequent disposal, is also still dominant in the literature.32 To date, only fluorescently labelled probes have been proposed as an alternative.33 In addition, the applicability of the method seems to be restricted to short and single-stranded amplicons as the analysis of double-stranded samples is rarely mentioned.30
Design of the split probes
Displaying typical electroanalytical signals, Fig. 2 provides important insights into the performance characteristics of the split probes developed in this study.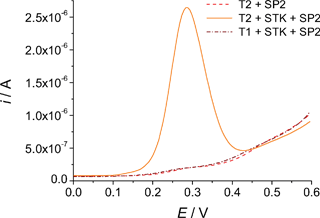 | ||
| Fig. 2 Voltammetric signals elucidating the features of the split probes assay. Both T1 and T2 were diluted to 20 nM using an 0.5 M sodium phosphate buffer; STK (when used) and SP2 concentrations were 100 nM. Other details as described in the Experimental section. | ||
The design of ASO probes (SP1 to SP4, Table 1) which were not thermodynamically allowed to directly bind the target even in conditions of perfect complementarity imparted highly stringent selectivity criteria to the assay. As one of the primary objectives of this work was to achieve full mismatch discrimination with no need to control the temperature, working at ambient conditions represented the most attractive option. The Tm of the ASO probes was thus adjusted in a way that, at 21–23 °C (the temperature at which most of the assays were performed), their hybrids were in a nearly fully dissociated state. No signal was thus observed as a consequence of the interaction of the sole ASO probes with any of the target sequences (e.g., Fig. 2, dash line). According to their GC content, these biotinylated probes were defined as 9 or 10-mer oligonucleotides. In this way, all of them possessed an identical theoretical Tm (30.0 °C, according to the MWG-Biotech ordering website) and were, therefore, expected to finally provide similar sensitivities.
The second of the two probes, STK, also played a crucial role, because of its ability to influence the thermodynamics of the duplex generated by the ASO probe. Hybridising the sample immediately upstream the region targeted by the ASO probe, the coaxial hydrophobic and electronic stacking of the bases at the conjunction between the two sequences mutually stabilised the corresponding hybrids.35,56 As a result, the Tm of the allele-specific oligonucleotide was enhanced by several °C, leading to the observation of strong analytical signals (Fig. 2, solid line). While the strength of the stacking interactions (which is base-dependent35) influenced the extent of such a stabilising effect, the length of the stacking probe can, in general, be defined with no particular restrictions. The stacking probe employed in this study was actually an 18-mer oligonucleotide. However, any sufficiently long sequence (typically longer than 15–17 bases) would have effectively served the scope.
The long and “universal” stacking probe was expected to bind equally strongly to both wild-type and mutant targets. However, in the presence of a mismatch occurring within the short ASO probe, two simultaneous phenomena inhibited mishybridisation.57 Firstly, the short labelled duplex was inherently destabilised by the mismatch. Secondly, the local geometrical distortion and greater dynamic motion to which the mispaired bases are subjected58 often diminished the degree of base overlap at the conjunction between stacking and ASO probe, thus substantially impeding stabilisation of the allele-specific oligonucleotide (Fig. 2, dash dot line).
Tests on synthetic oligonucleotide targets
Table 1 lists all synthetic oligonucleotides employed in this study. In particular, the sequences T1 and T2 represented the synthetic analogues of the only targets of real clinical interest (i.e., those carrying the wild-type and the mutant codon 257 of TP53, respectively). However, with the aim to further extend the study and explore the effect of other possible SNPs within the same sequence context [i.e., 5′C![[X with combining low line]](https://www.rsc.org/images/entities/char_0058_0332.gif) G3′/3′G
G3′/3′G![[Y with combining low line]](https://www.rsc.org/images/entities/char_0059_0332.gif) C5′], two more “artificial” mismatched targets (T3 and T4) were introduced. Consequently, besides the common stabilising stacking probe, 4 allele-specific oligonucleotide probes (SP1, SP2, SP3 and SP4) were also designed. For the sake of clarity, SP1 specifically recognised the wild-type TP53 sequence, while SP2 recognised the T>A mutation characteristic of those patients affected by LFS.
C5′], two more “artificial” mismatched targets (T3 and T4) were introduced. Consequently, besides the common stabilising stacking probe, 4 allele-specific oligonucleotide probes (SP1, SP2, SP3 and SP4) were also designed. For the sake of clarity, SP1 specifically recognised the wild-type TP53 sequence, while SP2 recognised the T>A mutation characteristic of those patients affected by LFS.
Selectivity and sensitivity of the split probes methodology are demonstrated by Fig. 3, which illustrates the analysis of the model oligonucleotide targets.
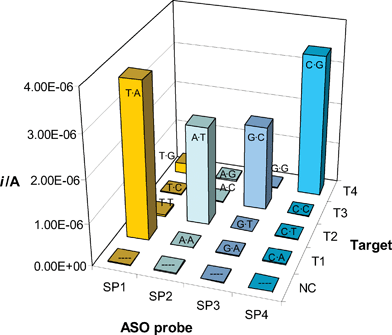 | ||
| Fig. 3 Electrochemical analysis of model oligonucleotide sequences by means of the split probes assay. The noncomplementary oligo (NC) and all targets (T1, T2, T3 and T4) were diluted to 20 nM using a 0.5 M sodium phosphate buffer that contained 100 nM of STK and 100 nM of a biotinylated ASO probe (SP1, SP2, SP3 or SP4). Other details as described in the Experimental section. The (mis)match type generated by each combination Target-ASO probe is indicated in each column. On average, the reproducibility of the above results is expressed by a relative standard deviation (RSD) of 5.1% (n = 3). | ||
With the exception of NC (that represented a fully noncomplementary sequence), each target Ti was specifically hybridised by the ASO probe SPi, while generating single-base mismatches in the presence of the remaining probes. Hence, only 4 out of the 16 combinations of target/ASO probe represented Watson–Crick pairs and yielded strong analytical signals. Barely detectable currents were observed only for the highly stabilised59T·G mismatch, while none of the other mispaired combinations yielded a measurable signal. The requirement that another probe (STK) hybridised adjacently to the allele-specific oligonucleotides to allow for their stabilisation and consequent binding, was thus shown to confer strong selectivity criteria to the assay. While circumventing the major specificity issues of the methods based on the use of very short allele-specific oligonucleotides,57 our specific design of the split probes led to sensitivity and selectivity levels previously unmet in our studies.19
Please note that the biotinylated probes employed here were designed to hybridise the samples in a way that the mismatched site corresponded to a nearly terminal base of the allele-specific oligonucleotide (the second from its 5′ end). As centrally placed mismatches are known to destabilise a duplex to a larger extent, this means that, in principle, there is still room for further improvement of the selectivity.
Table 2 further demonstrates the specificity of our split hybridisation probes.
| Sample | T2% | Electrochemical response/A | |
|---|---|---|---|
| [T2]/nM | [T1]/nM | ||
| a Details as described in the Experimental section. | |||
| 0 | 0 | — | (9 ± 1) × 10−9 |
| 5 | 0 | 100 | (9.3 ± 0.2) × 10−7 |
| 5 | 5 | 50 | (6.88 ± 0.08) × 10−7 |
| 5 | 20 | 20 | (6.1 ± 0.2) × 10−7 |
| 5 | 45 | 10 | (6.4 ± 0.1) × 10−7 |
| 5 | 95 | 5 | (4.22 ± 0.05) × 10−7 |
| 1 | 99 | 1 | (8.1 ± 0.5) × 10−8 |
| 0 | 100 | 0 | (2.0 ± 0.5) × 10−8 |
In particular, the STK/SP2 pair enabled clear identification of the mutant target (T2) even when it represented as few as 1% of the total hybridising sample. Concurrently, an excess amount of the single-base substituted (wild-type) sequence (T1, 100 nM) yielded negligible non-specific signals, thus suggesting that the mutant target could be still detected among larger quantities of “interfering” DNA.
Analysis of clinical samples
Our tests on clinically relevant samples specifically involved analysis of the DNA obtained from different members of a phenotypic Li–Fraumeni syndrome family. These LFS affected patients carried, in particular, a c.770T>A missense mutation at codon 257 (exon 7) of the TP53 tumour suppressor gene. Unexpectedly, when simply adopting the assay conditions developed for the analysis of the oligonucleotide targets, any attempt to assign the genotype of these clinical samples initially failed. The currents observed analysing such PCR products (169 bp) were, in fact, negligible.Generally speaking, these amplicons differed from the oligonucleotides T1–T4 in two aspects: their much bigger size (which increased the likelihood of the sequence to fold into thermodynamically stable secondary structures) and their double-stranded nature (with the strand complementary to the amplified target competing with the capture and the split probes for the target itself).
Indeed, analysis of the amplified sequence using the Mfold program60 highlighted one possible reason for hybridisation inefficiency/failure. Besides motifs variable from structure to structure, these simulations revealed the existence of two constantly present structured domains (which presumably strongly stabilised the solution-phase conformation of the amplicon). The first helix-loop motif largely involved the 3′ end of the sequence (where the capture probe binding site was located), thus potentially interfering with the interfacial hybridisation of the target. Concurrently, the second helix-loop system “masked” the hybridisation site of the short ASO probe. The most stable of these structures is shown in Figure S-3, in ESI.†
Possible adverse effects from the strand complementary to the amplified target were substantially excluded after performing further analyses on the synthetic and single-stranded sequence l-T2. If compared to its shorter analogue T2, l-T2 additionally replicated the region of the mutant amplicon that, according to the predictions of Mfold, contributed to the formation of the hairpin at the mutational site. Interestingly, these electrochemical analyses also gave no results (data not shown). As the ASO probes similarly failed to hybridise both the double-stranded amplicon and the single-stranded l-T2, competition from the “interfering” amplified sequence was assumed to play only a marginal role. Indeed, the genotyping assays were likely to be mostly hindered by stable intramolecular foldings of the PCR products. A combination of increased steric hindrance and unavailability of the regions “blocked” as folded domains probably concurred to inhibit all heterogeneous hybridisation processes and binding of the short ASO probes in particular. Similar intramolecular problems could not, clearly, be circumvented by employing asymmetric PCR to amplify patients DNA.
As a means to break the interfering hairpins predicted by the Mfold program, different unmodified oligonucleotides (referred to as “auxiliary oligos”, Table 1) were tested. Such sequences were designed to hybridise the samples immediately downstream of the sites targeted by the capture and ASO probes. Hence, binding of these oligos was anticipated to unfold any surrounding structured domain.20,61 As hypothesised, use of Aux (the auxiliary oligo which hybridised the target contiguously to the surface-tethered capture probe) enhanced the analytical signals by 3 to 5-fold. This effect, observed in the presence of the non-discriminating probe SP257 (Table 1) and using sensors modified with variable amounts of the capture oligonucleotide, clearly reflected the improved heterogeneous hybridisation of the amplicon (Figure S-4, ESI†).
The existence of a hairpin which blocked the mutational site was also confirmed when exploring the influence of the auxiliary oligonucleotides that hybridised the target downstream the ASO probe. In search for the optimal assay configuration, two different sequences (Aux2 and Aux3) were tested in parallel. The hybridisation site of the first one, Aux2, was immediately adjacent to the region bound by the ASO probe. As no gaps divided one sequence from the other, the additional stacking interaction at the 3′ end of the ASO probe was anticipated to impart further stability to the duplex generated by the allele-specific oligonucleotide. A gap of 3 bases was, in contrast, produced upon hybridising Aux3 (Fig. 1B), specifically with the intent to exclude establishment of new base stacks.
For both the homozygous mutant and the wild-type sample, both auxiliary oligonucleotides were used in parallel in the presence of SP1 (wild-type) or SP2 (mutant), while adopting the experimental setup in which use of such auxiliary sequences is omitted as the reference. The results obtained for the homozygous mutant sample are compared in Table 3.
| Auxiliary oligos | ||||
|---|---|---|---|---|
| none | Aux3 (3 bases gap) | Aux2 (no gap) | ||
| a The concentration of the TP53 homozygous mutant sample (169 bp) was 20 nM. Current values are expressed in amperes. Other details as described in the Experimental section. | ||||
| ASO probes | SP1 | (2.0 ± 0.9) × 10−8 | (3.0 ± 0.7) × 10−8 | (1.3 ± 0.3) × 10−7 |
| SP2 | (1.9 ± 0.7) × 10−8 | (9.6 ± 0.3) × 10−7 | (2.4 ± 0.2) × 10−6 | |
When no oligonucleotides assisted the hybridisation of the allele-specific probes, only negligible currents were measured, thus confirming earlier observations. In contrast, well defined signals were observed upon introducing either Aux3 or Aux2 into the hybridisation solution. These results clearly reflected the much easier binding of the ASO probes, which hybridised being stabilised and “protected” by the surrounding STK and Auxi (Fig. 1B). In the presence of Aux3, in particular, the split probes regained performances comparable with those previously observed with the model oligonucleotide targets. More importantly, the excellent selectivity of the assay was confirmed. As predicted, the additional stacking interaction at the 3′ end of the ASO probes established in the presence of Aux2, led to the allele-specific oligos in a double-stacked configuration (i.e., STK/SPi/Aux2). This peculiar arrangement of the oligonucleotides consistently allowed specific signals of remarkably higher intensity. Nevertheless, the specificity of the assay was also occasionally compromised. SP1, for example, was found to yield some non-specific hybridisation with the mutant target when interacted as STK/SP1/Aux2. Such a diminished selectivity of SP1 was thus ascribed to an excessive stabilisation of the ASO probe as a consequence of stacking forces acting on both ends of this 10-mer oligonucleotide. Therefore, use of Aux3 as the oligonucleotide auxiliary to the STK/ASO probe pairs was preferred, as the resulting higher selectivity prevented any ambiguous assignment of the TP53 samples genotype.
An example of analyses performed on samples from healthy (SFS27, wild-type) and affected (SFS26, heterozygous–SFS28, homozygous mutant) patients is shown by Fig. 4. In total agreement with sequencing results (Figure S-1, ESI†), the obtained electrochemical pattern allowed clear identification of each amplified sample, thus confirming that the method could be efficaciously employed to screen for patients affected by the Li–Fraumeni syndrome.
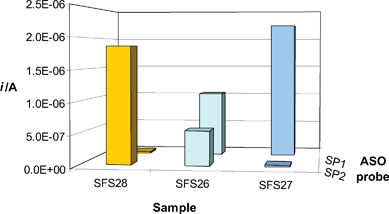 | ||
| Fig. 4 Electrochemical genotyping of samples from healthy (SFS27, wild-type) and affected (SFS26, heterozygous-SFS28, homozygous mutant) patients. Each sample was 20 nM; STK, ASO probes and Aux3 concentrations were 100 nM. Other details as described in the Experimental section. Mean RSD: 8.8% (n = 3). | ||
The analytical features of the assay for increasing concentrations of both wild-type and homozygous mutant amplicons are highlighted in Fig. 5. The electrochemical response rapidly increased with the mutant target concentration up to 10 nM and then more slowly, indicating the gradual saturation of capture probes immobilised at the electrode surface. Linear fit of the initial portion (0–10 nM) of the response/concentration profile (r2 = 0.99), evidenced a detection limit of about 250 pM, calculated as the concentration that yielded a signal higher than the mean of the blank plus 3 times its standard deviation. The response for concentrations of the wild-type sequence as high as 30 nM was, in contrast, comparable with the background current. On average, the relative standard deviation for these measurements was 4.7% (n = 3).
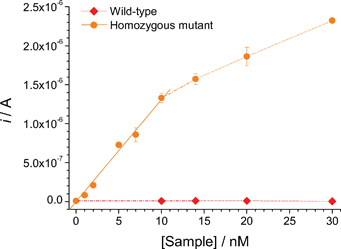 | ||
| Fig. 5 Calibration plots obtained for increasing concentrations of both wild-type and homozygous mutant amplicons in the presence of the STK/SP2 probes pair and Aux3. Other details as described in the Experimental section. | ||
Of great significance for the practical application of the method, this ability to unambiguously detect alterations at the single-nucleotide level was also checked against small variations of ambient temperature (up to 24–26 °C). As a result of the enhanced instability of the hybridised ASO probes at higher temperatures, the intensity of the specific electroanalytical signals observed at 26 °C was several fold lower than that observed at 21 °C (Figure S-5, ESI†). Nevertheless, the sensitivity of the method always remained sufficient to clearly identify the genotype of any given sample, thus confirming that there is not ultimately the need to strictly control the temperature during the assay.
Additional considerations and costs estimate
Levels of parallelisation similar to that of certain commercially available genotyping technologies can hardly be met by those approaches which rely on the use of stacked probes. However, related methodologies of stacking hybridisation have been already implemented into low density array formats and used for the analysis of actual patient samples.31–33 As our currently available arrays of electrodes62 allow the assay to be parallelised into an 8-plex format, work is currently in progress to check whether specificity and sensitivity of the method are kept when multiple probes must simultaneously face more complex samples. Preliminary data are encouraging; these tests are, however, still incomplete.Even taking all factors into account, to give an accurate estimate of what the cost per genotype would be using our electrochemical platform is rather difficult. As an advantageous feature of electrochemical readout, this detection technology is one of the cheapest, with hand-held multichannel potentiostats being relatively inexpensive (e.g., www.palmsens.com). Considering that a minimum of three oligomers (2 of which labelled) is needed to interrogate each SNP site and that, optimistically, the assay can be implemented as an 8-plex, a plausible cost for reagents, labour and consumables could be in the order of $2 per genotype (100 samples minimum). Even so, it could still represent a cost-effective solution for those labs with low/moderate genotyping requirements that, because of limited budgets, cannot afford the initial investment in highly sophisticated optical or mass spectrometer equipment.
Conclusions
This paper described a genotyping method based on the use of split hybridisation probes and demonstrated the concept through the electrochemical analysis of single-nucleotide polymorphisms in actual patient samples.Our analytical strategy presented a number of valuable advantages. The characteristics of the stacking/ASO probe pairs were tuned in a way that full mismatch discrimination was achieved with no need to strictly control the temperature during the assay. Indeed, all genotyping experiments were performed at room temperature, using the planar surface of disposable capture probe-modified screen-printed gold electrodes as the genosensing platform.
In combination with use of auxiliary oligonucleotides (which helped restore the availability of poorly accessible SNP sites), the assay showed the ability to rapidly (60 min) and fully discriminate against single-base mutations with detection limits in the high picomolar range.
Our specific probe design (with all sequences being conventional oligo-deoxyribonucleotides), hybridisation (a single step with no need to use thermostatic devices) and signal transduction paths thus simplified the whole analytical process, making it well suited for low throughput analysis of clinically relevant samples. Such preliminary screenings for mutations could help in the selection of more appropriate chemotherapy protocols, with important implications on the clinical outcomes of cancer patients.63,64
References
- L. Kruglyak, Nat. Genet., 1997, 17, 21–24 CrossRef CAS.
- http://www.affymetrix.com .
- http://www.sequenom.com/ .
- J. Mengel-Jorgensen, J. J. Sanchez, C. Borsting, F. Kirpekar and N. Morling, Anal Chem., 2005, 77, 5229–5235 CrossRef CAS.
- F. Patolsky, A. Lichtenstein and I. Willner, Nat. Biotechnol., 2001, 19, 253–257 CrossRef CAS.
- J. Li and W. Zhong, Anal. Chem., 2007, 79, 9030–9038 CrossRef CAS.
- Z. -S. Wu, J. -H. Jiang, G. -L. Shen and R. -Q. Yu, Hum. Mutat., 2007, 28, 630–637 CrossRef CAS.
- Y. Chen, M.R. Shortreed, M. Olivier and L.M. Smith, Anal. Chem., 2005, 77, 2400–2405 CrossRef CAS.
- P. Abad-Valle, M.T. Fernandez-Abedul and A. Costa-Garcia, Biosens. Bioelectron., 2007, 22, 1642–1650 CrossRef CAS.
- J. B. Fiche, J. Fuchs, A. Buhot, R. Calemczuk and T. Livache, Anal. Chem., 2008, 80, 1049–1057 CrossRef CAS.
- http://www.corbettlifescience.com .
- E. A. Barlaan, M. Sugimori, S. Furukawa and K. Takeuchi, J. Biotechnol., 2005, 115, 11–21 CrossRef CAS.
- F. Fixe, V. Chu, D. M. F. Prazeres and J. P. Conde, Biosens. Bioelectron., 2005, 21, 888–893 CrossRef CAS.
- F. Wei, C. Chen, L. Zhai, N. Zhang and X. S. Zhao, J. Am. Chem. Soc., 2005, 127, 5306–5307 CrossRef CAS.
- E. M. Boon, D. M. Ceres, T. G. Drummond, M. G. Hill and J. K. Barton, Nat. Biotechnol., 2000, 18, 1096–1100 CrossRef CAS.
- E. L. S. Wong and J. J. Gooding, Anal. Chem, 2006, 78, 2138–2144 CrossRef CAS.
- G. Liu, T. M. H. Lee and J. Wang, J. Am. Chem. Soc., 2005, 127, 38–39 CrossRef CAS.
- J. Zhang, S. Song, L. Zhang, L. Wang, H. Wu, D. Pan and C. Fan, J. Am. Chem. Soc, 2006, 128, 8575–8580 CrossRef CAS.
- F. Lucarelli, G. Marrazza and M. Mascini, Anal. Chim. Acta, 2007, 603, 82–86 CrossRef CAS.
- S. Sando, H. Abe and E. T. Kool, J. Am. Chem. Soc., 2004, 126, 1081–1087 CrossRef CAS.
- F. Patolsky, A. Lichtenstein and I. Willner, J. Am. Chem. Soc., 2001, 123, 5194–5205 CrossRef CAS.
- F. Ricci, R. Y. Lai, A. J. Heeger, K. W. Plaxco and J. J. Sumner, Langmuir, 2007, 23, 6827–6834 CrossRef CAS.
- R. Miranda-Castro, P. de-los-Santos-Alvarez, M. J. Lobo-Castaňón, A.J. Miranda-Ordieres and P. Tuňón-Blanco, Anal. Chem., 2007, 79, 4050–4055 CrossRef CAS.
- M. Steichen, Y. Decrem, E. Godfroid and C. Buess-Herman, Biosens. Bioelectron., 2007, 22, 2237–2243 CrossRef CAS.
- J. Liu, S. Tian, P. E. Nielsen and W. Knoll, Chem. Commun., 2005, 2969–2971 RSC.
- K. Kerman, M. Vestergaard, N. Nagatani, Y. Takamura and E. Tamiya, Anal. Chem., 2006, 78, 2182–2189 CrossRef CAS.
- L. Wang, C. J. Yang, C. D. Medley, S. A. Benner and W. Tan, J. Am. Chem. Soc., 2005, 127, 15664–15665 CrossRef CAS.
- D. M. Kolpashchikov, J. Am. Chem. Soc., 2008, 130, 2934–2935 CrossRef CAS.
- Y. Xu, N. B. Karalkar and E. T. Kool, Nat. Biotechnol., 2001, 19, 148–152 CrossRef CAS.
- R. Maldonado-Rodriguez and K. L. Beattie, Methods Mol. Biol., 2001, 170, 157–171 Search PubMed.
- R. Maldonado-Rodriguez, M. Espinosa-Lara, O. Barrera-Leon, C. Colin-Tovar, B. Gonzalez-Yebra, M. Salcedo-Vargas, J. C. Santiago-Hernandez, A. Mendez-Tenorio and K. L. Beattie, Mol. Biotechnol., 2003, 25, 113–126 Search PubMed.
- A. Rangel-Lopez, R. Maldonado-Rodriguez, M. Salcedo-Vargas, J. M. Espinosa-Lara, A. Mendez-Tenorio and K. L. Beattie, BMC Biotechnol., 2005, 5, 1–13 CrossRef.
- D. Wang, Y. Li, R. Zhang, D. Jiang, X. Ma, Y. Zhou and J. Cheng, Biotechnol. Lett., 2003, 25, 1613–1618 CrossRef CAS.
- S. Parinov, V Barsky, G Yershov, E Kirillov, E Timofeev, A Belgovskiy and A Mirzabekov, Nucleic Acids Res, 1996, 24, 2998–3004 CrossRef CAS.
- V. A. Vasiliskov, D. V. Prokopenko and A. D. Mirzabekov, Nucleic Acids Res., 2001, 29, 2303–2313 CrossRef CAS.
- A. L. Ghindilis, M. W. Smith, K. R. Schwarzkopf, K. M. Roth, K. Peyvan, S. B. Munro, M. J. Lodes, A. G. Stover, K. Bernards, K. Dill and A. McShea, Biosens. Bioelectron., 2007, 22, 1853–1860 CrossRef CAS.
- J. Wang, Biosens. Bioelectron., 2006, 21, 1887–1892 CrossRef CAS.
- J. Wang, G. Liu and A. Merkoci, J. Am. Chem. Soc., 2003, 125, 3214–3215 CrossRef CAS.
- M. T. Castañeda, A. Merkoçi, M. Pumera and S. Alegret, Biosens. Bioelectron., 2007, 22, 1961–1967 CrossRef CAS.
- M. Fojta, L. Havran, M. Vojtiskova and E. Palecek, J. Am. Chem. Soc., 2004, 126, 6532–6533 CrossRef CAS.
- D. Ozkan, A. Erdem, P. Kara, K. Kerman, B. Meric, J. Hassmann and M. Ozsoz, Anal. Chem., 2002, 74, 5931–5936 CrossRef CAS.
- K. Kerman, M. Saito, Y. Morita, Y. Takamura, M. Ozsoz and E. Tamiya, Anal. Chem., 2004, 76, 1877–1884 CrossRef CAS.
- D. M. Jenkins, B. Chami, M. Kreuzer, G. Presting, A. M. Alvarez and B.Y. Liaw, Anal. Chem., 2006, 78, 2314–2318 CrossRef CAS.
- M. L. Del Giallo, F. Lucarelli, E. Cosulich, E. Pistarino, B. Santamaria, G. Marrazza and M. Mascini, Anal. Chem., 2005, 77, 6324–6330 CrossRef CAS.
- J. Albers, T. Grunwald, E. Nebling, G. Piechotta and R. Hintsche, Anal. Bioanal. Chem., 2003, 377, 521–527 CrossRef CAS.
- M. Gabig-Ciminska, A. Holmgren, H. Andresen, K. Bundvig Barken, M. Wümpelmann, J. Albers, R. Hintsche, A. Breitenstein, P. Neubauer, M. Los, A. Czyz, G. Wegrzyn, G. Silfversparre, B. Jürgen, T. Schweder and S.-O. Enfors, Biosens. Bioelectron., 2004, 19, 537–546 CrossRef CAS.
- D. J. Caruana and A. Heller, J. Am. Chem. Soc., 1999, 121, 769–774 CrossRef CAS.
- Y. Zhang, H. -H. Kim and Adam Heller, Anal. Chem., 2003, 75, 3267–3269 CrossRef CAS.
- G. Hartwich, D. J. Caruana, T. de Lumley-Woodyear, Y. Wu, C. N. Campbell and A. Heller, J. Am. Chem. Soc., 1999, 121, 10803–10812 CrossRef.
- Y. Zhang, H. -H. Kim, N. Mano, M. Dequaire and A. Heller, Anal. Bioanal. Chem., 2002, 374, 1050–1055 CrossRef CAS.
- F. P. Li and J. F. Fraumeni Jr, Ann. Intern. Med., 1969, 71, 747–752 CAS.
- D. Malkin, F. P. Li, L. C. Strong, J. F. Fraumeni Jr., C. E. Nelson, D. H. Kim, J. Kassel, M. A. Gryka, F. Z. Bischoff and M. A. Tainsky, et al., Science, 1990, 250, 1233–1238 CrossRef CAS.
- F. P. Li, J.F. Fraumeni Jr., J. J. Mulvihill, W. A. Blattner, M. G. Dreyfus, M. A. Tucker and R. W. Miller, Cancer Res, 1988, 48, 5358–5362 CAS.
- S. F. Altschul, T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller and D.J. Lipman, Nucleic Acids Res, 1997, 25, 3389–3402 CrossRef CAS.
- G. Carpini, F. Lucarelli, G. Marrazza and M. Mascini, Biosens. Bioelectron., 2004, 20, 167–175 CrossRef CAS.
- M. J. Lane, T. Paner, I. Kashin, B. D. Faldasz, B. Li, F. J. Gallo and A. S. Benight, Nucleic Acids Res, 1997, 25, 611–616 CrossRef CAS.
- R. Maldonado-Rodriguez, M. Espinosa-Lara, P. Loyola-Abitia, W. G. Beattie and K. L. Beattie, Mol. Biotechnol., 1999, 11, 13–25 Search PubMed.
- H.T. Allawi and J. SantaLucia Jr., Biochem, 1997, 36, 10581–10594 CrossRef CAS.
- N. Peyret, A. Seneviratne, H. T. Allawi and J. SantaLucia Jr, Biochem., 1999, 38, 3468–3477 CrossRef CAS.
- M. Zuker, Nucleic Acids Res., 2003, 31, 3406–3415 CrossRef CAS.
- K. Metfies, S. Huljic, M. Lange and L.K. Medlin, Biosens. Bioelectron., 2005, 20, 1349–1357 CrossRef CAS.
- F. Bettazzi, F. Lucarelli, I. Palchetti, F. Berti, G. Marrazza and M. Mascini, Anal. Chim. Acta, 2008, 614, 93–102 CrossRef CAS.
- G. Blandino, A. J. Levine and M. Oren, Oncogene, 1999, 18, 477–485 CrossRef CAS.
- S. Capponcelli, E. Pedrini, M. A Cerone, V. Corti, S. Fontanesi, M. Alessio, A. Bachi, S. Soddu, D. Ribatti, P. Picci, L.J. Helman, G. Cantelli-Forti and L. Sangiorgi, Hum. Mutat, 2005, 26, 94–103 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Table S-1 and Fig. S-1 to S-5. See DOI: 10.1039/b806514d |
| This journal is © The Royal Society of Chemistry 2009 |