Peptide- and protein -mediated assembly of heparinized hydrogels
Kristi L.
Kiick
University of Delaware, Department of Materials Science & Engineering, 201 DuPont Hall, Newark, DE 19716 and the Delaware Biotechnology Institute, 15 Innovation Way, Newark DE 19711, USA. E-mail: kiick@udel.edu
First published on 27th November 2007
Abstract
Polymeric hydrogels have demonstrated significant promise in biomedical applications such as drug delivery and tissue engineering. A continued direction in hydrogel development includes the engineering of the biological responsiveness of these materials, via the inclusion of cell-binding domains and enzyme-sensitive domains. Ligand–receptor interactions offer additional opportunities in the design of responsive hydrogels, and strategies employing protein –polysaccharide interactions as a target may have unique relevance to materials intended to mimic the extracellular matrix (ECM). Accordingly, we have developed approaches for producing hydrogels via noncovalent interactions between heparin and heparin-binding peptides/proteins , and have demonstrated that such matrices are capable of both passive and receptor-mediated growth factor delivery. Further modification of these materials via the integration of these noncovalent strategies with chemical crosslinking methods will expand the range of their potential use and is under exploration. The combination of these approaches offers broad opportunities for the production of responsive matrices for biomedical applications.
 Kristi L. Kiick | Kristi Kiick is an Associate Professor of Materials Science and Engineering at the University of Delaware and joined the faculty in August 2001. She received a BS in Chemistry from the University of Delaware in 1989, and an MS in Chemistry as an NSF Predoctoral Fellow from the University of Georgia in 1991. After working in industry, she rejoined the academic ranks as a doctoral student in 1996. In 2001, she received a PhD in Polymer Science and Engineering from the University of Massachusetts Amherst under the direction of David Tirrell, after completing her doctoral research as an NDSEG Fellow at the California Institute of Technology. Her current research programs are focused on combining biosynthetic techniques, chemical methods, and bioinspired assembly strategies for the production of novel polymers with advanced multifunctional behaviors. These research programs have been funded in part by a Camille and Henry Dreyfus Foundation New Faculty Award, a Beckman Young Investigator Award, an NSF CAREER Award, and a DuPont Young Professor Award. |
Introduction
Polymeric hydrogels have been investigated in a wide range of technologies – including tissue regeneration, drug delivery, actuation, and sensors – because of the facile synthesis of crosslinked, hydrophilic polymer networks of many compositions and the responsiveness of many of these hydrogels to various stimuli.1–11 The high water content, biocompatibility, non-fouling properties, and mechanically robust nature of many of these hydrogels has also continued to motivate their investigation. Synthetic polymers such as poly(ethylene glycol) (PEG), poly(vinyl alcohol), poly(N-isoproprylacrylamide), poly(lactic-co-glycolic acid) (PLGA), and copolymers of these and other polymers have provided many useful systems for application in biomedical areas, but are often hindered by their lack of biological activity. Natural polymers such as gelatin, collagen, alginate, and hyaluronic acid have been widely explored owing to their biological activity, although the range of mechanical properties accessible via the use of the natural polymers can be limited.The ability to integrate synthetic versatility, desirable bulk mechanical properties, controlled degradation, and controlled drug release in hydrogel materials has stimulated continued research. Both covalent and noncovalent approaches to crosslinking have been employed, with noncovalent methods of particular research interest owing to the potential to produce responsive gels with reversible gelation on the basis of changes in shear rate, pH, temperature, salt concentration, and ligand concentration. Protein –protein interactions have been employed in such assembly, with the use of molecular recognition processes such as those involved in coiled-coil formation12–18 and antibody–antigen recognition,19 and many of these gels show useful stimuli-responsive changes in mechanical properties on the basis of these interactions. Similarly, supramolecular hydrogel systems employing β-sheet-forming and α-helical peptides,20–29 block copolypeptides and peptide-polymer conjugates,30–36 and peptide amphiphiles37–43 have also gained prominence, owing to the synthetic ease of making the building blocks, and the range of responsive behaviors afforded in these materials. Protein –saccharide interactions have also been used to produce noncovalently assembled materials that are responsive to stimuli and have been of interest in sensor development,44–46 with assembly via glucose–ConA (concanavalin A) interactions representing a widely employed strategy. The use of cells as crosslinks in materials has also been introduced.47,48 An area of research that remains of interest for all of these types of hydrogels is their functionalization with integrin-binding domains, MMP (matrix metalloproteinase)-degradation domains, and heparin-binding domains, in order to manipulate cell adhesion, cell-mediated matrix degradation, and protein -binding functions and to set a desired biological outcome.8,9,49–52
Of hydrogels with targeted applications in affinity-based protein delivery, polysaccharide-derivatized materials have been widely investigated.5,7,53–55 Heparin-functionalized hydrogels, in particular, have been increasingly used, owing to the importance of interactions between highly anionic, sulfated glycosaminoglycans (GAGs) and proteins in the extracellular matrix (ECM). The highly anionic charge and heterogeneous sulfation patterns of heparin mediates its important role as a binding partner for many proteins ,56,57 such as antithrombin III (to mediate blood clotting) and various growth factors (to protect against degradation and potentiate receptor binding). This GAG has been widely employed in covalent hydrogels, surfaces, and fibrous materials to generate antithrombogenic materials, growth factor delivery vehicles, and materials to modulate stem cell differentiation.58–68 In addition, our group and others have been exploring the noncovalent assembly of heparinized materials as a route to responsive, reversible, and injectable drug delivery systems that may provide controlled protein delivery profiles and that may serve as models for the extracellular matrix.69–74 In initial work of this kind, Panitch and co-workers employed poly(ethylene glycol) star polymers functionalized with heparin-binding peptides (HBPs) for the formation of viscoelastic solutions that showed tunable properties upon interaction with heparin.71 These materials show frequency-responsive rheological behavior and are able to bind to and release heparin-binding molecules over several days with rates that depend on heparin affinity. The combined use of covalent and noncovalent crosslinking strategies72 yielded materials with more complicated and also tunable rheological behavior that may better mimic the mechanical properties of the ECM. The covalent crosslinks exhibit the expected frequency- and temperature-independent behavior, while the presence of the noncovalent crosslinks in these systems increases the rate of gelation and imparts frequency- and temperature-responsive rheological behavior.
Our investigations, described below, have focused on the use of heparinized polymers for protein - and peptide-mediated assembly; the materials have been shown to form hydrogels via the interactions of various heparin-binding peptides and proteins (Fig. 1). The hydrogels summarized below are composed of heparinized, four-arm star PEG–based polymers and star PEGs functionalized with heparin-binding peptides, are shear thinning (injectable), and are competent for the binding and controlled release of multiple growth factors that retain their bioactivity. The use of the heparinized polymer as an assembly partner has also afforded opportunities for the assembly of hydrogels via interactions with growth factors. The hydrogels can selectively erode in the presence of growth factor receptors, and may therefore offer new opportunities in targeted delivery and cell-mediated, selective erosion.
 | ||
| Fig. 1 Schematic of described hydrogel assembly and erosion strategies. Assembly via two different methods can be mediated by noncovalent interactions with four-arm star, poly(ethylene glycol) polymers modified with low molecular weight heparin (PEG–LMWH). In one case, star PEGs modified with heparin-binding peptides (PEG–HBP) can be used in the noncovalent assembly of hydrogels via the interaction with PEG–LMWH. Heparin-binding growth factors (GF) can be loaded into these materials and released as the hydrogel erodes. In the second case, the direct interaction of dimeric GF with PEG–LMWH results in a viscoelastic hydrogel. Upon interaction with GF receptors, the GF crosslinks are removed from the network, causing receptor-responsive erosion of the material. | ||
Heparin binding peptide-crosslinked, heparinized hydrogels
Our original investigations in this area focused on star PEGs functionalized with low molecular weight heparin (LMWH). PEG–LMWH copolymers are desirable targets owing to their hydrophilicity, biocompatibility, low immunogenicity, and ability to bind growth factors and other proteins and peptides. The PEG–LMWH conjugates have been produced by functionalization of the LMWH (Mr = 3000) with maleimide groups, followed by their reaction with a commercially available, thiol -terminated, four-arm star PEG (Scheme 1). LMWHs modified via the reactions in Scheme 1 were illustrated, via surface plasmon resonance studies, to be competent for binding to HBPs. NMR analysis of the purified PEG–LMWH products suggested that approximately 75% of the PEG termini (3 out of 4) were modified with LMWH, indicating their suitability in the formation of noncovalently crosslinked networks (i.e., the number of crosslink-active termini (f) is >2). The use of LMWH ensured production of a soluble macromolecule, rather than a chemically crosslinked network, as the LMWH on average carries only one maleimide group per chain after functionalization.70 In addition, previous investigations indicated that heparin conjugated to PEG retains its ability to simultaneously bind antithrombin III (ATIII) and thrombin, which suggested that the PEG–LMWH conjugates would remain competent for binding to heparin-binding peptides and proteins . Furthermore, the binding of LMWH to growth factors, including bFGF (basic fibroblastgrowth factor), VEGF (vascular endothelial growth factor), and HGF (hepatocytegrowth factor), has been reported,75,76 which is consistent with the fact that the bFGF and VEGF binding sequences in heparin have been indicated to be penta- or hexasaccharide sequences.77–79 | ||
| Scheme 1 Chemical strategies for synthesis of the PEG–LMWH conjugate. (a) Chemical reaction conditions for modification of LMWH with SMCC (succinimidyl 4-(N-maleimidomethyl) cyclohexane-1-carboxylate). A few drops of anhydrous triethylamine were used as the catalyst and the reaction mixture in DMF (dimethylformamide) was stirred at 60 °C for 20 h. (b) Reaction of SMCC–LMWH with four-arm star, thiolated PEG. The reaction mixture was stirred in degassed aqueous buffer (PBS, pH 6.5) at room temperature for 2 h. | ||
Polymers modified with heparin-binding peptides (HBPs) were the macromolecular species initially exploited for the noncovalent assembly of networks upon association with PEG–LMWH. There are a myriad of HBPs available, which offers multiple strategies for producing hydrogels of various mechanical properties. Star PEG–HBPs are readily produced by reaction of cysteine-terminated HBPs with vinyl sulfone -modified star PEG (Scheme 2); this useful bioconjugation strategy72,80,81 yielded PEG–HBPs with degrees of substitution ranging from approximately 70 to nearly 100%. Three different PEG–HBPs have been employed to date; those derived from antithrombin III (ATIII), from the heparin interacting protein (HIP), and from human platelet factor 4 (PF4ZIP), although a variety of other PEG–HBPs should be similarly useful in the assembly of hydrogels.
 | ||
| Scheme 2 Chemical strategies for synthesis of the PEG–HBP conjugate. (a) Chemical modification of four-arm star PEG in order to prepare PEG–VS. The reaction mixture in DCM was stirred at room temperature for 2 d. (b) Reaction of PEG–VS with Cys-HBP to yield PEG–HBP. The reaction mixture was stirred in degassed aqueous buffer (PBS, pH 6.5) at room temperature for 16 h. | ||
Self-supporting hydrogels are easily and immediately formed from these soluble macromolecular species upon mixing of viscous solutions of PEG–LMWH with PEG–HBP, even at reasonably low polymer concentration (2.5 wt%). Vortexing of the solutions prior to and directly upon addition is employed to improve hydrogel homogeneity. In contrast, mixing of PEG–HBPs with LMWH does not result in hydrogel formation, and preformed hydrogels can be readily liquefied upon addition of soluble LWMH or HBP. Representative oscillatory rheological data for one of the hydrogels (PEG–LMWH/PEG–HIP) is shown in Fig. 2; the hydrogels exhibit a bulk storage modulus that exceeds the loss modulus at all frequencies studied,70 indicating the formation of a crosslinked network, in contrast to the viscoelastic solutions produced by the mixing of PEG–HBPs with high molecular weight heparin (HMWH). The origin of these properties arises at least in part from the association of the LMWH termini prior to addition of the HBP,82 as an initial elastic response is observed even in the absence of PEG–HBP. The increase in storage modulus upon addition of various amounts of PEG–HBP correlates with the amount of HBP in the gel,73 further confirming that the crosslinking occurs through the interaction of the LMWH with the HBP. The moduli of the PEG–HIP-based hydrogels ranged from approximately 80 Pa to 200 Pa; higher moduli were possible with variations in HBP identity, HBP : LMWH ratios, and polymer concentration. Plots of the normalized elastic modulus for the gels versus HBP concentration69,73 show a linear increase in the elastic modulus as a function of increasing peptide concentration, confirming the role of the LMWH–HBP interactions as crosslinks, although a distinct plateau is observed at higher peptide concentrations in the case of HIP and PF4ZIP. Given the similarity in the off-rates of the LMWH–HBP interactions (ca. 1 × 10−3 s−1),69,73 which should make the lifetimes of the crosslinks and therefore resulting rheological behavior similar for a given crosslink density, the plateaus suggest variations in the numbers of HBP binding sites available on the LMWH for a given HBP. Limitations in the number of crosslinking sites for a given HBP may in certain cases limit the maximum possible elastic modulus for a given LMWH : HBP pair; evaluation of specific target binding partners may therefore be required for hydrogel design via these methods.
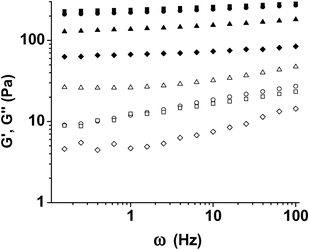 | ||
| Fig. 2 Storage moduli (closed symbols) and loss moduli (open symbols) of the PEG–LMWH/PEG–HIP hydrogels for varying molar ratios of LMWH : HIP (squares, 6 : 4; circles, 8 : 2, triangles, 9 : 1). The response of 7.1 wt% of the PEG–LMWH (diamonds) is shown for comparison. | ||
The mechanical properties of the PEG–LMWH : PEG–HBP hydrogels were consistent with those expected on the basis of the kinetics of the LMWH–HBP binding event. Elastic moduli of 100's Pa were reproducibly obtained from hydrogels produced from PEG–LMWH with any of the three PEG–HBPs investigated; the similarity in the mechanical properties is consistent with the measured similarity in the off-rates of the binding between LMWH and the HBPs (mentioned above), as confirmed via SPR.69,73 Moduli of these values were observed at both room temperature and 37 °C, which is also consistent with SPR data that show that the off-rates do not change significantly over this temperature range.69 In addition, the moduli obtained are consistent with those obtained from crosslinked polymer networks assembled via the interactions of metal complexes that have similar off-rates (ca. 10 −3 s−1),83 as would be anticipated if the measured LMWH–HBP interactions mediate assembly.
The noncovalent nature of the hydrogel assembly offers opportunities for the reversible manipulation of the gels. Indeed, all of the hydrogels show shear-thinning behavior and are injectable after formation. Bulk oscillatory rheology provided a more quantitative assessment of this behavior, and illustrated the very rapid and essentially immediate recovery of the gels after shear thinning.69 The recovery of hydrogel properties for the stronger gels is immediate, and the recovery time for weaker gels is essentially complete within 30 min. The shear thinning and recovery properties of these gels offers opportunities for the encapsulation of cells and the administration and handling of these hydrogels via injection methods. All of the PEG–LMWH hydrogels studied to date show this rheological behavior, indicating the general use of these protocols in hydrogel assembly with various binding partners.
Matrix erosion-mediated growth factor delivery
The ability of the LMWH to bind to multiple partners has also been exploited for the binding of growth factors (or other desired heparin-binding peptides or proteins ) to the noncovalently assembled matrices. For loading of growth factors (GF), a high LMWH : HBP ratio (at least 8 : 2) is generally employed in gel formation, so that a large excess of LMWH is free for GF binding. Indeed, in the noncovalent gels employed to date, LMWH : GF ratios generally exceed 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 : 1, although such high ratios are likely not necessary for the sequestration of the growth factor. The growth factors are incubated with the PEG–LMWH in PBS prior to the addition of the PEG–HBP, in order to ensure more homogeneous incorporation of the growth factor in the matrix; multiple types of growth factors should be readily immobilized in the matrices in this fashion. The release of the growth factor under quiescent conditions is then monitored via immunochemical assays; in these assays, small aliquots of supernatant (PBS) are removed and replenished on a daily basis, and the growth factor concentration in the supernatant is quantified immunochemically.70 As shown in Fig. 3, PEG–LMWH/PEG–HBP matrices loaded with basic fibroblastgrowth factor (bFGF) are able to passively release the GF over a period of two weeks or longer, with an initial release of approximately 2–30% followed by very slow delivery with a release of approximately 5–40% over a two-week period. The initial release and release rates are reduced significantly via the production of hydrogels of higher storage modulus (e.g., the lowest release rates in Fig. 3); the correlation of total GF release at approximately one week, relative to the initial elastic modulus of the given gel (ranging from ∼200–1000 Pa), is shown in Fig. 4.
000000 : 1, although such high ratios are likely not necessary for the sequestration of the growth factor. The growth factors are incubated with the PEG–LMWH in PBS prior to the addition of the PEG–HBP, in order to ensure more homogeneous incorporation of the growth factor in the matrix; multiple types of growth factors should be readily immobilized in the matrices in this fashion. The release of the growth factor under quiescent conditions is then monitored via immunochemical assays; in these assays, small aliquots of supernatant (PBS) are removed and replenished on a daily basis, and the growth factor concentration in the supernatant is quantified immunochemically.70 As shown in Fig. 3, PEG–LMWH/PEG–HBP matrices loaded with basic fibroblastgrowth factor (bFGF) are able to passively release the GF over a period of two weeks or longer, with an initial release of approximately 2–30% followed by very slow delivery with a release of approximately 5–40% over a two-week period. The initial release and release rates are reduced significantly via the production of hydrogels of higher storage modulus (e.g., the lowest release rates in Fig. 3); the correlation of total GF release at approximately one week, relative to the initial elastic modulus of the given gel (ranging from ∼200–1000 Pa), is shown in Fig. 4.
 | ||
| Fig. 3 bFGF delivery profiles for PEG–LMWH/PEG–PF4ZIP and PEG–LMWH/PEG–HIP hydrogels at LMWH to PF4ZIP or HIP ratios (■, LMWH : PF4ZIP = 8 : 1, G′ = 70 Pa; ●, LMWH : HIP = 8 : 2, G′ = 207 Pa; ▲, LMWH : PF4ZIP = 9 : 0.5; ▼, LMWH : PF4ZIP = 3 : 2; ◆, LMWH : PF4ZIP = 3 : 1; ◀, LMWH : PF4ZIP = 8 : 1, G′ = 1114 Pa). | ||
 | ||
| Fig. 4 Comparison of the percentage of bFGF release after 8 days, the percentage of hydrogel erosion after 8 days and the storage modulus, G′ (ω = 0.1 rad s−1) (normalized by the storage modulus of the PEG–LMWH) for various hydrogels. The errors are derived from the average of duplicate measurements. | ||
The extremely high ratios of LMWH : GF employed in these hydrogels, coupled with previous investigations showing essentially complete minimization of GF release at much lower heparin : GF ratios (≤13000 : 1),54,58,60,70,84 suggested that the GF delivery from these materials must result from the slow dissolution of the matrix as the molecules diffuse from the matrix. Indeed, a plot of the percentage of growth factor released versus the percentage of PEG–LMWH released (assessed via fluorimetry measurements of PEG–LMWH labeled with Alexa Fluor 350 or via gravimetric analysis of released polymer) shows mainly a linear relationship (Fig. 5), indicating that the release of the bFGF is a matrix-erosion-mediated process (Fig. 6) and is not a result of overloading or heterogeneous loading of the bFGF. The faster initial release observed in these hydrogels may indicate some heterogeneity in their structure that results in two regions of different erosion profiles. Although the GF release is not triggered by any specific cellular event or solution condition, the release can be altered by varying the mechanical properties of the network (Fig. 3 and Fig. 4). Hydrogels that release GF over these timescales would be potentially useful in the treatment of ischemic conditions or in tissue engineering applications; indeed, other heparin-containing delivery systems with similar GF release profiles have demonstrated utility for stimulation of endothelial cell growth in vitro and neovascularization in vivo.61,85,86 These binding and release results, coupled with the successful noncovalent assembly of hydrogels upon interaction of PEG–LMWH with multiple PEG–HBPs, indicates that PEG–LMWH is capable of binding multiple types of heparin-binding molecules, and suggests that the bioconjugate may be generally useful for hydrogel assembly mediated by other heparin-binding peptides and proteins . The use of these approaches in the noncovalent assembly of hydrogels offers multiple opportunities for the production of fully erodible matrices with engineered erosion and delivery profiles.
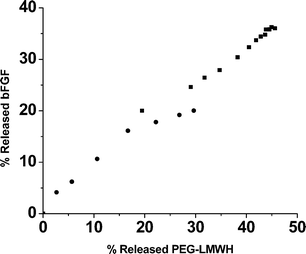 | ||
| Fig. 5 The percentage of released bFGF vs. the percentage of released PEG–LMWH for the PEG–LMWH/PEG–PF4ZIP hydrogel (■, slope of 0.77) and the PEG–LMWH/PEG–HIP hydrogel (●, slope of 0.81). | ||
 | ||
| Fig. 6 Schematic of co-release of bFGF with PEG–LMWH from hydrogels formed via the crosslinking of polysaccharide-derivatized star copolymers by HBP-derivatized star copolymers. | ||
Growth factor-crosslinked hydrogels
The utility of LMWH–peptide interactions for the noncovalent assembly of hydrogels suggested additional opportunities for the generation of cell surface receptor-responsive hydrogels via the crosslinking of PEG–LMWH with therapeutic proteins . Because LMWH modified with star PEG is competent for growth factor binding, and since growth factors such as bFGF, VEGF and HGF are thought to bind to short sequences in heparin, we reasoned that the dimeric heparin-binding growth factors (f = 2) may therefore serve as functional crosslinks upon interaction with PEG–LMWH (f > 2) as a new strategy for hydrogel formation. Furthermore, these crosslinks may be sensitive to ligand-exchange interactions that would promote their selective release from the hydrogels, and therefore the selective erosion of the network, in the presence of GF receptors. Given that overexpression of growth factor receptors plays a key role in both normal healing and pathological conditions,87–89 hydrogels assembled via such strategies may provide unique opportunities for stimuli-responsive delivery and erosion via biologically relevant, ligand-exchange mechanisms.Our initial studies have focused on assembly of these hydrogels with the dimeric heparin-binding growth factor VEGF (vascular endothelial growth factor). A schematic of the assembly and erosion strategy is illustrated in the lower pathway in Fig. 1. The VEGF was expressed from E. coli and purified via heparin-affinity chromatography as previously described.90 Hydrogels were formed via the mixing of homogeneous, low-viscosity solutions of each component in phosphate buffered saline (PBS) at a final polymer concentration of 4 wt%. As observed for the hydrogels above, addition of a solution of VEGF to a solution of PEG–LMWH immediately resulted in the formation of a self-supporting, viscoelastic hydrogel. These hydrogel networks were characterized via optical tweezer microrheology, as shown in Fig. 7.74 The apparently low-viscosity PEG–LMWH solutions exhibit storage moduli, G(ω) ≈ 0.7 Pa, in excess of the loss moduli G(ω) at low frequencies, indicating a weak viscoelastic material, consistent with our previous observations of the PEG–LMWH by oscillatory rheology (above). An increase in elastic modulus was observed upon the addition of VEGF, with no statistically significant increase observed upon the addition of a control protein , BSA (bovine serum albumin) (Fig. 7(a) and (b)), clearly demonstrating the effective crosslinking by VEGF in the VEGF-containing PEG–LMWH samples. That the crosslinking is mediated by VEGF–LMWH interactions was also confirmed by the addition of free LMWH to the PEG–LMWH/VEGF gels, which immediately liquefied the samples. Similarly, VEGF-containing samples with polymer concentrations of 8 wt% resulted in elastic gels in which probe particles could not be moved by the optical trap, indicating a G(ω) >10 Pa, whereas the modulus in the absence of VEGF was G(ω) ≈ 1 Pa.
 | ||
| Fig. 7 (a, b) Optical tweezer microrheological characterization of the viscoelastic properties of VEGF-crosslinked hydrogels. (c) Release of vascular endothelial growth factor from VEGF-crosslinked hydrogels as a function of time in the absence and presence of the VEGFR-2 receptor. The average of duplicate measurements of separate samples is shown. Reprinted with permission from ref. 74. Copyright 2007, American Chemical Society. | ||
Receptor-mediated erosion of hydrogels for responsive growth factor delivery
An important outcome of the production of hydrogel networks via these strategies is the potential for receptor-mediated gel erosion; VEGF crosslinks may be selectively removed in the presence of VEGF receptors that control proliferation and migration of vascular endothelial cells.91 Many contemporary drug delivery strategies have demonstrated the feasibility of employing such receptor-mediated targeting, including targeting based on growth factorreceptor binding. For example, receptor-mediated targeting of VEGF-modified drug vehicles to VEGFR-2-expressing cells has been previously demonstrated,92–94 and a VEGFR-1 binding peptide has been shown to target a chlorin-type photosensitizer to human endothelial cells.95 VEGFR-2 plays a primary role in controlling cell proliferation and migration,91 so in our initial investigations, we probed the ability of the hydrogels to release VEGF in a VEGFR-2-responsive manner.74 In these studies, a PEG–LMWH/VEGF hydrogel was incubated in buffer, and the cumulative amount of 125I-labeled VEGF released from the hydrogels at specific timepoints was quantified via gamma counting.74 Polystyrene particles (1 µm, PS) were used as receptor carriers and were incubated with the hydrogels to study the potential for targeted erosion. In one case, the particles were passivated via covalent attachment of anti-IgG (negative control), and in the second case, the anti-IgG nanoparticles were modified with a VEGFR–2-IgG conjugate. Growth factor release was monitored in a similar format as described above.As observed in the data in Fig. 7(c), the PEG–LMWH/VEGF hydrogels incubated in PBS demonstrate a total cumulative release of approximately 30% over the 10-day time period, while those incubated in the presence of PS/anti-IgG show a cumulative passive release of 40%. The slight increase in overall release may be a result of incomplete passivation of the PS particles with the anti-IgG, which may result in residual sulfate groups on the PS particle surface. In contrast, however, hydrogel samples incubated in the presence of PS/VEGFR-2 demonstrated increased rates of VEGF release and a total release of nearly 80%. Only hydrogels incubated with PS/VEGFR-2 eroded visibly after day 4 (Fig. 7(c), asterisk). In all other cases, the hydrogels remained intact over the course of the experiment. Although the precise extent of erosion of the networks could not be quantified owing to the very small amounts of sample employed, these results clearly illustrate the potential for receptor-mediated VEGF delivery and erosion of these hydrogels, and suggest opportunities for targeted erosion in response to other VEGF-binding receptors (e.g. VEGFR-1). It is likely that such erosion will require cell contact with the matrix (as in the release experiments described here), as passive diffusion at the interface of the gel and solution may otherwise dominate the response. This diffusion would cause the release of a limited amount of VEGF, as indicated by the control release experiments in Fig. 7(c), with the extent limited by the LMWH : VEGF ratio and the passive erosion of the network.
The activity of the VEGF released from the erodible hydrogels was assessed in assays of the proliferation of porcine aortic endothelial cells (PAE KDR) in the presence of gels over time via cytometry methods. In these assays, the gels were incubated in a transwell insert that was placed in wells in which the PAE KDR cells were cultured. The gels remain intact in the presence of the serum-containing media, suggesting their use under physiologically relevant conditions. The results of the proliferation assays are shown in Fig. 8. A plot of the normalized number of cells at each timepoint, is presented in Fig. 8(a) for the different hydrogels and controls. As shown in Fig. 8(a), the PEG–LMWH alone does not cause any increase in cell proliferation over the control (no polymer, no VEGF), while the PEG–LMWH/VEGF hydrogels show a statistically significant increase in proliferation (noted by an asterisk, p <0.05) over the PEG–LMWH samples by as early as day 1. Different amounts of VEGF were included in the two gels in order to probe a range of proliferative responses, with dose-dependent proliferation observed; the gel with the greater amount of VEGF shows decreased proliferation, likely owing to the minimization of the VEGFR-2 dimerization that is necessary for signaling. Confocal images illustrating the overlaid live/dead fluorescent staining of the cells in the presence of PEG–LMWH and PEG–LMWH/VEGF are shown in Fig. 8(b) and (c). The very low number of dead cells (red) observed via confocal microscopy confirms the lack of cytotoxicity of these materials. The assessment of cell proliferation and gel erosion with cells encapsulated directly in the gels was not possible given the nonadhesive character of these PEG–based materials. While this lack of adhesivity would be advantageous in applications in which fouling is undesired (e.g. targeted delivery from particles), the cell adhesivity of these materials could be easily modulated by the inclusion of appropriate cell-binding peptides for expansion of their use in other targeted applications. Erosion and proliferation assays with cell-adhesive, GF-crosslinked hydrogels are underway.
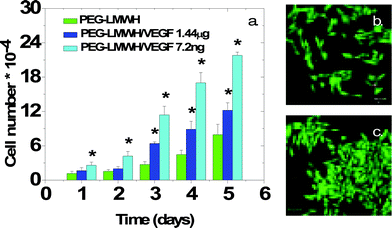 | ||
| Fig. 8 (a) Proliferation of PAE KDR cells in the presence of PEG–LMWH or hydrogels crosslinked with vascular endothelial growth factor. The asterisk denotes statistical differences between cell numbers (p <0.05). (b) Confocal microscopy image of cells, incubated with PEG–LMWH, in a live/dead assay . (c) Confocal microscopy image of cells, incubated with PEG–LMWH/VEGF, in a live/dead assay . Reprinted with permission from ref. 74. Copyright 2007, American Chemical Society. | ||
These results clearly demonstrate that therapeutically relevant growth factors can serve as elastic crosslinks in noncovalently assembled hydrogel networks, and that these networks can be selectively eroded in the presence of the growth factor receptors upon the release of the bioactive crosslinks. While the therapeutic potential for these VEGF-crosslinked hydrogels has yet to be demonstrated directly, the therapeutic relevance of other dimeric heparin-binding growth factors (e.g. PDGF, HGF), as well as opportunities for designing peptides with specific affinities and therapeutic action, suggests broader opportunities for these strategies in the production of responsive matrices for biomedical applications.
Conclusions and outlook
Our studies to date have demonstrated that a variety of PEG–HBP or heparin-binding proteins can be utilized in the noncovalent assembly of heparinized hydrogels. The viscoelastic properties of these materials can be varied by modulating the LMWH : HBP ratios and polymer concentration, and the use of other glycosaminoglycans or heparin-mimics such as sulfated peptides in such materials should also be useful in the immobilization of other therapeutic proteins and would likely result in an expanded range of mechanical and biological properties. The LMWH-functionalized hydrogels exhibit elastic moduli that would be useful for a range of soft-tissue engineering applications, and it is likely that the moduli could be readily manipulated via the use of other branched and multi-arm polymer architectures. They also exhibit passive or receptor-mediated erosion that may have benefits in producing materials that could be readily cleared from the body and would not produce chemical degradation products. In addition, multiple growth factors can be bound and released from these materials at rates that have been previously demonstrated to be useful in neovascularization applications.Our desire to employ these materials in a broader range of formats (e.g., injectable gels, particles, monolithic sheets, fibers) has prompted the engineering of their properties with the addition of covalent crosslinks. As previously demonstrated in related heparinized gels,72 the combination of covalent and noncovalent crosslinks can be employed to increase modulus but retain frequency- and temperature-responsive behavior. Indeed, in our recent investigations, crosslinking of maleimide-functionalized heparin by thiol -terminated, linear PEGs of various molecular weights and compositions, affords gels with moduli ranging from 100s Pa to greater than 10 kPa, of appropriate range for a variety of soft tissue engineering applications.84 These crosslinked, heparinized materials can bind GF and release them in a controlled fashion. These results suggest promising opportunities to combine these covalent strategies with the noncovalent crosslinking by GF to produce hydrogels that are responsive to a variety of biological stimuli, including proteolytic remodeling and ligand–receptor interactions. In addition to their applications in growth factor delivery, multiple other applications for these materials are also possible, including pro-coagulative materials that bind heparin, anticoagulant therapeutics, and receptor-responsive matrices for the delivery of other peptide- or small-molecule therapeutics.
Acknowledgements
The author's work in this area has been supported by grants from the National Institutes of Health (1 R01 EB00317201), the National Science Foundation (DBE-0221651), and the Arnold and Mabel Beckman Foundation. Le Zhang and Ann Kim are thanked for assistance with manuscript preparation.References
- R. V. Ulijn, N. Bibi, V. Jayawarna, P. D. Thornton, S. J. Todd, R. J. Mart, A. M. Smith and J. E. Gough, Mater. Today, 2007, 10, 40–48 CrossRef CAS.
- J. L. Drury and D. J. Mooney, Biomaterials, 2003, 24, 4337–4351 CrossRef CAS.
- F. Brandl, F. Sommer and A. Goepferich, Biomaterials, 2007, 28, 134–146 CrossRef CAS.
- T. Matsumoto and D. J. Mooney, Tissue Eng. I: Scaffold Syst. Tissue Eng., 2006, 102, 113–137 Search PubMed.
- A. D. Augst, H. J. Kong and D. J. Mooney, Macromol. Biosci., 2006, 6, 623–633 CrossRef CAS.
- J. K. Leach, Regen. Med., 2006, 1, 447–455 Search PubMed.
- G. D. Prestwich, X. Z. Shu, Y. C. Liu, S. S. Cai, J. F. Walsh, C. W. Hughes, S. Ahmad, K. R. Kirker, B. L. Yu, R. R. Orlandi, A. H. Park, S. L. Thibeault, S. Duflo and M. E. Smith, Tissue Eng., 2006, 585, 125–133 CAS.
- M. P. Lutolf, G. P. Raeber, A. H. Zisch, N. Tirelli and J. A. Hubbell, Adv. Mater., 2003, 15, 888 CrossRef CAS.
- S. C. Rizzi, M. Ehrbar, S. Halstenberg, G. P. Raeber, H. G. Schmoekel, H. Hagenmuller, R. Muller, F. E. Weber and J. A. Hubbell, Biomacromolecules, 2006, 7, 3019–3029 CrossRef CAS.
- L. S. Ferreira, S. Gerecht, J. Fuller, H. F. Shieh, G. Vunjak-Novakovic and R. Langer, Biomaterials, 2007, 28, 2706–2717 CrossRef CAS.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- S. B. Kennedy, K. Littrell, P. Thiyagarajan, D. A. Tirrell and T. P. Russell, Macromolecules, 2005, 38, 7470–7475 CrossRef CAS.
- W. A. Petka, J. L. Hardin, K. P. McGrath, D. Wirtz and D. A. Tirrell, Science, 1998, 281, 389–392 CrossRef CAS.
- W. Shen, R. G. H. Lammertink, J. K. Sakata, J. A. Kornfield and D. A. Tirrell, Macromolecules, 2005, 38, 3909–3916 CrossRef CAS.
- W. Shen, K. Zhang, J. A. Kornfield and D. A Tirrell, Nat. Mater., 2006, 5, 153–158 CrossRef CAS.
- J. Kopecek, Eur. J. Pharm. Sci., 2003, 20, 1–16 CrossRef CAS.
- A. Tang, C. Wang, R. J. Stewart and J. Kopecek, J. Controlled Release, 2001, 72, 57–70 CrossRef CAS.
- C. Wang, R. J. Stewart and J. Kopecek, Nature, 1999, 397, 417–420 CrossRef CAS.
- T. Miyata, N. Asami and T. Uragami, Nature, 1999, 399, 766–769 CrossRef CAS.
- X. J. Zhao and S. G. Zhang, Macromol. Biosci., 2007, 7, 13–22 CrossRef CAS.
- S. G. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS.
- L. A. Haines, K. Rajagopal, B. Ozbas, D. A. Salick, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2005, 127, 17025–17029 CrossRef CAS.
- J. P. Schneider, D. J. Pochan, B. Ozbas, K. Rajagopal, L. Pakstis and J. Kretsinger, J. Am. Chem. Soc., 2002, 124, 15030–15037 CrossRef CAS.
- D. N. Woolfson and M. G. Ryadnov, Curr. Opin. Chem. Biol., 2006, 10, 559–567 CrossRef CAS.
- A. M. Smith, S. F. A. Acquah, N. Bone, H. W. Kroto, M. G. Ryadnov, M. S. P. Stevens, D. R. M. Walton and D. N. Woolfson, Angew. Chem., Int. Ed., 2005, 44, 325–328 CrossRef CAS.
- M. G. Ryadnov, B. Ceyhan, C. M. Niemeyer and D. N. Woolfson, J. Am. Chem. Soc., 2003, 125, 9388–9394 CrossRef CAS.
- A. Mahler, M. Reches, M. Rechter, S. Cohen and E. Gazit, Adv. Mater., 2006, 18, 1365–1370 CrossRef CAS.
- J. H. Collier and P. B. Messersmith, Bioconjugate Chem., 2003, 14, 748–755 CrossRef CAS.
- J. H. Collier, B. H. Hu, J. W. Ruberti, J. Zhang, P. Shum, D. H. Thompson and P. B. Messersmith, J. Am. Chem. Soc., 2001, 123, 9463–9464 CrossRef CAS.
- T. J. Deming, Soft Matter, 2005, 1, 28–35 RSC.
- V. Breedveld, A. P. Nowak, J. Sato, T. J. Deming and D. J. Pine, Macromolecules, 2004, 37, 3943–3953 CrossRef CAS.
- H. A. Klok and S. Lecommandoux, Adv. Polym. Sci., 2006, 202, 75–111 CAS.
- G. W. M. Vandermeulen and H. A. Klok, Macromol. Biosci., 2004, 4, 383–398 CrossRef CAS.
- H. A. Klok, J. F. Langenwalter and S. Lecommandoux, Macromolecules, 2000, 33, 7819–7826 CrossRef CAS.
- S. E. Paramonov, V. Gauba and J. D. Hartgerink, Macromolecules, 2005, 38, 7555–7561 CrossRef CAS.
- J. H. Collier and P. B. Messersmith, Adv. Mater., 2004, 16, 907–910 CrossRef CAS.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5133–5138 CrossRef CAS.
- M. O. Guler, S. Soukasene, J. F. Hulvat and S. I. Stupp, Nano Lett., 2005, 5, 249–252 CrossRef CAS.
- E. Kokkoli, A. Mardilovich, A. Wedekind, E. L. Rexeisen, A. Garg and J. A. Craig, Soft Matter, 2006, 2, 1015–1024 RSC.
- K. Rajangam, H. A. Behanna, M. J. Hui, X. Q. Han, J. F. Hulvat, J. W. Lomasney and S. I. Stupp, Nano Lett., 2006, 6, 2086–2090 CrossRef CAS.
- S. J. Yang and S. G. Zhang, Supramol. Chem., 2006, 18, 389–396 CrossRef CAS.
- S. E. Paramonov, H. W. Jun and J. D. Hartgerink, J. Am. Chem. Soc., 2006, 128, 7291–7298 CrossRef CAS.
- G. A. Silva, C. Czeisler, K. L. Niece, E. Beniash, D. A. Harrington, J. A. Kessler and S. I. Stupp, Science, 2004, 303, 1352–1355 CrossRef CAS.
- K. Nakamae, T. Miyata, A. Jikihara and A. S. Hoffman, J. Biomater. Sci., Polym. Ed., 1994, 6, 79–90 CAS.
- T. Miyata, T. Uragami and K. Nakamae, Adv. Drug Delivery Rev., 2002, 54, 79–98 CrossRef CAS.
- T. Miyata, M. Jige, T. Nakaminami and T. Uragami, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1190–1193 CrossRef CAS.
- K. Y. Lee, H. J. Kong, R. G. Larson and D. J. Mooney, Adv. Mater., 2003, 15, 1828–1832 CrossRef CAS.
- J. L. Drury, T. Boontheeku and D. J. Mooney, J. Biomech. Eng., 2005, 127, 220–228 CrossRef.
- E. H. Chung, M. Gilbert, A. S. Virdi, K. Sena, D. R. Sumner and K. E. Healy, J. Biomed. Mater. Res. A, 2006, 79A, 815–826 Search PubMed.
- J. R. Tauro and R. A. Gemeinhart, Bioconjugate Chem., 2005, 16, 1133–1139 CrossRef CAS.
- G. P. Raeber, M. P. Lutolf and J. A. Hubbell, Biophys. J., 2005, 89, 1374–1388 CrossRef CAS.
- M. P. Lutolf, J. L. Lauer-Fields, H. G. Schmoekel, A. T. Metters, F. E. Weber, G. B. Fields and J. A. Hubbell, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5413–5418 CrossRef CAS.
- D. A. Wang, S. Varghese, B. Sharma, I. Strehin, S. Fermanian, J. Gorham, D. H. Fairbrother, B. Cascio and J. H. Elisseeff, Nat. Mater., 2007, 6, 385–392 CrossRef CAS.
- D. B. Pike, S. S. Cai, K. R. Pomraning, M. A. Firpo, R. J. Fisher, X. Z. Shu, G. D. Prestwich and R. A. Peattie, Biomaterials, 2006, 27, 5242–5251 CrossRef CAS.
- C. Huin-Amargier, P. Marchal, E. Payan, P. Netter and E. Dellacherie, J. Biomed. Mater. Res. A, 2006, 76A, 416–424 Search PubMed.
- I. Capila and R. J. Linhardt, Angew. Chem., Int. Ed., 2002, 41, 390–412 CrossRef CAS.
- J. Chen, S. Jo and K. Park, Carbohydr. Polym., 1995, 28, 69–76 CrossRef CAS.
- S. E. Sakiyama-Elbert and J. A. Hubbell, J. Controlled Release, 2000, 65, 389–402 CrossRef CAS.
- S. Cai, Y. Liu, X. Zheng Shu and G. D. Prestwich, Biomaterials, 2005, 26, 6054–6067 CrossRef CAS.
- G. Y. Tae, M. Scatena, P. Stayton and A. S. Hoffman, J. Biomater. Sci., Polym. Ed., 2006, 17, 187–197 CrossRef CAS.
- M. J. B. Wissink, R. Beernink, A. A. Poot, G. H. M. Engbers, T. Beugeling, W. G. van Aken and J. Feijen, J. Controlled Release, 2000, 64, 103–114 CrossRef CAS.
- L.-S. Liu, C.-K. Ng, A. Y. Thompson, J. W. Poser and R. C. Spiro, J. Biomed. Mater. Res., 2002, 62, 128–135 CrossRef CAS.
- N. Chinen, M. Tanihara, M. Nakagawa, K. Shinozaki, E. Yamamoto, M. Mizushima and Y. Suzuki, J. Biomed. Mater. Res. A, 2003, 67A, 61–68 Search PubMed.
- D. S. W. Benoit, A. R. Durney and K. S. Anseth, Biomaterials, 2007, 28, 66–77 CrossRef CAS.
- D. S. W. Benoit and K. S. Anseth, Acta Biomater., 2005, 1, 461–470 Search PubMed.
- A. B. Pratt, F. E. Weber, H. G. Schmoekel, R. Muller and J. A. Hubbell, Biotechnol. Bioeng., 2004, 86, 27–36 CrossRef CAS.
- S. Halstenberg, A. Panitch, S. Rizzi, H. Hall and J. A. Hubbell, Biomacromolecules, 2002, 3, 710–723 CrossRef CAS.
- B. Olander, A. Wirsen and A. C. Albertsson, Biomacromolecules, 2003, 4, 145–148 CrossRef CAS.
- L. Zhang, E. M. Furst and K. L. Kiick, J. Controlled Release, 2006, 114, 130–142 CrossRef CAS.
- N. Yamaguchi and K. L. Kiick, Biomacromolecules, 2005, 6, 1921–1930 CrossRef CAS.
- B. L. Seal and A. Panitch, Biomacromolecules, 2003, 4, 1572–1582 CrossRef CAS.
- B. L. Seal and A. Panitch, Macromolecules, 2006, 39, 2268–2274 CrossRef CAS.
- N. Yamaguchi, B.-S. Chae, L. Zhang, K. L. Kiick and E. M. Furst, Biomacromolecules, 2005, 6, 1931–1940 CrossRef CAS.
- N. Yamaguchi, L. Zhang, B.-S. Chae, C. S. Palla, E. M. Furst and K. L. Kiick, J. Am. Chem. Soc., 2007, 129, 3040–3041 CrossRef CAS.
- M. Ishihara, K. Ono, K. Ishikawa, H. Hattori, Y. Saito, H. Yura, T. Akaike, Y. Ozeki, S. Tanaka, H. Mochizuki and A. Kurita, J. Biochem., 2000, 127, 797–803 CAS.
- M. Ishihara, M. Sato, H. Hattori, Y. Saito, H. Yura, K. Ono, K. Masuoka, M. Kikuchi, K. Fujikawa and A. Kurita, J. Biomed. Mater. Res., 2001, 56, 536–544 CrossRef CAS.
- J. E. Turnbull, D. G. Fernig, Y. Q. Ke, M. C. Wilkinson and J. T.M Gallagher, J. Biol. Chem., 1992, 267, 10337–10341 CAS.
- M. Maccarana, B. Casu and U. Lindahl, J. Biol. Chem., 1993, 268, 23898–23905 CAS.
- W. J. Fairbrother, M. A. Champe, H. W. Christinger, B. A. Keyt and M. A. Starovasnik, Struct. Fold. Des., 1998, 6, 637–648 Search PubMed.
- M. P. Lutolf, N. Tirelli, S. Cerritelli, L. Cavalli and J. A. Hubbell, Bioconjugate Chem., 2001, 12, 1051–1056 CrossRef CAS.
- D. L. Elbert, A. B. Pratt, M. P. Lutolf, S. Halstenberg and J. A. Hubbell, J. Controlled Release, 2001, 76, 11–25 CrossRef CAS.
- F. Spinelli, K. L. Kiick and E. M. Furst, Biomaterials Search PubMed , in press.
- W. C. Yount, D. M. Loveless and S. L. Craig, J. Am. Chem. Soc., 2005, 127, 14488–14496 CrossRef CAS.
- T. Nie, A. Baldwin, N. Yamaguchi and K. L. Kiick, J. Controlled Release, 2007, 122, 287–296 CrossRef CAS.
- M. Ishihara, K. Obara, T. Ishizuka, M. Fujita, M. Sato, K. Masuoka, Y. Saito, H. Yura, T. Matsui, H. Hattori, M. Kikuchi and A. Kurita, J. Biomed. Mater. Res. A, 2003, 64A, 551–559 Search PubMed.
- N. Chinen, M. Tanihara, M. Nakagawa, K. Shinozaki, E. Yamamoto, Y. Mizushima and Y. Suzuki, J. Biomed. Mater. Res. A, 2003, 67A, 61–68 Search PubMed.
- J. A. Rodriguez, B. Nespereira, M. Perez-Ilzarbe, E. Eguinoa and J. A. Paramo, Cardiovasc. Res., 2005, 65, 665–673 CrossRef CAS.
- W. Y. Shen, C. M. Lai, C. E. Graham, N. Binz, Y. K. Y. Lai, J. Eade, D. Guidolin, D. Ribatti, S. A. Dunlop and P. E. Rakoczy, Diabetologia, 2006, 49, 1690–1701 CrossRef CAS.
- L. L. Zhang, J. A. F. Hannay, J. H. Liu, P. Das, M. C. Zhan, T. Nguyen, D. J. Hicklin, D. H. Yu, R. E. Pollock and D. Lev, Cancer Res., 2006, 66, 8770–8778 CrossRef CAS.
- A. H. Zisch, M. P. Lutolf, M. Ehrbar, G. P. Raeber, S. C. Rizzi, N. Davies, H. Schmokel, D. Bezuidenhout, V. Djonov, P. Zilla and J. A. Hubbell, FASEB J., 2003, 17, 2260–2262 CAS.
- G. Neufeld, T. Cohen, S. Gengrinovitch and Z. Poltorak, FASEB J., 1999, 13, 9–22 CAS.
- M. V. Backer and J. M. Backer, Protein Expression Purif., 2001, 23, 1–7 CrossRef CAS.
- M. V. Backer and J. M. Backer, Bioconjugate Chem., 2001, 12, 1066–1073 CrossRef CAS.
- M. V. Backer, V. Patel, B. T. Jehning and J. M. Backer, Bioconjugate Chem., 2006, 17, 912–919 CrossRef CAS.
- L. Tirand, C. Frochot, R. Vanderesse, N. Thornas, E. Trinquet, S. Pinel, M. L. Viriot, F. Guillemin and M. Barberi-Heyob, J. Controlled Release, 2006, 111, 153–164 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2008 |