Helix self-assembly through the coiling of cylindrical micelles†
Sheng
Zhong
a,
Honggang
Cui
a,
Zhiyun
Chen‡
b,
Karen L.
Wooley
b and
Darrin J.
Pochan
*a
aDepartment of Materials Science and Engineering and Delaware Biotechnology Institute, University of Delaware, Newark, DE 19716, USA. E-mail: pochan@udel.edu; Fax: +1 302 831 4545; Tel: +1 302 831 3569
bCenter for Materials Innovation, Department of Chemistry and Department of Radiology, Washington University in Saint Louis, Saint Louis, Missouri 63130, USA
First published on 8th November 2007
Abstract
Both single and double helical superstructures have been created through solution self-assembly of cylindrical micelles for the first time. Helical micelles were formed from the co-assembly of poly(acrylic acid)-block-poly(methyl acrylate)-block-polystyrene (PAA-b-PMA-b-PS) triblock copolymers with different multiamines. The helix pitch could be adjusted by adjusting the amount and type of multiamine added. The helical structure exhibits unprecedented regularity for a nanostructure self-assembled from solution indicating the presence of strong, synergistic forces underlying the helix formation.
The helix nanostructure, a common molecular1–3 and supramolecular4–6 motif in biological materials, has also been produced molecularly in many synthetic polymers7–9 and supramolecularly through the self-assembly of many synthetic small molecules and polymers including peptides,10peptide amphiphiles,11polymer single crystals,12 and bolaamphiphiles.13 Supramolecular helices generally can be produced through the intermolecular assembly of chiral molecules or the co-assembly of chiral and achiral molecules.14Peptide motifs are a common chiral chemistry to induce helix formation. For example, the Nolte group prepared helical superstructures from the assembly of peptide hybrid poly(styrene)–poly(isocyanodipeptide).15 The helical aggregation is due to the presence of a hydrogen bonded network among peptidic side chains within the hydrophilic block. The Stupp group has also observed supramolecular helical assembly by attaching a β-sheet forming peptide to a series of short hydrocarbon chains.11 This system provided tunability in the final helix structures with simple changes in molecule chemistry; the supramolecular helical pitch could be modified by changing the bulkiness of chemical substituents connected to the peptide. The above referenced supramolecular helix formers share one common aspect on the molecular level: well-defined, local, intermolecular interactions exist between neighboring molecules along the helix, either from hydrogen bonding (e.g.peptide–amphiphile helices11) or from covalent chain torsion and steric interaction (e.g. achiral polyheterocyclic strands16), that dictate the nanoscopic intermolecular assembly and, thus, the micro and macroscopic helical morphologies that are formed.
Interestingly, there are many other predicted or experimentally observed examples of helices due to completely different inter or intramolecular (or inter/intraparticle) interactions. For example, mechanical forces are known to induce helicity in rod-like objects. Very long carbon nanotubes are predicted to buckle into a helical superstructure on application of a compressive load.17 Experimentally, many rodlike objects ranging from spaghetti and Teflon rods18 to supramolecular tubes such as microtubules19 are experimentally observed to exhibit sinusoidal buckling, a structure similar to a helix, due to uniaxial compression. Multilamellar lipid tubes, otherwise known as myelin, are known to bend into helices and double helices.20,21 This myelin helix formation has been modelled to be a result of lateral tension within the fluid bilayers that constitute the multilamellar tube,22 a mechanism analogous to the buckling of rods under compression. Much more complicated bilayer tubes, such as the cell wall membrane of the rod-like prokaryotic bacterium Bacillus subtilis, have been observed to make helices partially because of mechanical forces; as the bacteria grows from a common nucleation site of a spore, the rod-shaped cells elongate into a double helix due to local helical growth of the cell wall that is tension restricted from uncoiling into a straight rod due to the tethering of the double helix ends to the original spore coat.23 A very different example of a non-intermolecular interaction or non-intermolecular packing-induced helix formation mechanism is the recent idea of entropically driven helix formation.24 The model predicts that interactions between a molecular or supramolecular rod with appropriately sized depleting spheres can induce helix formation of the rod even in complex environments such as the cell. An intriguing concept is to create the ability to design these types of long-range interactions and forces into a self-assembly system for new nanostructure formation.
One underlying goal of self-assembly is to be able to construct increasingly complex nanostructures in a simple manner. For example, there have been many recent successes in complex, multicompartment, cylindrical nanostructure formation through block copolymer solution self-assembly mechanisms.25–27 We feel that a manifestation of what constitutes a “simple manner” of complex nanostructure construction is our effort to make a variety of different nanostructures including spheres, cylinders, and disks,28–30 toroids,31 and multicompartment cylinders25 all from the same underlying triblock copolymer chemistry and binary solvent mixtures. The key to the ability to construct various assembly geometries and structures with the same triblock copolymer chemistry is both the use of solvent mixtures and, more importantly, the addition of multivalent, organic counterions to interact with the charged, hydrophilic block of the amphiphilic triblock copolymer. In this communication, we introduce the ability to construct cylindrical micelles that are uniformly coiled into single or double helices with tunable pitch dependent on the amount and type of multivalent organic counterion within the self-assembling system. The local cylindrical structure contains no chiral molecular components or specific block copolymer intermolecular packing other than the classical hydrophobic–hydrophyllic core–shell micelle structure. We propose that the driving force for helix formation from a supramolecular cylinder is a combination of long-range interactions including electrostatic repulsion of a charged cylinder along its own length combined with uniaxial compression of the same cylinder. The key to the coexistence of both the electrostatic and mechanical forces to form the helices is the presence of a large excess of multivalent, organic counterions to interact with oppositely charged corona blocks of the triblock copolymer cylinders.
The helix forming experimental protocol is as follows: first the triblock copolymer poly(acrylic acid)-block-poly(methyl acrylate)-block-polystyrene (PAA94-b-PMA103-b-PS88) was dissolved in 3 ml of pure tetrahydrofuran (THF) to form 0.1 wt% polymer–THF solution. Next, organic multiamine triethylenetetramine [NH2(CH2CH2NH)2CH2CH2NH2] was added to produce the molar ratio of amine group : acid group of 10 : 1 or 15 : 1. Then the mixture was stirred overnight to allow complexation of the multiamines with the PAA block. Water was subsequently added viatitration at a rate of 1 millilitre per hour. The low solubility of PMA and PS in water resulted in hydrophobic PMA and PS collapsing into a cylindrical micellar core swollen by THF while the PAA–amine complexes transform into a water-compatible corona. When either 67% or 90% by volume water content was reached, the suspension was sealed tightly and subsequently aged for 20 d at room temperature. In addition to triethylenetetramine, ethylenediamine (NH2CH2CH2NH2) and diethylenetriamine (NH2CH2CH2NHCH2CH2NH2) were also used at an amine to acid molar ratio of 10 : 1. Cryogenic transmission electron microscopy (cryoTEM) and conventional transmission electron microscopy (TEM) were both performed to examine the assembled structure.
In Fig. 1A CryoTEM revealed that well-defined, cylindrical micelle helices with extreme long-range order dominated the assembled morphology when triethylenetetramine formed a 10 : 1 ratio of amine group to PAA carboxylic acid group at a water volume ratio of 67% in THF. A schematic of the cylinder helix superstructure, as well as the local block copolymer and multiamine molecular packing in a cross-section of the cylinder helix, is shown in Fig. 2. AFM images of helices cast from solution (please see ESI Fig. S1†) clearly show the coexistence of both left-handed and right-handed helices. This mixture of handedness is reasonable since there were no molecules with specific chirality used during the assembly. Therefore, the cylinder micelle helix superstructure is fundamentally different from other helical systems that exhibit specific handedness owing to precise molecular packing of chiral molecules. Another aspect of handedness worth noting is that the handedness of any single helix is persistent throughout the entire helix length due to a complete lack of defects except for the helix termini. Other structural parameters of the helix such as cylinder width, helix pitch, helix width, and helix core channel diameter, labeled in the higher magnification insert of Fig. 1A, were constant along the multimicrometre-long assembled structure as well as between different helical strands, suggesting the forces that underlie cylinder helix formation are uniform across the whole system.
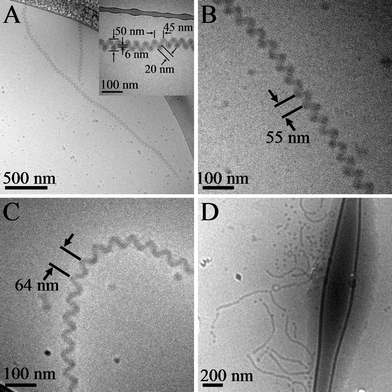 | ||
| Fig. 1 Cryogenic TEM images for (A) amine = triethylenetetramine, amine group : acid group molar ratio is 10 : 1, H2O vol% = 67% in THF and after 20 d aging; insert figure is a high magnification image of a helix; pitch distance is 45 ± 3 nm; (B) amine = triethylenetetramine, amine group : acid group molar ratio is 15 : 1, H2O vol% = 67% in THF and after 20 d aging; pitch distance is 58 ± 5 nm; (C) amine = diethylenetriamine, amine group : acid group molar ratio is 10 : 1, H2O vol% = 67% in THF and after 20 d aging; pitch distance is 64 ± 3 nm; (D) amine = triethylenetetramine, amine group : acid group molar ratio is 10 : 1, H2O vol% = 90% in THF and after 20 d aging; only unwound cylindrical micelles and spherical micelles were found. | ||
 | ||
| Fig. 2 Schematic of helical cylinder. | ||
We observed in earlier work that micelle structure could be altered simply by changing the amount and type of multiamine used in the assembly process.28,29 Similarly, we observed that we could control helix structural attributes by altering the type and amount of multiamine used in the assembly process. For example, the pitch distance was easily tuned by varying the amount of the triethylenetetramine multiamine. Fig. 1B shows an example of a cylindrical helix made from triethylenetetramine with a molar ratio of amine group to acid group of 15 : 1 and water volume ratio of 67% in THF. The larger amount of amine produced a cylindrical micelle helix pitch of 58 ± 5 nm as compared to approximately 45 ± 3 nm observed for an amine to acid ratio of 10 : 1 in Fig. 1A. Another example of helix tunability is shown in Fig. 1C in which a different type of multiamine was used in the assembly process. When triethylenetetramine was replaced by diethylenetriamine and added to produce an amine to acid ratio of 10 : 1 and water volume ratio 67% in THF, identical to the solution conditions created with tetrafunctional amine in Fig. 1A, cylindrical helices were produced with a pitch of approximately 64 ± 3 nm as measured from the cryoTEM image in Fig. 1C. All of these helix superstructures were stable in solution for more than two months. Interestingly, when ethylenediamine was used in the same solution conditions with an amine to acid ratio of 10 : 1 and 67% water in THF, helical cylinders were not formed but rather disk-like micelles were observed (please see ESI Fig. S2†). This result clearly indicates that the valency of the organic counterion is important for helix formation.
The solvent composition also plays an important role in controlling the assembled morphology. The co-assembly of PAA94-b-PMA103-b-PS88 with triethylenetetramine at an amine to acid molar ratio of 10 : 1 but at a higher water volume content of 90% in THF produced cylindrical and spherical micelles as shown in Fig. 1D. At this higher water content, hydrophilic PAA chains are more solubilized by the higher water component of the solvent thus expanding the corona volume. In addition, the hydrophobic PMA and PS blocks in the core tend to be more tightly packed because of decreasing organic solvent content in solution. Both of these effects lead to an increase in the interfacial curvature between the corona and hydrophobic core and lead to the introduction of spheres.29 Importantly, the cylindrical micelles observed at 90% water content in Fig. 1D do not form helices. Therefore, the forces coiling the cylinders at lower water content are either screened or not strong enough to achieve the helix structure at higher water content.
As the data in Fig. 1 reveals, self-assembly of PAA94-b-PMA103-b-PS88 into helical cylinder superstructures occurs over a broad range of amine to acid molar ratios, and the pitch distance can be easily tuned by simply changing the quantity and type of multiamine. In our previous studies of micelle formation we observed that different types and amounts of multiamines, combined with PAA-b-PMA-b-PS copolymers and mixtures of THF and water,29–31 could be used to define local interfacial curvature and micelle geometry viaPAA corona block electrostatic complexation with multiamine counterions. Fourier transform infrared spectroscopy (FTIR)25 indicated that the carboxylic acid groups of PAA are predominantly charged in the presence of an equal or slightly greater number of amine groups contained in multiamines (that are, consequently, equally and oppositely charged) in solutions with a water content greater than 40%. Therefore, in the cylindrical helices presented herein, PAA chains should be 100% deprotonated and strongly interacting electrostatically with oppositely charged multiamines at a 10 : 1 amine to acid molar ratio in a 67% by volume water suspension. However, in the current system there is a 9-fold excess of amine functionality. While, on average, one amine on every 2.5 triethylenetetramine molecules is electrostatically interacting with a PAA acid side group, the remaining amine funcationality is free to form hydrogen bonds with the prevalent carbonyl groups of PAA. The strong electrostatic association and weaker hydrogen bonding between the multiamine molecules and PAA side chains give rise to excessive amine packing within the PAA corona as well as on the surface of the micelle, as schematically represented in Fig. 2.
The presence of the high amount of amine, both within the cylinder corona and coating the helix cylinder surface, could lead to two important effects that lead to cylinder helix formation. First, the high amount of electrostatic and hydrogen bonding complexation between the multiamines and PAA corona blocks will lead to a contraction of the corona volume both perpendicular and parallel to the cylinder axis. The corona contraction parallel to the cylinder axis effectively could produce a significant uniaxial compressive force that is alleviated by buckling/coiling of the cylinder into a helix, similar to the coiling of other rod-like structures under uniaxial compression.17–19 Second, the perfection of the cylinder helices formed may be due to long-range electrostatic interactions from excess amines distributed on the surface of the cylinder helix micelles. While the fluid cylinders want to buckle and collapse due to the corona complexation and uniaxial compression, excess surface charge produces intracylinder repulsion that stabilizes a particular pitch. Also, the buckling cylinders may be quite stiff due to the significant amount of multiamine that is complexed within the corona. This corona crowding would also fight against complete cylinder collapse and, perhaps, help to stabilize a particular helix pitch. The importance of this corona crowding effect can be see by comparing helices made with same 10 : 1 amine to acid ratio but with a different multiamine, triethylentetramine in Fig. 1Avs. diethylenetriamine in Fig. 1C. Both systems produce cylindrical helices but with different pitches, 45 ± 3 nm vs. 64 ± 3 nm with triethylenetetraminevs. diethylenetriamine, respectively. This is because an approximately 17% larger volume (due to an approximately 25% higher concentration) of diethylenetriamine is needed to achieve an identical molar ratio of amine to acid as the more highly functionalized triethylenetetramine. This larger volume of diethylenetriamine complexed within the micelle corona resists the cylinder buckling more than the lower corona volume triethylenetetramine system. Thus, the helices formed in Fig. 1C have a longer pitch than those in Fig. 1A.
However, a compelling result indicating the importance of possible long-range electrostatic interactions is shown in Fig. 3. Double-stranded cylindrical helix superstructures, studied by both cryoTEM and conventional TEM in Fig. 3, were found to coexist with single cylinder helices at the same solution conditions using triethylenetetramine at an amine to acid molar ratio of 15 : 1 and water content of 67% by volume in THF. The pitch of the individual helical cylinders that comprise the double-stranded helix surprisingly exhibit double the pitch (∼116 ± 7 nm) of neighboring single helical cylinders (∼58 ± 5 nm). This larger pitch within the double helix produces a characteristic intercylinder distance of approximately 58 ± 5 nm across the width of the double helix, exactly the pitch of the neighboring single cylindrical helix. Therefore, there exists a characteristic distance, either within a single cylindrical helix or between two cylinders comprising a double helix, at which cylinder interactions, such as intra and intercylinder electrostatic repulsions, are optimal. This electrostatic repulsion may be the same reason that the single cylinder helix pitch distance in Fig. 1B was enlarged from that observed in Fig. 1A as triethylenetetramine was increased in concentration to an amine to acid ratio of 15 : 1 in Fig. 1B from 10 : 1 in Fig. 1A. The higher excess of amine functionality at 15 : 1 may lead to a higher surface charge on the cylinder that, in turn, more strongly resists coiling of the cylinder and, therefore, produces a helix with a larger pitch. At higher water content in Fig. 1D, the corona is more solvated by water that would resist compressive forces due to multiamine complexation with PAA chains, and the solvent has a higher dielectric constant that is more able to screen the electrostatic association and interactions. Therefore, non-helical cylindrical micelles were again observed at a water volume ratio of 90% in THF.
 | ||
| Fig. 3 (A) Cryogenic TEM image of double-stranded helices; the pitch distance of a single cylinder helix is the same as the distance between the cylinders of the double-stranded helix. The pitch distance of the double-strand helix, 116 ± 7 nm, is approximately twice that of the single cylinder helix, 58 ± 5 nm; (B) conventional TEM for double-stranded helices. Amine = triethylenetetramine, amine group : acid group molar ratio of 15 : 1, H2O vol% = 67% in THF and after 20 d aging. | ||
We have discovered a new strategy for supramolecular helix formation based on simple solution block copolymer self-assembly methods. The resultant helices can be single-stranded or double-stranded but, importantly, consist of cylindrical micelles that can coil into extremely regular, helical superstructures. By altering the type or amount of multiamine in the system, the pitch of the cylinder helices can be tuned. Even though we are just beginning our exploration of this new nanostructure, it is clear that the key to helix formation is the use of a triblock copolymer with an acidic corona block that can complex with a large excess of organic multiamine. We propose that this complexation induces both uniaxial compression along the long axis of the cylinders as well as long-range electrostatic repulsion along the cylinder surface. The competition of buckling due to uniaxial compression and electrostatic repulsion of the charged cylinder surface causes a stable, regular helical superstructure many microns in length to form. It is important to note that the helical superstructures do not result from specific intermolecular, oriented packing due to molecular chirality.
Acknowledgements
We thank NSF for funding, specifically the Nanoscale Interdisciplinary Research Teams program under grant DMR-0210247. Any opinions, findings, conclusions, or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of NSF. We also thank the W. M. Keck College of Engineering electron microscopy laboratory at the University of Delaware, and the nuclear magnetic resonance facilities of the Department of Chemistry at Washington University in Saint Louis.Notes and references
- H. B. Liu, J. M. Gao, S. R. Lynch, Y. D. Saito, L. Maynard and E. T. Kool, Science, 2003, 302, 868–871 CrossRef CAS.
- J. M. Mason and K. M. Arndt, ChemBioChem, 2004, 5, 170–176 CrossRef CAS.
- V. Ottani, D. Martini, M. Franchi, A. Ruggeri and M. Raspanti, Micron, 2002, 33, 587–596 CrossRef CAS.
- J. W. Kelly, Curr. Opin. Struct. Biol., 1998, 8, 101–106 CrossRef CAS.
- C. W. G. Fishwick, A. J. Beevers, L. M. Carrick, C. D. Whitehouse, A. Aggeli and N. Boden, Nano Lett., 2003, 3, 1475–1479 CrossRef CAS.
- C. Sachse, J. Z. Chen, P. D. Coureux, M. E. Stroupe, M. Fandrich and N. Grigorieff, J. Mol. Biol., 2007, 371, 812–835 CrossRef CAS.
- M. M. Green, N. C. Peterson, T. Sato, A. Teramoto, R. Cook and S. Lifson, Science, 1995, 268, 1860–1866 CrossRef CAS.
- J. J. L. M. Cornelissen, J. J. J. M. Donners, R. Gelder, W. S. Graswinckel, G. A. Metselaar, A. E. Rowan, N. A. J. M. Sommerdijk and R. J. M. Nolte, Science, 2001, 293, 676–680 CAS.
- V. Berl, I. Huc, R. G. Khoury, M. J. Krische and J. M. Lehn, Nature, 2000, 407, 720–723 CrossRef CAS.
- A. Aggeli, I. A. Nyrkova, M. Bell, R. Harding, L. Carrick, T. C. B. McLeish, A. N. Semenov and N. Boden, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 11857–11862 CrossRef CAS.
- L. S. Li, H. Z. Jiang, B. W. Messmore, S. R. Bull and S. I. Stupp, Angew. Chem., Int. Ed., 2007, 46, 5873–5876 CrossRef CAS.
- C. Y. Li, S. Z. D. Cheng, J. J. Ge, F. Bai, J. Z. Zhang, I. K. Mann, F. W. Harris, L. C. Chien, D. H. Yan, T. B. He and B. Lotz, Phys. Rev. Lett., 1999, 83, 4558–4561 CrossRef CAS.
- T. Shimizu, R. Iwaura, M. Masuda, T. Hanada and K. Yase, J. Am. Chem. Soc., 2001, 123, 5947–5955 CrossRef CAS.
- E. Yashima, K. Maeda and Y. Okamoto, Nature, 1999, 399, 449–451 CrossRef CAS.
- J. J. L. M. Cornelissen, M. Fischer, N. A. J. M. Sommerdijk and R. J. M. Nolte, Science, 1998, 280, 1427–1430 CrossRef CAS.
- D. M. Bassani, J. M. Lehn, G. Baum and D. Fenske, Angew. Chem., Int. Ed. Engl., 1997, 36, 1845–1847 CrossRef CAS.
- M. J. Buehler, Y. Kong and H. J. Gao, J. Eng. Mater. Technol., 2004, 126, 245–249 CrossRef CAS.
- J. R. Gladden, N. Z. Handzy, A. Belmonte and E. Villermaux, Phys. Rev. Lett., 2005, 94, 035503 CrossRef CAS.
- C. P. Brangwynne, F. C. MacKintosh, S. Kumar, N. A. Geisse, J. Talbot, L. Mahadevan, K. K. Parker, D. E. Ingber and D. A. Weitz, J. Cell Biol., 2006, 173, 733–741 CrossRef CAS.
- K. C. Lin, R. M. Weis and H. M. McConnell, Nature, 1982, 296, 164–165 CrossRef CAS.
- H. Dave, M. Surve, C. Manohar and J. Bellare, J. Colloid Interface Sci., 2003, 264, 76–81 CrossRef CAS.
- J. R. Huang, Eur. Phys. J. E, 2006, 19, 399–412 CrossRef CAS.
- N. H. Mendelson, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 1740–1744 CAS.
- Y. Snir and R. D. Kamien, Science, 2005, 307, 1067 CrossRef CAS.
- H. G. Cui, Z. Y. Chen, S. Zhong, K. L. Wooley and D. J. Pochan, Science, 2007, 317, 647–650 CrossRef CAS.
- Z. B. Li, E. Kesselman, Y. Talmon, M. A. Hillmyer and T. P. Lodge, Science, 2004, 306, 98–101 CrossRef CAS.
- X. S. Wang, G. Guerin, H. Wang, Y. S. Wang, I. Manners and M. A. Winnik, Science, 2007, 317, 644–647 CrossRef CAS.
- H. G. Cui, Z. Y. Chen, K. L. Wooley and D. J. Pochan, Macromolecules, 2006, 39, 6599–6607 CrossRef CAS.
- Z. B. Li, Z. Y. Chen, H. G. Cui, K. Hales, K. Qi, K. L. Wooley and D. J. Pochan, Langmuir, 2005, 21, 7533–7539 CrossRef CAS.
- Z. B. Li, Z. Y. Chen, H. G. Cui, K. Hales, K. L. Wooley and D. J. Pochan, Langmuir, 2007, 23, 4689–4694 CrossRef CAS.
- D. J. Pochan, Z. Y. Chen, H. G. Cui, K. Hales, K. Qi and K. L. Wooley, Science, 2004, 306, 94–97 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and methods; AFM. See DOI: 10.1039/b715459c |
| ‡ Current address: Rhodia Inc. 350 George Patterson Drive, Bristol, PA 19007, USA. |
| This journal is © The Royal Society of Chemistry 2008 |