Controlling cell adhesion on polyurethanes†
T. Joseph
Dennes
and
Jeffrey
Schwartz
*
Department of Chemistry, Princeton University, Princeton, NJ 08544, U. S. A. E-mail: jschwartz@princeton.edu
First published on 14th November 2007
Abstract
Cell attractive or non-attractive surface properties of polyurethane devices can be controlled by treating them with zirconium tetra(tert-butoxide). This gives reactive interfacial zirconium complex species that can be used subsequently to bond cell attractive peptides such as arg-gly-asp (RGD) or cell non-attractive organics such as polyethylene glycol (PEG) to the device surface. Control of the surface properties of the polyurethane occurs on the nanoscale, and does not compromise the physical properties of the polymer. Interfacial Zr complex formation occurs at N–H sites of the polyurethane; therefore surface loadings of the Zr complex depend on the spatial separation of these N–H groups in the polymer backbone. A complex loading of 110 ± 15 pmol cm–2 is achieved on poly(hexamethylenehexylene)urethane, and 40 ± 10 pmol cm–2 is bound on the medically relevant polyurethane, tecoflex®. About 25% and 10% of these polymer surfaces, respectively, can be covered by RGDvia the zirconium complex interface; because of its greater size, about 100% of both polymer surfaces is covered by PEG. The response of 3T3 fibroblasts to surface-treated and untreated tecoflex® is described.
Atherosclerosis, which is characterized by localized reduction in arterial blood flow, is a leading cause of death in the western world.1,2 Therapeutically, a diseased vessel may be bypassed using a saphenous vein autograft,3 but the required additional procedures to harvest the autograft can increase patient pain and procedure morbidity,4 and a significant fraction of patients in need of vessel grafts do not have veins that are healthy enough for harvesting.3 Synthetic vascular grafts are intriguing replacements for autografts, and polyurethanes are an especially important class of materials in this context.5 They have been examined for use in other blood-contacting devices, including aneurysm patches, cardiac stents, and left-ventricular assist devices.6 Although biostability and mechanical properties of such polyurethanes have been expertly crafted, a persistent problem is their hydrophobicity, which causes significant biofouling. As a result, the surface of a polyurethane device can be thrombogenic, which can lead to device failure, or, in severe cases, death.7 The development of polyurethane surfaces that can resist nonspecific biofouling or can direct the colonization of cells might enhance device performance.8
Approaches to adjust surface properties of polyurethane devices include making composites with inorganic materials,9 or coating the polymer with organics such as cell adhesion peptides6 to foster tissue integration or polyethylene glycol10 to reduce fibrous encapsulation. Unfortunately most organic modification procedures yield only very low surface coverage of the polymer;11–20 indeed, polyurethanes are especially challenging materials for surface modification with organics6 because they have both amorphous and hard segments, and neither segment contains functional groups that are amenable for chemical derivatization using common synthetic organic methods. Furthermore, changes in the physical properties of the polymer that accompany surface modification with organics or composite formation with inorganics9 may yield undesirable side effects.20
We have reported that zirconium tetra(tert-butoxide) (1) can react with the surface of simple nylon devices by reaction with amide N–H bonds to give surface metallic complex derivatives;21 therefore surface loadings of the Zr complex depend on the spatial separation of these N–H groups in the polymer backbone. These surface complexes can then anchor reagents that are commonly used to cross-link biomolecules. We now report that this methodology succeeds for polyurethane devices, which we illustrate through reaction of 1 with the surface carbamate groups of a simple polyurethane, poly(hexamethylenehexylene)urethane, and of medically relevant tecoflex® polyetherurethane; both give Zr surface complexes that can be easily treated to bond organics.
We show by fluorescence spectroscopy that ca. 25% of the surface of poly(hexamethylenehexylene)urethane and about 10% of the tecoflex® surface can be covered by the cell adhesive peptide RGDvia Zr surface complex 2 (Scheme 2). This is, to date, the highest yield reported for the surface functionalization of a polyurethane with peptide moieties; it is comparable to amounts we have previously attained on polyamides.21 Because of its greater size, about 100% of both polymer surfaces can be covered by PEG via2. We also show that significant control of fibroblastcell surface attachment to tecoflex® can be effected, with substantially increased adhesion on RGD-derivatized surfaces and with significantly decreased adhesion on PEG-derivatized analogs.
Polyamides and polyurethanes are similar in that both expose polymer “backbone” N–H groups of surface amide and carbamate groups, respectively; however, they are different in a key way. Zirconium alkoxide 1 reacts readily with polyamides, apparently due to protolytic lability of the alkoxide ligands in the Zr coordination sphere22 and the fact that the high oxyphilicity of Zr acidifies the polyamide N–H groups concomitant with amide carbonyl group coordination to it. But, whereas comparable carbonyl group coordination is possible for polyurethanes, it was not clear if attendant N–H acidification in a polyurethane would be sufficient to enable protolytic cleavage of a Zr-alkoxide bond: the carbamate oxygen moiety could attenuate the necessary increase in electrophilicity of the urethane carbonyl group on coordination to 1 and preclude the important proton transfer step (Scheme 1).
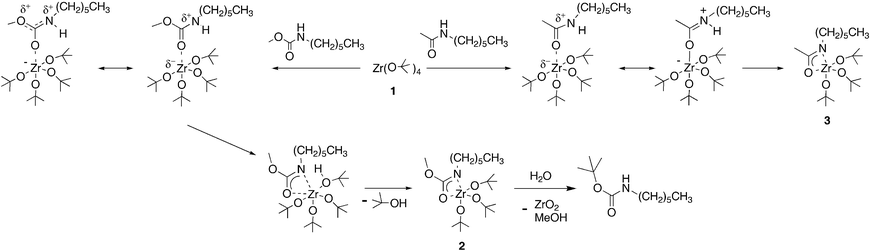 | ||
| Scheme 1 Coordination of the amide vs. the carbamate carbonyl, which results in acidification of N–H; formation of Zr carbamate complex 2, and hydrolysis of 2. | ||
We first examined a small molecule model system. In a typical experiment, methyl N-hexylcarbamate (0.16 g, 1.0 mmol) was treated with 1 (0.40 g, 1.0 mmol) to yield complex 2 [ca. 50% by 1H NMR (CDCl3): δ 0.9 (t, 3H); 1.3 (m, 35 H); 3.1 (t, 2H); 3.6 (s, 3H)] (Scheme 1). For Zr-treated amide,21 an 8 ppm downfield shift for the acyl carbon in 3versus the free amide was measured in the 13C NMR;21 for 2, the acyl carbon is shifted downfield by only 3 ppm vs. the free carbamate [13C NMR (CDCl3): δ 156.1 for methyl N-hexylcarbamate; δ 159.1 for 3]. Thus the presence of the carbamate oxygen apparently does reduce the electrophilicity of the coordinated carbonyl (Scheme 1). Whereas hydrolysis of 3 simply returns the amide and precipitates zirconia, a surprising finding was that when 2 is hydrolyzed, zirconia and tert-butyl N-hexyl carbamate were formed [>95% by 1H NMR (CDCl3): δ 0.8 (t, 3H); 1.2 (m, 6 H); 1.4 (m, 11H); 3.1 (quartet, 2H); 4.5 (s, 1H)]. Hydrolysis of 2 in the presence of 10.0 mmol ethanol, ethanolamine, or hexylamine gave zirconia and only tert-butyl N-hexyl carbamate, suggesting that transesterification is intramolecular.
Surface derivatization of preformed polyurethane (4) and tecoflex® polyurethane (5) proceeded as for our model system. Films of 4 [R = (CH2)6O; R′ = (CH2)6NHCO] and of 5 {R = [(CH2)4O]12; R′ = C6H12CH2C6H12NHCO} were cast from formic acid or THF solution, respectively, onto glass microscope slides, and were treated with vapor of 1 as previously described.21 The IR spectra of polymer surface-bound Zr complexes 6 and 7 showed νC–H = 2976 cm–1, indicative of tert-butoxide groups.
Slides of 6 and 7 were immersed in a 0.1 mM solution of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Mw (polyethylene glycol)carboxylic acid (Laysan) in dry acetonitrile for 16 h, which gave PEGylated surfaces 8 and 9, respectively. Immersion of 6 and 7 in a 0.1 mM solution of 3-maleimidopropionic acid in dry acetonitrile gave surface complexes 10 and 11, which have been shown to be active for bonding RGDC peptides21 (Scheme 2) (for 11, νC
000 Mw (polyethylene glycol)carboxylic acid (Laysan) in dry acetonitrile for 16 h, which gave PEGylated surfaces 8 and 9, respectively. Immersion of 6 and 7 in a 0.1 mM solution of 3-maleimidopropionic acid in dry acetonitrile gave surface complexes 10 and 11, which have been shown to be active for bonding RGDC peptides21 (Scheme 2) (for 11, νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O = 1707 cm–1). RGDC-modified polyurethanes 12a and 13a were prepared by immersing 10 or 11 in a 0.1 mM aqueous solution of RGDC for 16 h. RGD-derivatized surface 13a and PEG-derivatized surface 9 showed a substantial change in surface hydrophilicity compared to 5, which was confirmed by contact angle hysteresis (θadv = 102 ± 3° and θrec = 69 ± 2° for 5; θadv = 68 ± 2° and θrec = 38 ± 2° for 9; θadv = 70 ± 3° and θrec = 40 ± 3° for 13a) (Scheme 2).
O = 1707 cm–1). RGDC-modified polyurethanes 12a and 13a were prepared by immersing 10 or 11 in a 0.1 mM aqueous solution of RGDC for 16 h. RGD-derivatized surface 13a and PEG-derivatized surface 9 showed a substantial change in surface hydrophilicity compared to 5, which was confirmed by contact angle hysteresis (θadv = 102 ± 3° and θrec = 69 ± 2° for 5; θadv = 68 ± 2° and θrec = 38 ± 2° for 9; θadv = 70 ± 3° and θrec = 40 ± 3° for 13a) (Scheme 2).
![Bonding RGDC, PEG, or DANSYL-Cys to polyurethane surfaces. For 6, R = (CH2)6O; R′ = (CH2)6NHCO; for 7, R = [(CH2)4O]12; R′ = C6H12CH2C6H12NHCO.](/image/article/2008/SM/b714947f/b714947f-s2.gif) | ||
| Scheme 2 Bonding RGDC, PEG, or DANSYL-Cys to polyurethane surfaces. For 6, R = (CH2)6O; R′ = (CH2)6NHCO; for 7, R = [(CH2)4O]12; R′ = C6H12CH2C6H12NHCO. | ||
Surface loadings for 12 and 13 were measured by fluorescence spectroscopy using an RGDC analog as previously described:23 here, DANSYL-cysteine (DANSYL = (dimethylaminonaphthyl)sulfonyl) was added to the reactive termini of 10 and 11 (Scheme 2). DANSYL-Cys was chosen as a probe molecule because its reactivity with the termini of 10 and 11 should be very similar to that of RGDC; therefore, the molar surface loading of DANSYL-Cys accurately approximates that of RGDC. Briefly, samples of 12b and 13b were immersed in water (adjusted to pH 7.5 with NaOH) for 6 d at 37 °C. Fluorescence intensities of the supernatants from 12b and 13b were monitored and compared to control samples 4 or 5, which allowed calculation of any release of DANSYL groups from the surface. Excess DANSYL reagent desorbed rapidly from the polyurethane surfaces, in less than one day. In contrast, no release of surface-bound DANSYL material occurred from either 12b or 13b over 6 d (see ESI† ), showing the stability to hydrolysis of carboxylate derivatized, surface-bound Zr-carbamate complexes under physiological conditions. To determine surface coverages for 12b and 13b (which correlate with 12a and 13a), they were immersed in water (at pH 12.5) for 3 h at room temperature; this cleaves the Zr-carbamate complexes from the surface and releases DANSYL-cys into solution. The amount of DANSYL surface-bound to 12b was measured to be 110 ± 15 pmol cm–2, which corresponds to a surface coverage of 25% (see ESI† ). Based on their different polymer stoichiometries, tecoflex® polyurethane (5), with a much smaller surface ratio of carbamate linkages to aliphatic (and ethyleneoxy) groups than 4, would be expected to have only about one-fourth the number of carbamate linkages per unit area at its surface than 5. In fact DANSYL surface-bound to 13b was measured to be 40 ± 10 pmol cm–2, or 10% spatial surface coverage.
Tecoflex® activated by our procedure and terminated with RGDC peptides is highly active for supporting cell adhesion; an in vitro study was conducted as previously described.21 Briefly, fibroblasts (3.3 × 105 NIH 3T3 cells in serum-free DMEM) were added to 35 mm tissue culture wells containing untreated or derivatized polyurethane surfaces. At 3 h cells were fixed, permeabilized, and stained with FITC-phalloidin (for actin filaments), anti-vinculin antibody (Sigma) followed by rhodamine-IgG secondary antibody (for focal adhesions), and DAPI (for DNA). Significant numbers of cells (average from nine 10× fields) attached to the RGDC-modified surface (13a) (Fig. 1A, D) compared to control untreated tecoflex® (5) (Fig. 1B, D). Furthermore, PEG-derivatized tecoflex® (9) showed significant cell resistant properties (Fig. 1C, D). A one way analysis of variance (ANOVA) test showed a statistically significant increase in cell adhesion for 13a (p = 2.6 × 10–9) and a significant decrease in cell adhesion for 9 (p = 8.7 × 10–5) when compared to 5. Cells were well-spread on the RGD-modified surface showing organized actin filaments and focal adhesions when stained with fluorescent phalloidin and anti-vinculin antibodies (Fig. 1A), while the cells on untreated 5 (Fig. 1B) and PEG-treated 9 (Fig. 1C) remained round.
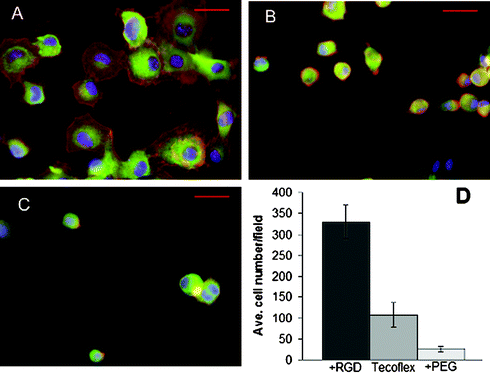 | ||
| Fig. 1 NIH3T3 cell attachment on derivatized tecoflex® polyurethane. Cells on RGD-modified tecoflex® (A), unmodified tecoflex control (B), or PEG-modified tecoflex® (C) surfaces after 3 h, fixed and stained for DNA, vinculin, and actin. Scale bars are 50 µm. (D) Number of cells per 10× microscope field counted for untreated, RGD-derivatized, and PEG-derivatized tecoflex®. For (D), average values for at least nine fields are shown with error bars representing ±1 standard deviation. | ||
In conclusion, we have found that the surface cell adhesive properties of polyurethanes can be controlled through the intermediacy of surface-bound Zr-carbamate complexes, which are synthesized in high yield. Cell adhesive peptide RGD can be bound to poly(hexamethylenehexylene)urethane and tecoflex® at levels of about 110 and 40 pmol cm–2, or ca. 25% and 10% surface coverage, respectively. These are the highest yields yet reported on any polyurethane surface6,10,24,25 and are comparable to loadings we previously attained on nylon.21 Significantly, treated polyurethane surfaces have increased hydrophilicity compared to untreated controls, and can bond significant amounts of the cell adhesive peptide RGD, or cell non-adhesive PEG to enable effective control of cell seeding or to reduce biofouling of biomedically important polymers.
Acknowledgements
The authors thank the National Science Foundation for support of this work. They also thank Dr Nancy Karuri and Prof. Jean E. Schwarzbauer, both of the Department of Molecular Biology, Princeton University, for helpful advice and commentary.Notes and references
- B. L. Seal, T. C. Otero and A. Panitch, Mater. Sci. Eng., R, 2001, 34, 147–230 CrossRef.
- J. Mustard and M. Packham, Thromb. Diath. Haemorrh., 1975, 33, 444–456 Search PubMed.
- T. Chandy, G. Gladwin, R. Wilson and G. Rao, Biomaterials, 2000, 21, 699–712 CrossRef CAS.
- R. Hopkins, Prog. Pediatr. Cardiol., 2006, 21, 137–152 CrossRef.
- U. Hersel, C. Dahmen and H. Kessler, Biomaterials, 2003, 24, 4385–4415 CrossRef CAS.
- H. Salacinski, G. Hamilton and A. Seifalian, J. Biomed. Mater. Res., Part A, 2002, 66, 688–697 Search PubMed.
- D. Vara, H. Salacinski, R. Kannan, L. Bordenave, G. Hamilton and A. Seifalian, Pathol. Biol., 2005, 53, 599–612 Search PubMed.
- S. E. Sakiyama-Elbert and J. A. Hubbell, Annu. Rev. Mater. Res., 2001, 31, 183–201 CrossRef CAS.
- D. K. Chattopadhyay and K. V. S. N. Raju, Prog. Polym. Sci., 2007, 32, 352–418 CrossRef CAS.
- J. P. Santerre, K. Woodhouse, G. Laroche and R. S. Labow, Biomaterials, 2005, 26, 7457–7470 CrossRef CAS.
- R. A. Quirk, W. C. Chan, M. C. Davies, S. B. J. Tendler and K. M. Shakesheff, Biomaterials, 2001, 22, 865–872 CrossRef CAS.
- K. P. Walluscheck, G. Steinhoff, S. Kelm and A. Haverich, Eur. J. Vasc. Endovasc., 1996, 12, 321–330 Search PubMed.
- J. J. Yoona, S. H. Songa, D. S. Leeb and T. G. Park, Biomaterials, 2004, 25, 5613–5620 CrossRef.
- P. Dankers, M. Harmsen, L. Brouwer, M. Van Luyn and E. Meijer, Nat. Mater., 2005, 4, 568–574 CrossRef CAS.
- A. D. Cook, J. S. Hrkach, N. S. Gao, I. M. Johnson, U. B. Pajvani, S. M. Cannizzaro and R. Langer, J. Biomed. Mater. Res., 1997, 35, 513–523 CrossRef CAS.
- T. Yamaoka, Y. Hotta, K. Kobayashi and Y. Kimura, Int. J. Biol. Macromol., 1999, 25, 265–271 CrossRef CAS.
- E. Smith, J. Yang, L. McGann, W. Sebald and H. Uludag, Biomaterials, 2005, 26, 7329–7338 CrossRef CAS.
- H. D. Maynard, S. Y. Okada and R. H. Grubbs, J. Am. Chem. Soc., 2001, 123, 1275–1279 CrossRef CAS.
- Y. Hu, S. Winn, I. Krajbich and J. Hollinger, J. Biomed. Mater. Res., Part A, 2003, 64, 583–590 Search PubMed.
- A. Sanghvi, K. Miller, A. Belcher and C. Schmidt, Nat. Mater., 2005, 4, 496–502 CrossRef CAS.
- T. J. Dennes, G. C. Hunt, J. E. Schwarzbauer and J. Schwartz, J. Am. Chem. Soc., 2007, 129, 93–97 CrossRef CAS.
- J. B. Miller and J. Schwartz, Acta Chem. Scand., 1993, 47, 292–295 CrossRef CAS.
- M. P. Danahy, M. J. Avaltroni, K. S. Midwood, J. E. Schwarzbauer and J. Schwartz, Langmuir, 2004, 20, 5333–5337 CrossRef CAS.
- D. Wang, L. Feng, J. Ji, Y. Sun, X. Zheng and J. Elisseeff, J. Biomed. Mater. Res., Part A, 2003, 65, 498–510 Search PubMed.
- H. B. Lin, C. Garcia-Echeverria, S. Asakura, W. Sun, D. Mosher and S. Cooper, J. Biomed. Mater. Res., 1994, 28, 329–342 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Hydrolysis plots for (a) 12b and (b) 13b, and (c) calculation of sample surface loading. See DOI: 10.1039/b714947f |
| This journal is © The Royal Society of Chemistry 2008 |