Flexible macromolecules attached to lipid bilayers: impact on fluidity, curvature, permeability and stability of the membranes
Christophe
Tribet
and
Florent
Vial
Physico-chimie des Polymères et Milieux Dispersés, CNRS UMR 7615 and Université Paris 6, ESPCI, 10 rue Vauquelin, F-75005 Paris, France
First published on 11th October 2007
Abstract
This review summarizes recent investigations on the association of macromolecules on lipid bilayers. Hydrophilic and flexible polymers can form soft coronae tenuously adsorbed or anchored on the lipid membrane. Other synthetic macromolecules are embedded in the apolar region of the membrane. Recent experimental and theoretical works focus on the perturbation of lipid properties achieved depending on the nature and strength of binding. Of importance to biomimicry, to tethered model membranes, and drug carriers, the effects achievable include modulation of the lateral diffusivity of lipids, shape distortions, lateral segregations, formation of well-defined nanopores and ultimately the stimuli responsive disruption of the membrane.
 Christophe Tribet | Christophe Tribet received his Ph.D. in chemistry from the University Paris VI (1993) and pursued post-doctoral research at ULB (Center of Non-Linear Phenomena, Bruxelles) and at the Institute of Biology and Physical Chemistry, Paris. He joined the Dept of polymer science at ESPCI in 1994, and since 2005 has been visiting professor at the University de Montreal. His current research interests relate to stimuli- and light-responsive polymers for the handling of lipid membranes and proteins, sp. integral membrane proteins. |
 Florent Vial | Florent Vial studied chemistry at the Ecole Normale Superieure de Cachan & University Paris XI Orsay, France. He completed his Ph.D. at the University Paris VI, on the controlled permeabilization of lipid membranes by stimuli-responsive amphiphilic copolymers. He is now teaching physics and chemistry. |
Introduction
Lipid membranes in cells form amphipathic barriers that isolate the cellular content and cell compartments from the external environment. Given the multiple and essential role of membranes in the regulation of exchanges, the control and understanding of the ubiquitous functions of lipid bilayers are among the most fundamental and practical challenges in membrane biophysics. The development of novel molecules and macromolecules that affect the properties of membranes is of enormous importance, from both fundamental and practical points of view. Pharmaceutical prospects include for instance antimicrobial compounds, cost-effective and robust mimics of natural peptides capable of permeabilizing the outer membrane of bacteria or fungus.1,2 Substantial challenges are also identified to appropriately design “drug cargoes”3,4 and liposomal formulations that prevail in the currently approved drug-delivery systems.5,6 Macromolecular coronae often regulate the circulating time in vivo of such cargoes, and help to target cells. Recognition of these challenges is leading to new approaches that provide liposomes with the ability to respond to stimuli, and to trigger endocytosis,7 or transmembrane delivery on demand.8The association of macromolecules with membranes has proven to be an efficient means of achieving remarkable effects. Most often, polymers are expected to form a protective (repulsive) corona that enhances the circulation time of liposomal formulations in vivo, and substitutes for glycolipids and glycoproteins. The adsorbed macromolecules confer also on the layer above the membrane, high viscosity and/or elastic properties that differ markedly from those of bulk water. Experimental evidence9 and theoretical models10 show that yet at low coverage (isolated chains) anchored polymers modify the mechanical properties of the lipid membrane (e.g. increase the bending rigidity). Not surprisingly, obvious effects are reported on the mechanical response of vesicles,11,12 buddings and deformations of soft bilayers.13 The mechanical coupling between complex flows in a gel-like layer and the deformation of membranes enables one to mimic with giant vesicles the behaviour of red-blood cells in vessels14 or their disk-like shapes.15,16 To the benefit of the development of biosensors, tethered lipid membranes can be deposited on a cushion of hydrophilic polymers improving mechanical stability, but also reducing mobilities of ions in the cushion as compared to water.17 When it is tightly anchored in a membrane, a polymer may also control the formation of lateral lipid domains,18 transversal asymmetry in the composition of the leaflets,19 permeabilization and formation of pores.20,21
Ringsdorf et al.22 have pioneered the chemistry of polymerized lipids and liposomes, aiming at the stabilization of hollow capsules against rupture and de-assembly (e.g. in presence of other amphiphiles or at high dilution below the critical association concentration). Since that time, the expectation for new medical applications based on liposomes has motivated most studies of drug delivery by polymer-containing vesicles. Polymerization of lipids,23 a wide variety of hydrophilic macromolecules anchored on lipid vesicles,3,24 and novel polymer self-assemblies and polymeric membranes (polymersome)5,25,26 have been designed as promising nano-carriers with enhanced circulation-time in vivo. The reviews cited above discuss the formulation and potentialities of such systems for the purpose of therapeutic application. Likewise it is emphasized on the protection against aggregation and resistance to disruption/digestion of the carriers in biological fluids. Slow kinetics of dissociation provide for instance an apparent “stability” that is not distinguished from a low free energy of formation (e.g. kinetically frozen assemblies of diblock copolymers and polymersomes).25 The chemistry of such nano-carriers and delivery issues are not the purpose of the present review, and the reader is referred to the papers cited above. Here we focus on the interaction of polymers with bilayers and the resulting changes in the energy and shape of the membranes.
The main features of macromolecules compared to molecular additives come from the covalent string of monomers whose global behaviour averages the properties of each “molecular” subunit together with constraints in their accessible spatial distributions. As a consequence, one domain of the chain with enough affinity for the lipids would bring the rest of the chains in the vicinity of the membrane irrespective of the monomer–membrane (possibly repulsive) interaction. The reduced translational entropy of the monomers leads to abrupt adsorption and collapse of polymer chains as soon as the monomer–surface or monomer–monomer interactions are weakly attractive.27 Some practical consequences are listed below: (i) water-soluble polymers are usually too hydrophilic to pass through the bilayer, and most often stick to a single leaflet. This may explain their mild effect on membranes and the low toxicity of chains made of a majority of hydrophilic monomers. Likewise, “cushions” of hydrophilic polymers appeared as promising mild supports to deposit tethered bilayers.28,29,30 (ii) A few monomers with an affinity for a surface provide enough driving force for tight adsorption of a polymer chain. The multipoint attachment by a few anchors achieves dilution-independent binding. In addition, a possible inter-polymer connectivity would impart the layer with new elastic properties. (iii) An isolated polymer chain may perturb (or protect) a large area containing tens to several thousands of lipids. The typical size of a polymer coil can be increased from ∼5 nm2 to 104 nm2 with increasing the polymer chain length. (iv) The properties of polymers, including hydrophobicity, can be abruptly switched. Parameters such as pH, salt concentration, or temperature can easily trigger the monomer–monomer intrachain interaction between (weakly) attractive and repulsive conditions. Such a slight change in solvent conditions results in collapse of a chain or inter-chain aggregation, which in turn makes mixed membranes responsive to stimuli.8,31
We illustrate and summarize the various systems recently investigated as follows. Section 1 summarizes the nature of polymer–lipid interactions and the structures of macromolecules that have been shown to be efficient at anchoring the chains onto/into membranes. In section 2, we illustrate practical aspects of controlling the association on flat cushions of macromolecules (tethered model bilayers) or in solution to obtain liposome-filled gels. At conditions of tighter association, polymers induce the destabilization of liposomes. The coupling with membrane stability is described in section 3, including general thermodynamic considerations as recently proposed by Marsh et al32 to clarify the magnitude of perturbations achieved by an hydrophilic polymer layer and the case of stimuli-responsive chains. The last two sections present the effects of polymers on the shape of liposomes, their curvature (section 4), and the stabilization of complex inclusions (domains and pores, sections 5). Over the last few years, progress in the micromanipulation and the direct microscopic visualization of both supported membranes and giant vesicles has made possible new investigations on domain formation, protrusion, adhesion, and fusion events. The works summarized here illustrate representative use of polymer–lipid mixed assemblies in such experimental set-ups, and the present understanding that has emerged about the coupling between membrane properties and polymer properties. We will particularly emphasize the impact and characteristic features of synthetic polymers that trigger non-specific effects. Indeed, peptide and protein interactions with membranes have recently been reviewed.33
1. Association modes between polymers and lipid membranes
The surface of a lipid membrane is a complex environment of intermediate polarity and may contain diverse functional groups, for instance saccharides, zwitterions, anions (carboxylate, sulfonate) or cations (mostly ammonium). The diversity of the interfacial composition is likely to display all common modes of non-covalent association that can be formed in water. Depending on both the polymer and lipid structure, hydrogen binding,34 anion–cation bridging,35,36 as well as oligosaccharides recognition37 have been shown to primarily control the association. However, most studies have been performed with zwitterionic phosphorylcholine heads, since they are the main constituents of natural membranes. Except when specified below, the phosphocholine lipids will be the major component of membranes with possibly fluorescent lipid probes and/or anionic lipids (e.g.phosphatidic acid). Polymer structures that bind to phospholipids are grouped below depending on the type of interaction (hydrophobic, coulombic, etc.).Hydrophobicity
The deeper penetration of the polymer into the lipid bilayer is achieved by hydrophobic interactions with the tails of the lipid molecules. The hydrophobic nature of the association with polymers bearing n-alkyl side groups has been clearly demonstrated by using fluorescent probes (i.e. polarity reporters) as side groups in macromolecules.38,39 In addition, macromolecules and liposomes of like charges (e.g. anionic) form complexes when the polymer is amphiphilic, but do not bind in the absence of hydrophobic moieties on the chain.31 Hydrophobic association rapidly overcomes other interactions, including strong coulombic repulsions. The binding strength can be estimated using the average free energy of transfer of a methene group from water to apolar solvents (∼3 kJ mol−1 of CH2group)40 and the free energy of ca. one kT per ion entering in the Debye layer of the vesicle (with k the Boltzman constant and T the temperature). Accordingly, the binding of a single dihexadecyl anchor (∼−39 kT at ambient temperature) balances the coulombic repulsion of an ionized chain, as typically about 40 monomers penetrate into the 1–3 nm thick Debye layer (n.b. the majority of the ionic groups in the macromolecule could be screened by Manning condensation). Even relatively short alkyl groups, such as ethyl, if present at high density in the macromolecule, can force the binding against charge repulsion.41 However, long hydrophobes or cholesterol groups provide more stable anchoring. A single cholesteryl end-group, or a pair of dodecyl groups, can maintain the attachment of a polyethylene oxide (PEO) chain containing about 200 ethylene oxide (EO) units (Fig. 1(a)).42 The free energy of hydrophobic anchoring balances here the loss of translational and configurational entropy of the hydrophilic PEO block. Further reinforcement of the association can be achieved by covalent attachment of several amphiphilic motives, each containing one hydrophobe and one PEO block (comb-like amphiphilic polymers), because the translational entropy is not recovered upon detachment of a single anchor.43 With a number of amphiphilic motives increasing by a factor of 5, Auguste et al. have measured the increase over more than a decade of the partition constant of polymer between water and the lipid membrane. A low degree of dissociation upon dilution (tight association) is of crucial importance to in vivo injection and circulation of liposomal drug carriers. Cholesterol groups are often preferred because their presence improves membrane fluidity and decreases permeability,44 but this effect is not specific to the polymeric nature of the compounds and is obtained as well with molecular cholesterol additives. | ||
| Fig. 1 Some chemical structures representative of macromolecules forming associates with lipid membranes. (a) PEO with a hydrophobic end, (b) PEO–PPO–PEO triblock, (c, d) polycations of adjustable content of ionic groups, alkyl chains, and hydrophilic segments, (e,f) amphiphilic derivatives of acrylic or glycolic acids (g) complexes of organic phosphate and polyarginin. | ||
Hydrogen bonds
In comparison to amphiphilic polymers, reports on associations viahydrogen bonds or ionic complementarities are rather rare. Polozova et al. have found that hydrogen binding drives the adsorption of poly(N-isopropylacrylamide-co-glycidylacrylamide) (Fig. 1(f)) irrespective of the charge of the membranes, although the lipid was in this case a nonphosphocholine lipid (ethylene oxide polar group) combined with either an anionic or a cationic co-surfactant.45 In the case of phosphocholine lipids, in contrast, hydrogen bridges did not significantly contribute to the adsorption. Kawakami et al. have cautiously characterized the interaction with representative hydrocolloids (PEO, Dextran, cellulose derivatives) by isothermal titration calorimetry and infra-red spectrometry.46 The interaction of phosphocholine lipids with these hydrophilic chains is very weak in water, and was tentatively ascribed to hydrogen bonds. The presence of salt weakens further the association (alginate, PEO) below the limit of detection. Hydrogen-bond complexation between donors and the phosphate group in phosphorylcholine heads has however been identified as a major determinant of the binding of small molecules, especially in the case of acidic amide groups.3447 Rich density of amide groups and/or hydroxyl side groups capable of reaching the phosphate group embedded in the interfacial layer are structural features that can form surprisingly tight bonds. Accordingly, hydrophilic macromolecules containing amidoethanol side groups trigger obvious perturbation of liposomes, and even the disruption of the membrane.48Coulombic association
Polycations strongly adhere to lipid membranes that are most typically negatively charged (Fig. 1(c,d)).19,35,36,49,50 Similarly, polyanions bind to cationic lipids (e.g.DNA and lipopolyamines).51 Of practical importance, polycationic particles are among the most efficient DNA carriers for the transfection of living cells.52–54 The visualization of spontaneous DNA endocytosis in giant cationic vesicles has also been reported (with sphingosine cationic lipids).55 The mechanism of cell penetration and of delivery of the genetic material into the nucleus is still debated, but an initial stage of tight binding of the positively charged particles leaves no doubt. Highly charged polycations disrupt the membranes.56 It is worth pointing out, however, that the toxicity of polycations and amphiphilic cations may have several origins in vivo, and electrostatic binding is only one of them. For example, a chemical effect of adsorbed cations has been recently shown to catalyze the hydrolysis of lipids into lysolipids (single-tailed lipids) that destabilize the bilayer structures.57The physical origin of predominantly electrostatic binding is the entropic gain of translational freedom of the counter ions released upon association of the polymer and the membrane. This free-energy gain balances the loss of freedom of multi-charged assemblies having limited conformational space even in their unbound state, namely the lipids in a membrane and monomers of the polyelectrolyte chains. Charged peptides and proteins with even less intrachain mobility of the charges bind to membranes because of similar effects, and detailed calculations of the electric potential enabled a quantitative estimation of the association constant in this case.58,59 A polyelectrolyte (or a protein) adsorbed in the surface of charged vesicles could be completely removed from the membrane by an increase in simple salt concentration or by competition with a soluble polyelectrolyte of opposite charge.35,60 On almost neutral membranes, the electrostatic interaction may not be strong enough to provide attachment. The zwitterionic heads of phospholipids, though neutral, are oriented by self-assembly and their negative phosphate ions lay closer to the hydrophobic core than their positive ammonium moieties. This preferred orientation does not however favour the interaction with anionic surfaces. Pure phosphocholine liposomes are not adsorbed in cationic surfaces, as shown on multilayers of polystyrensulfonate and polyallylamine hydrochloride, but in contrast adhere to the anionic ones, possibly because of the hydrophobicity of the polystyrene backbone.61 The generic binding of polycation on biomembranes is presumably not due to electrostatic association with phospholipids, but most likely driven by the presence of anionic lipids or negatively charged proteins.
Of practical relevance to construct vesicles with controlled shapes and loading, lipids can be adsorbed onto colloidal particles coated with polyelectrolytes and polyelectrolyte multilayers.62 Charged lipids, such as DPPA, form bilayers while zwitterionic lipids, such as DPPC, may adsorb in the form of more than one bilayer. The reduced permeability imparted to those complex particles by the lipid layer, and the reduced mobility of lipids in contact with polyelectrolyte cushion have been extensively studied by Mohwald and colleagues.63,64 Somehow surprisingly, the diffusivity of zwitterionic lipids (phosphocholine heads) was more hindered than that of charged lipids. An obvious strong coupling between head groups and polyelectrolyte cushions (PHA/PSS) was therefore revealed irrespective of the total charge of the lipid head.63
Partition of ions and polar groups in the apolar core of bilayers
Indirect attractive forces can be mediated by multivalent ions or complexing agents, especially calcium ions. A complicated balance between the effects of polymer length, screening of coulombic interaction and calcium bridges modulates for instance the association of dextran sulfate, a chain that does not bind phosphocholine lipids in the absence of the multivalent ions.65,66 Other complexes between ions and polymers generate associates that become capable of penetrating the hydrophobic interior of the membrane. Recalling that the free energy of transfer of one ion between water and oil is of the order of +60 kJ mol−1 (mostly due to dehydration), spontaneous penetration of charged phosphate groups is clearly unexpected. Nevertheless, two flexible macromolecules devoid of known tertiary structure have been found in vivo to help the translocation of ions, namely polyarginine (Fig. 1(g))67,68 and polyhydroxybutyric acid in the presence of polyphosphate.69,70 These linear homopolymers, namely inorganic polyphosphate, poly-(R)-3-hydroxybutyrate, and the polypeptide polyarginine are ubiquitous constituents of biological cells. All three macromolecules are hydrophilic and water-soluble, in the form of polyelectrolytes, and none of them can cross a lipid bilayer as an isolated chain. Furthermore, high pH conditions, which ionize the polyhydroxybutyric acid are required to observe ion channels in the presence of polyphosphate. Presumably neutral complexes with polyphosphate and/or other multivalent ions (Ca2+, Ba2+, etc) generate polyzwitterionic species prone to entering the membrane core.71 Similarly, the presence of hydrophobic anions (for instance a lipid with a phosphorylglycerol head group) is required for polyarginine efficiency, in synergism with phosphate ions (Fig. 1(g)). Poly(lysine) in similar conditions does not penetrate into the membrane, which validates a mechanism based on a specific complex and not of electrostatic nature.67 It could be considered that the formation of complexes leads to dramatic changes in the intrachain hydrophilic–lipophilic balance, and to water insolubility, which in turn favours the partition of the all associated chains in the core of the lipid membrane. Weak loss of hydrophilicity and concomitant abrupt switch toward water insolubility of a macromolecule is well-documented in the case of a polymer showing a low critical solubility temperature (LCST), like poly(N-isopropylacrylamide) and pluronics (Fig. 1(b,f), cf. section 3.). Interestingly, the two latter polymers do not interact with lipids at low temperature, but bind tightly to membranes at T above their LCST and can even translocate across the bilayer.39,72,732. Weak polymer–lipid attractions: tethered membranes and liposome-filled gels
Below are listed applications of the coupling of polymers and membranes by weak forces that minimize the perturbation on the lipid leaflets. First, lipid bilayers supported on a solid plate are now widely used as cell-surface models and for the development of biosensors.74,75 Ultrathin and hydrated polymer supports (5–50 nm thick) mimic the role of the extracellular matrix and intercalate a soft water reservoir that protects the supported layer against the deleterious effects of a solid support (e.g. denaturation or adhesion of proteins intracellular domains, lack of fluidity). The first successful constructs of an hydrophilic spacer between the membrane and its solid support were achieved by covalent attachment of functional poly(ethylene oxide) heads of lipids on electrodes.76 This technique form membranes with high electrical resistivity, a criterion for the absence of defect, although the PEO spacer may contribute to reducing the mobilities of ions below the membrane.17 It has been shown that non covalent deposition on a water-swollen cushion of polymers also forms membranes with a higher electrical resistance than those deposited on glassware, and more than likely reduces the density of defects.77But, on neutral and hydrophilic cushions, lipid membranes have been shown to be unstable, presumably because of very weak adhesion.78 In contrast, cushions made of cationic macromolecules,78,79 and/or containing hydrophobic groups,28,29,75,80 specifically lipid-like anchors, are very effective at rapidly adsorbing liposomes (in less than one minute) and at preserving the stability of the membrane afterward, even against rinsing and drying (Fig. 2).81 Reversibility of the adhesion and its exquisite control has been achieved by modulating the (anionic) charge density of the polymer, using thermo-responsive macromolecules,82,83 or other polymers imparted with stimuli-triggered hydrophobicity.84 The “sticky” cholesterol or lipid tails can be, for instance, replaced by a photo-responsive chain that partitions into lipid bilayers in the dark but, when exposed to UV light, switches to a more polar state and releases the attached vesicles. It seems reasonable that the lateral density of hydrophobic anchors affects the properties and stability of supported membranes. Well-defined telechelic polymers (i.e. one hydrophobe at one end, and functionalized to be adsorbed into the substrate at the other end) provide good orientation of the anchors, toward the upper phase, and good control of their lateral density. A density of 5 mol% anchors is sufficient to maintain attachment of a lipid bilayer.
 | ||
| Fig. 2 Schematic sketch of the adsorption of a vesicle and formation of tethered bilayers on a cushion of amphiphilic polymer. Reprinted with permission from ref. 79, copyright 2000, American Chemical Society. | ||
Low surface densities of the hydrophobes plunged in the membrane do not hinder the diffusivity of lipids (up to ∼20 mol% anchors relative to lipids). Low density of anchors are also compatible with high diffusivity of integral membrane proteins in tethered bilayers.85,86 However, higher density may freeze the Brownian motion in membranes.80,87,88 Naumann et al.89 have studied and quantified the obstructed diffusion of both lipids and membrane proteins by particle-tracking experiments. They show that the diffusivity decreases almost linearly with the density of anchors, down to zero mobility at a threshold that depends on the size of the mobile species (Fig. 3). This threshold and the complex signature of lateral diffusion at intermediate densities match well with hard–core repulsion with obstacles (the anchors) that are randomly distributed at low density (typically <10 mol% of lipid anchors) but gradually form small aggregates reaching percolation at higher densities. Note that the length of the polymer, which ultimately controls the swelling, the thickness, and roughness86 of the tether also appears critical to avoid the aggregation of protein inclusions in the membranes and maintain fluidity.77 The method of deposition (vesicle adsorption/fusion, langmuir–blodget, etc.) is in addition a major determinant of the long-range diffusion in tethered membranes, because it affects the continuity or the “patchy” morphology of the layer.30
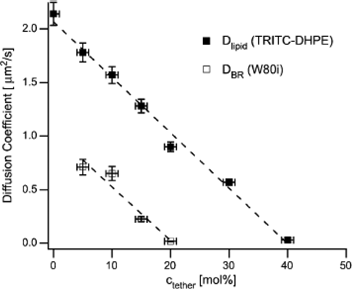 | ||
| Fig. 3 Lateral diffusivity of a labelled lipid or of the transmembrane protein Bacteriorhodopsin, as a function of the molar fraction of polymer-attached lipid anchors in a polymer supported bilayer. Zero-diffusivity matches with the percolation threshold of obstacles and hard core interactions with the probes, assuming that the anchors are immobile. Reprinted with permission from ref. 89, copyright 2005, Biophysical Society. | ||
In bulk solution, similar attraction between long macromolecules and liposomes may produce aggregates or stacks of membranes,39,90 and finally gelation of the mixtures. Liposome-loaded gels are tentative candidates for the encapsulation and the (slow) release of drugs.91,92 The formation of a network of polymer-bridged membranes is generally accepted to explain the sharp increase in the viscosity upon supplementation of a solution of vesicles with macromolecules (Fig. 4).93–96 In the more dilute solutions investigated, the distance between the vesicles may, however, be larger than the typical radius of the polymers. there is no doubt that the exact origin of viscosification is more complicated and could involve partial disruption of the initial liposomes, and contribution of a polymer–micelle mixed network.93,95 The capacity for drug entrapment and release in such gels would accordingly depend on membrane barriers against free diffusion, rather than encapsulation in liposomes. Alternatively, the conjugation of intact liposomes to a gel phase such as Sepharose is investigated for chromatography across modified phases that contain native integral membrane proteins as functional receptors.97,98 The function of the macromolecules in this case is to immobilize the vesicles by covalent attachment onto the stationary phase (e.g.disulfide bridges). Polymers also act as versatile spacers that prevent the adsorption of lipids in the phase (repulsion of the polymer corona) and preserve the accessibility to small substrates. Hydrophobic anchoring of the polymer is expected to be compatible with the functionality of proteins embedded in immobilized proteoliposomes.97
 | ||
| Fig. 4 Sketch of the physical gels and polymer–membrane bridges that may form in concentrated mixtures of amphiphilic macromolecules and liposomes. Reprinted with permission from ref. 96, copyright 2005, American Chemical Society. | ||
3. Destabilization of the bilayers: hydrophilic coronae or stimuli-responsive chains
In the literature, one can find two classes of liposomes whose stability is perturbed by polymer chains. Vesicles have been associated with stimuli-responsive macromolecules designed to break or dissolve the bilayers at specific pH or temperature conditions (e.g. stimulation by pH conditions may help membrane permeabilization/breakage in late endosomes; alternatively, sensitivity to temperature may target the effect on infected tissues, or tissues treated by hyperthermia).8,108 The stability of “decorated” membranes is also affected by a corona of hydrophilic macromolecules (e.g. liposomes called “stealth” and usually decorated with polyoxyethylene, PEO). In the latter case, it is generally believed that the repulsive interaction between the hydrophilic segments of lipid-grafted PEO contributes to membrane stretching and therefore lowers the cohesion of the lipid in bilayers.We discuss first the non-specific effect brought by repulsions between hydrophilic moieties of anchored chains. It is important to determine the magnitude of free energy added by PEO layers, and whether this could contribute to membrane disruption or not. To this aim, the energetics of polymer coupling with a soft membrane has been extensively examined by Marsh et al.11,32,99 A minimal surface density is needed to bring into “contact” the hydrophilic moieties of the anchored chains and develop significant lateral perturbation. The PEO coils have a typical radius of gyration of 4–11 nm (Rg∼a×N0.6, in the “mushroom” regime with the monomer size a = 0.4 nm and N the number of monomers in the range 50–250).11,32,100 Assuming that the surface per lipid head is Alipid = 70 Å2, the PEO chains are brought into contact (overlap) when their density reaches 0.2–1.5 mol% of anchor (relative to the lipids, Fig. 5). Note that even well below the overlap density, isolated hydrophilic chains repel the membrane. The perturbation by isolated chains has been treated theoretically by many authors.9,10,32,101 In particular, the bending stiffness and spontaneous curvature of the membrane are modified to an extent that does not afford the disruption of membrane (free-energy change < kT per lipid), but would considerably affect the membrane's curvature9,10 and thermal fluctuations.102 Changes in the shape properties are discussed in section 4. Experimentally, Ligoure et al. showed that anchored polymers cause an effective rigidification and a decrease in thickness of surfactant/co-surfactant membranes (see ref. 103 and references therein).
 | ||
| Fig. 5 The mushroom regime of non-interacting end-grafted polymers (upper panel) and brush regime at higher molar fraction Xp of polymer anchors (lower panel). Reprinted with permission from ref. 32, copyright 2003, Elsevier. | ||
Most preparations usually contain more than 5 mol% anchors and therefore the PEO chains are close to or above their overlap density and are stretched in the direction perpendicular to the membrane. It is often assumed that the PEO layers have reached the brush regime. Parsegian et al. however noted that for short chains, typically of Mw below 5000 g mol−1, this regime is reached well above the overlap density, above 10–20 mol% of anchors in the membrane.104 Energy terms as given by scaling laws for PEO brushes provide nevertheless a useful order of magnitude.9,11,32 The lateral pressure exerted by polymers on the lipids is written:
| Πbrush∼mkTNa2m(X/Alipid)m+1 | (1) |
With X being the molar fraction of anchors in the lipids, N is the average number of monomers per chain, m = 5/6 (scaling theory) or 2/3 (mean field theory).9,11 At a molar fraction of anchors X = 0.2, and with N = 100–200 monomers, a considerable lateral pressure Πbrush (up to 20 mN m−1) contributes to the stretching of the membrane. The free energy evolved upon increasing the surface of the membrane by a few percent (ΠbrushΔAlipid per lipid molecule) is of the order of the free energy of hydrophobic association (ΓΔA2lipid/Alipid with Γ being the oil–water interfacial tension = 50 mJ m−2).40 Therefore, the self-assembly as a flat lamellar phase can be unstable and the membrane would break at high densities of PEO. To decrease its free energy, the membrane may however offer more space to the PEO corona by forming buds (local high positive curvature), or being partially solubilized in mixed micelles. Experimental data match well with the calculated impact of PEO on lamellae–micelle coexistence.99 Upon increasing the amount of PEO in samples, a critical composition is reached, above which the lamellar phase with a low polymer–lipid ratio has the same free energy as a micellar structure with a high fraction of polymer. The destabilization of PEO-decorated bilayers has been reported both with hydrophobically anchored chains99 and electrostatically bound ones.105 The excess free energy due to PEO repulsion also decreases the temperature of fluid–gel transition by 1–2 °C.32,99 Inter-PEO repulsion makes the transition less favourable because of either the lower area of the membrane in the gel phase (increasing density of chain upon phase transition) or the expulsion of the anchors from the gel phase that would increase the density of PEO chains in fluid domains (in equilibrium with a pure gel phase). Experimentally, the temperature of the fluid–gel transition shifts by typically −1 to −2 °C, a result in relative agreement with the model developed by Marsh et al.
Although thermodynamically destabilized, the PEO-coated lipids are kinetically protected against attack from proteins or micelles. Penetration in the brush layer of a micelle of radius R would push away the polymer chains, which requires additional free energy of the order of Πbrush.R2. Needham et al. have estimated the effect of this energy cost on the kinetics of membrane disruption by micelles of a lysolipid.100 The drop in the frequency of contacts between micelles and PEO-coated membranes protects the surface of the lipids. Only the monomers of the lysolipids can spread across the polymer brush, which in practice increases the time required to saturate the membrane with the surfactant, and in turn increases the lifetime of the membrane. Altogether, the results above show that conventional anchor densities of highly hydrophilic chains do not usually break the membrane, but make it more fragile.
In contrast, obvious destabilization and permeabilization have been achieved with stimuli-responsive polymers. These macromolecules contain hydrophobic anchors to bind to the membranes and most importantly are close to bad solvent conditions. A wide variety of stimuli-responsive macromolecular structures have been tested in liposome formulations under the form of amphiphilic copolymers. 8,106,107 The membrane breakage is generally obtained near critical pH or temperature conditions that make the polymer insoluble in bulk water, and would trigger coil–globule transition of the conformation of the chains in solution.8,108 Most typically, pH-responsive macromolecules are hydrophilic at high pH (>7) in a polyanion form (Fig. 1(e,f), as salts of carboxylic acid side groups. Below a critical pH value, or in vivo at the pH of late endosomes, those polymers become abruptly insoluble because they are partially or totally neutralized.8,41,109 On the other hand, temperature-responsive systems contain N-isopropylacrylamide39,110,111 units, or propylene oxide units,73,112 or organophosphazene.113 These monomers become less hydrated at temperatures above 35–40 °C (i.e. the low critical solubility temperature of the corresponding polymer chains, LCST). The principle of permeabilization at critical solubility conditions appears remarkably independent on the chemical structure. Biocompatibility issues have, however, motivated the development of diverse compounds, to provide stability in serum (low adsorption of protein in the “hydrophilic” conditions of high pH or low T),114,115 and efficient haemolytic activities or permeabilization of mammalian cells.116,117,118,119,120 But the exact origin of membrane disruption in vivo is largely conjectural. Within the limits of weak interactions, an analogy should exist with the above mentioned PEO-induced lateral pressure. The switch from the repulsive regime toward a (weak) attractive regime is accordingly expected to stabilize the planar geometry and to “compress” the membrane, making it less permeable.121 The validity of the above theory however fails in poor solvent conditions, when strong attraction between monomers leads to chain collapse and the formation of globular aggregates. It is likely that insoluble segments of the chains penetrate inside the bilayers and introduce defects in their organization by a deep inclusion of ethoxy or carboxy groups in the apolar lipid core.122,123 This mechanism obviously differs from the (weak) destabilization of the membrane achieved by hydrophilic coronae of chains. In practice, it has been observed that flat membrane fragments, with disk-like shapes and radii below 40 nm, can be obtained by disruption of small vesicles.124,125Polymers forming micelles in water are presumably capable of stabilizing the rim of fragments of lipid bilayers with local curvatures below 3–4 nm. Compared to surfactant-induced disruptions, slow kinetics of reorganization (often days long) can be observed close to the critical conditions for disruption. The slow rates may be due to a slow adsorption and reorganization of the chains.31 (the slow kinetics of adsorption due to repulsions between polymers is now well documented).126,127 Even after several days incubation, the equilibrium sorption of charged amphiphilic polyelectrolytes is not reached on some hydrophobic surfaces.
4. Polymer-induced shape distortion
On flat membranes with no tension, a polymer layer imparting one leaflet of the membrane with additional lateral pressure or an excess area is expected and actually shown to trigger curvature and shape transitions. The response of a bilayer to small variations of lateral pressure is especially dramatic in membranes with no tension. Giant vesicles with diameters above a few microns, can be prepared with low membrane tension and are accordingly ideal models to investigate the polymer-controlled shape transitions. Early observations by Sackmann and coworkers report the shape distortion induced by thermo-responsive macromolecules (hydrophobic derivatives of poly (N-isopropylacrylamide)).128 The thermo-responsive polymer was anchored by alkyl side groups on the outer leaflet of giant vesicles, and transient buddings were observed on a temperature sweep ranging from below to above the LCST (35 °C) of the chains. Kinosita et al. have also observed tubular protrusions of vesicles and sphere–dumbell transitions upon polymerization and depolymerization, respectively, of actin gel inside the vesicle.129,130 In the latter case of actin-loaded GUVs, the interaction with the membrane is likely to be of a repulsive nature with no anchoring of the polymer. This shows that growth of a polymer chains attached to a gel (polymerization case) or the formation of a bundle of actin filaments (depolymerization case) can push on soft lipid membranes.Transient local budding and tubulation have been observed by Tsafrir et al. in floppy oblate lipid vesicles prepared by the swelling of a lipid reservoir to obtain membrane with zero tension.131 Injection of a weakly hydrophobic dextran in the vicinity of the vesicle triggers the growth of long tubes (Fig. 6(a)). Shrinkage and re-absorption of the tubes immediately follow the end of supplementation with polymers, which is consistent with an adsorption-induced spontaneous curvature followed by diffusive homogenization of the polymer concentration in the reservoir of lipids. In the case of isolated giant vesicles close to spherical shapes, amphiphilic macromolecules have also been shown to trigger the formation of persistent buds and tubes, even in the absence of a concentration gradient in the outer medium (Fig. 6(b)).132 The injection of chains that adsorb onto the vesicle do not afford a good control of the polymer density and eventually homogeneity. To overcome this experimental limit, Nikolov et al. have recently proposed a three-stage construct of membrane associates with long DNA molecules, via tight but non-covalent associations on biotinylated lipid heads.9 Their results confirmed the polymer-induced budding, even at rather low surface density, in the mushroom regime with essentially isolated chains on the membrane. The shape instabilities as triggered by polymer adsorption depend on the initial geometry of the vesicles. Hollow tubular vesicles relax the trend to form curved structures by the so called “pearling” instability, which results in a string of smaller spherical vesicles connected by extremely narrow necks (Fig. 6(d)).133 Multilamellar tubes of stacked membranes slowly form coils (single strand or double strands) upon swelling in the presence of amphiphilic polymers (Fig. 6(c)).134 The relaxation toward these geometries can be accounted for within a framework in which polymer chains affect the curvature of the bilayers. However, the obviously large range of curvatures present in some geometries (e.g. heterogeneous pearl radii) suggests the additional requirement for large gradients of the polymer surface density. The coiling of multilamellar tubes can also be explained by a transversal gradient of polymer concentration across the stack of membranes.134
 | ||
| Fig. 6 Typical shapes formed upon adsorption of amphiphilic polymers on lipid membranes: (a) transient tube growth/resorption from the rim of an oblate vesicle connected to a reservoir of lipids, scale bar = 10 μm (reprinted with permission from ref. 131, copyright, APS 2003). (b) Day-long stable buds and fluctuating tubes protruding from a giant unilamellar vesicle (reprinted with permission from ref. 132, copyright 2002, American Chemical Society, vesicle diameter = 40 μm). (c) Coiling of multilamellar stacks of membranes swollen in a solution of polymer, scale bar = 20 μm (with permission from ref. 134, copyright 2003, APS). (d) Pearling instability of a tube connected to a reservoir of lipids, the left-hand “pearl” diameter is = 20 μm (with permission from ref. 133, copyright 2001, American Physical Society). | ||
At low polymer coverage, the curvature can be induced by two mechanisms:135–137 i) an effective repulsion between the polymer segments and the bilayer over distances of the order of the polymer radius, the bending energy of the membrane balances the gain in free energy of a polymer chain that is allowed more conformational freedom when the impermeable membrane is pushed away; ii) an increase in the area of the anchor(s)-containing leaflet with respect to the other. Both effects i) and ii) result in an increase of the spontaneous curvature of the leaflet on which the polymer is stuck (Fig. 7). Theoretical treatment of the polymer as a perturbation of the membrane mechanical properties predicts in addition that effect i) increases the bending stiffness.10,101,102 This would lead in practice to increase of the membrane curvature, while decreasing the thermal fluctuations (more rigid membranes) as observed and quantified by Nikolov et al.9 The magnitude of variation of the bending rigidity can be surprisingly high and clearly beyond small perturbations effects (increase of the modulus by more than 10 kT)9,103 Theories capture nevertheless correct trends with concentration and polymer size. At high coverage, the lateral pressure due to interpolymer interactions dominates, and an increase in the outer surface of the membrane with increasing the curvature relaxes the excess free energy brought by the effective interchain repulsions.138 Long range lateral interactions between different polymers are also mediated by the membrane deformation. The pair potential due to local membrane distortion close to the anchors is attractive for chains grafted on the same side of the membrane,139 and may favor the lateral segregation of the chains.140 Accounting for thermal modes of fluctuation of both polymer conformations and membrane bends, theoretical approaches (sp. by Lipowsky et al. and Auth and Gompper) predict the order of magnitude of the observed curvatures and the possible shapes of vesicles.10,101,141,142 Finally, the phenomena are mostly driven by repulsions between the hydrophilic segments of the chains and an impenetrable soft membrane, which in turn triggers the membrane's protrusions toward the side of higher polymer density.
 | ||
| Fig. 7 Schematic representation of the origin of polymer-coupled curvature of a membrane: (a) increase of the spontaneous curvature due to the gain in conformation freedom of the hydrophilic corona; (b) swelling of the outer leaflet by hydrophobic inclusions, which is balanced by the bending of both leaflets in order to match their surface of contact. | ||
Transverse density gradients of the polymer coat has accordingly a marked impact on curvature and it has been shown that when liposomes are prepared from a mixture of polymer and lipids at a particular ratio, a spontaneous vesiculation occurs with a well-defined radius and a lower internal polymer density.143,144 The contribution of hydrophobic anchors plays a marginal role by (slightly) increasing the volume of a leaflet, but the local structure, and homogeneity of the bilayer is essentially not modified provided that the macromolecules bear a low density of hydrophobic side groups. In contrast, if attractive forces drive the adsorption of most segments of the polymer chains, one would expect membrane invaginations. Angelova et al. showed that coulombic binding of DNA and other synthetic polyanions onto positively charged giant vesicles (sphingosine-containing membranes) induces endocytosis-like phenomena55,145 In this case, the mechanism involves the formation of lateral domains whose composition differs from the average one, and surface and charge asymmetries between the two leaflets.
5. Inclusion, domains and pores
As a general feature of strong hydrophobic and/or coulombic associations, polymers induce the formation of domains whose properties differ markedly from the rest of the membrane. Effects of peptides and proteins are well documented including the stabilization of channels, pores, and lateral segregations.33 The purpose of this section is to point out that flexible polymers devoid of secondary structures also trigger similar effects. The type of response to binding depends on whether the polymer attaches itself to the polar or apolar region of the membrane. First we consider the disorder produced upon tight hydrophobic binding. In this case, the local disorder affects the vesicle permeability and/or the rate of translocation of labelled lipids. Krylova et al. have studied pluronics (copolymers of oxyethylene and oxypropylene units, Fig. 1(b)) that accelerate the flip-flop of lipids and increase permeability to doxorubicin.146 A good correlation between the flipase activity and a linear combination of copolymer bulk hydrophobicity and the van der Waals volume of its hydrophobic block was found.72 Similar effects have been reported by Ringsdorf et al in the case of poly(N-isopropylacrylamide).147 Investigations on amphiphilic polypeptides have now established that hydrophobic inclusions perturb the transversal organization of the membrane, which may result in the formation of well-defined pores.33In vivo, these peptides are essential classes of antimicrobial compounds and venoms. Our recent investigations on amphiphilic copolymers binding onto giant lipid vesicles showed similarly well-defined pores stabilized by macromolecules devoid of even a secondary structure.31 When they are partially neutralized (pH lower than ∼7), amphiphilic acrylic copolymers impart the bilayers with the ability to transiently accommodate nano-channels with no disruption of membrane. The measured pore sizes are similar to those of channel-forming peptides in membranes, and display a similar sensitivity to polymer and salt concentrations (Fig. 8). We also obtained the mild permeabilization of periplasmic membranes of mammalian cells (HEK, COS, unpublished data with D. Massotte). Previously, Tirrell and co-workers had noticed the ability of poly(2-ethylacrylic acid) to form cation-selective pathways in patch-clamped membranes, although the stability of membranes was a difficult issue (the very narrow range of conditions and the presence of highly flickering burst of permeability are in that case indicative of a nearly ruptured membrane).20 The remarkable properties of amphiphilic polymers pave the way for the development of new tools for handling membranesin vivo and for the control of cell population, specifically with polymers that can be made stimuli-responsive. Coulombic association with polycations also accelerates the translocation of anionic lipids, but the relative contribution of hydrophobic vs. coulombic effects is difficult to determine in this case.19,60Copolymers of ethylvinylpyridinium and vinylpyridine do not make the membrane more permeable, neither do they change the flip-flop rate, when the degree of cationic group is below 50 mol%, a fraction however sufficient to tightly attach anionic lipids and to induce lateral demixing of neutral and charged lipids.19,60 It has been proposed that in the absence of pores, the flip-flop and permeability could be (slightly) enhanced because of defects created at the edges of the rigid domains formed in polycation-rich regions. | ||
| Fig. 8 Well-defined channels formed in the presence of amphiphilic polyacrylate (a) schematic drawing of transversal pores whose size depends on ionic strength conditions and polymer density (from ref. 21, copyright 2007, Royal Society of Chemistry ); (b) discrete current steps recorded by patch-clamp experiments in the presence of poly(2-ethylacrylic acid) at low pH (reprinted with permission from ref. 173, copyright 1996, American Chemical Society) | ||
The origin of the stability of macromolecule-containing pores is still debated, specifically the stabilization of a well-defined radius. A common feature of all pore-forming molecules is the presence of a significant fraction of lipo-soluble segments and their capacity to promote a curvature strain at the rim of pores (i.e. an effective decrease of the line tension, which stabilizes the pores).148 The free energy of a lipid pore combines the gain of relaxing the tension of the membrane over the area of the pore with the energy needed to create the rim of a pore of radius R:
| Fpore = 2ΠRγ−ΠR2σ. | (2) |
Where γ is the line tension (of the order of 10−11 J m−1 in pure lipid bilayers, see ref. 40,149 and references therein), and σ the tension of the membrane (ranging from zero to almost 10 mN m−1 in an experimentally stretched membrane close to rupture).150 If both of the tensions are invariant with R, Fpore reaches a maximum at a critical nucleation radius, Rc= γ/σ, and decreases beyond Rc. In other words, small pores are expected to rapidly close, and large ones should grow with no size limit. The presence of inclusions other than lipids contributes to a dependence of the tension parameters on the size of the pores. The additional (internal) tension due to lateral repulsions between inclusions on the membrane favours the formation of a pore (stretching effect) and can be partially decreased by the additional space offered in the pore. For instance, repulsions between charged surfactants is proposed to increase the membrane tension, until the surfactants are gathered in the pore and form a structure with high average curvature and with more accessible space for the cloud of their counter-ions.151,152 In the case of peptides, Lee et al have accounted for inter-peptide repulsions by assuming a linear variation of σ with the radius of the pore, i.e. in proportion to the amount of peptides transferred in the rim of the pore.153 The line tension should similarly reach an optimal value if the average curvature in the pore is matched with the spontaneous curvature of the additives covering the pore edges.151 Amphiphilic polyelectrolytes such as polyacrylates presumably affect the tension parameters at a similar magnitude as do amphiphilic polypeptides. The surface charge density due to such polymers can be as high as that created by charged peptides (assuming a typical density of 1 mg m−2 of macromolecules)132 and they share with biocide peptides the ability to decrease the line tension and to self-assemble in paucimolecular particles with high positive curvature.154 Although the origin of pore stability is not elucidated, it appears clear that non-specific parameters as used in eqn (2) should capture the main features of synthetic pores, irrespective of the molecular arrangements and possible polydispersity of the macromolecular inclusions.
We focus now on the case of tight attractions occurring in the polar part of the membranes, on the hydrophilic heads of the lipids. As pointed in section 1, coulombic attraction with charged lipid heads is a common feature of such a case, both with synthetic polymers and proteins or peptides. The electrostatic affinity of peptide and proteins for oppositely charged lipids have been analyzed in great details showing that multicharged peptides may trigger sequestration of lipids and lateral domain formation (e.g. by McLaughlin et al.).59,155,156 The organization in the apolar region though not directly affected may betray the modification in polar headgroups, since a relative increase of head–head attraction compared to the thermal energy favours the gel phase of the lipid layer, and more generally triggers phase transitions in membranes. The neutralization of ionic lipids with polymers of opposite charge induces accordingly a significant rise in the temperature of fluid–gel transition. Experimental signatures of the conversion of a fluid state to a patchy solid state have been found since 1978 in poly(lysine)-coated small vesicles.157 Shifts in the melting temperature of faceted catanionic vesicles (mixture of anionic and cationic single-tailed surfactants) have been reported in the presence of an amphiphilic polyanion, but not with an hydrophilic polyanion.93,158,159 When anionic phospholipids membranes are mixed with a polycation, calorimetric measurements clearly demonstrate that the binding causes domains (so called “rafts”) of anionic lipids to form in the outer leaflet, and it is these domains that electrostatically bind the polymer (Fig. 9).50,60,160 It is possible to regulate the trend for clustering of the ionized lipids by using polyanions attached to a voluminous neutral block. An increase in the lateral pressure (cf. supra) due to the bulky neutral block balances the trend for anion–cation complexation.161 In other cases, the adsorption of a polyelectrolyte increases the local density of lipids with the opposite charge, which eventually generates complexes that are not in a lamellar phase. For example, hexagonal phases of DNA and cationic lipids have been extensively characterized162,163 but similar phases were also obtained with more flexible polyanions. Vivares and Ramos164 have shown that the formation of such dense complexes stretches the membrane of a multilamellar vesicle (made of didodecyl dimethyl ammonium cations) beyond the critical tension that can open a pore. Further complexation then rapidly peels off the stack of membranes, one by one.
 | ||
| Fig. 9 Rigid domains formed by coulombic association of a polyelectrolyte on a fluid membrane. Ionized lipids are concentrated in the vicinity of the adsorbed chain(s) by lateral diffusion, and in some cases translocation. | ||
Other interactions in the polar region that are not of a coulombic nature also trigger similar formation of domains and phase transitions. Hydrogen bonds with the phosphorylcholine heads and/or dehydration are presumably involved in the shift of fluid–gel temperature achieved in the presence of poly(methacrylic acid).165Poly(2-ethylacrylate) adsorbed on small lipid vesicles upon a decrease of pH (i.e. under a partially acid form) triggers the formation of domains with a shifted melting temperature as revealed by the presence of two sharp peaks in differential scanning calorimetry.166 We have recently demonstrated that a rise in the melting temperature by almost 20 °C can be achieved by association with hydrophobically modified polyacrylic acid. The domains consisting of fluorescent and non-fluorescent regions were observed by optical microscopy in giant vesicles.132 Their melting above 25 °C, and several hours of reconstitution at 8 °C was also confirmed by the same technique with various fluorophores-labelled lipids (Fig. 10).167
 | ||
| Fig. 10 Polymer-induced domains on egg-PC giant unilamellar vesicles (a) upper view of a faceted vesicle with fluorescent lipid probes segregated in the fluid regions; (b) diametral view showing the faceted shape. (reprinted with permission from ref. 132, copyright 2002, American Chemical Society) | ||
Lateral head–head interactions play an essential role in the homogeneity of mixed membranes that contain different lipids. In the simple case of binary mixtures of lipids, segregation effects and clustering have been triggered by coupling lipid head groups with bulky hydrophilic moieties.18 The lateral diffusivity of lipids enables rapid variation of the local composition, which in turn switches cooperative transition and formation of “raft-like” domains. For instance, the formation of highly charged patches efficiently strengthen the binding of polyelectrolytes; at low average charge densities, it has been accordingly reported that only fluid liposomes were able to form an association with polymers of the opposite charge, whereas below the temperature of fluid–gel transition, no association took place.168 The growth of a polymer-induced domain has also been proposed as a generic driving force for fusion between two vesicles brought into contact.169 When depletion forces have expelled the polymer out of the contact, the adsorption and domain formation at the opposite pole of a vesicle would attract lipids of the outer leaflets (marangoni flow) and weaken lipid–lipid cohesion in the contact zone. Profound change in membrane features are expected as a consequence of patch and domain formation and shape modification. Lipowsky and Dimova have formalized the geometric and energetic constraints applied to a vesicle whose membrane becomes laterally heterogeneous.170 For instance, when a giant vesicle develops facets, the fluid part of the membrane develops tension in order to accommodate the shape variation at constant inner volume. In vesicles initially close to a spherical shape, the increase of tension would stop the phase separation (frustration) and favour multiple faceting. On polymer-coated vesicles, the formation of 4–6 facets was the most common feature (Fig. 10).
An intriguing effect observed in polymer-controlled domains was the strong inter-leaflet coupling of lipid composition.132 Fluorescent-labelled lipids are similarly and simultaneously depleted from both the inner and outer leaflet by the growth of label-enriched facets, despite the fact that the polymer did not cross the membrane.31 Transbilayer coupling drives the co-location of domains formed in both leaflets of a giant vesicle.171 Similar coupling, so-called “domain registration”, is observed on polymer-tethered membranes, as well as in fluid membranes (liquid ordered–liquid disordered phase separation in the presence of cholesterol).172 Through building asymmetric bilayers, Naumann et al. have shown in this system the absence of flip-flop and domain induction in the upper leaflet by domains present in the lower leaflet. The transversal coupling occurs even if the upper composition should not be thermodynamically biphasic. The only requirement for transversal coupling was the possibility for domains to diffuse freely in both leaflets as suggested by the loss of coupling when the lipids interact with a glassy substrate (across a thin polymer tether).172 As a possible origin of strong inter-leaflet coupling, even in pure lipid layers, Lipowsky and Dimova suggest that a transverse bi-phasic organization would display a transverse gradient of the packing density of lipids, which creates tension and a driving force for co-localization.170
Conclusion
By simple mixing in solution, synthetic macromolecules and lipid membranes form associations whose properties alloy those of both partners. Owing to the resources of polymer chemistry, the variety of combinations have presumably not been explored up to now. This review has selected model systems that have recently brought a clearer understanding of the impact of association strength, type (hydrophobic, hydrogen bonds, ion bridges) and to a lesser extent the polymer structure (density of stickers for instance). Polymers were mostly used a decade ago as a protective layer against coagulation and adsorption, as a conventional extension of the applications of polymers as surface modifiers and dispersing agents. Other important polymer-specific applications in solution include the control of flow, visco-elastic properties, and responsiveness to small variations of solvent conditions. These properties are now promising tools to tune and control lipid membranes and to trigger biomimetic responses, including budding and invaginations, formation of lateral patches, or selective channels. The critical sensitivity of some macromolecular chains to solvent quality, pH, temperature, or light stimuli has opened new ways to specifically destabilize and break membranes, artificial ones as well as living cells. At variance with the effect of molecular additives, macromolecules most typically can affect the membrane mechanical properties with very few direct interactions on the lipids (a few mol% of anchors relative to the lipid). Nevertheless, the lateral pressure developed by macromolecular hydrophilic segments can markedly modify the bending and rigidity. Further developments in this direction will certainly consider visco-elastic properties of polymer layers and the complex coupling with other dynamic responses of membranes and liposomes. Compartmentalization and gelation in vesicles are also two recent topics with a promising future for the design of more complex, though well-controlled biomimetic assemblies.References
- M. Zasloff, Nature, 2002, 415, 389–395 CrossRef CAS.
- A. L. Sisson, M. R. Shah, S. Bhosale and S. Matile, Chem. Soc. Rev., 2006, 35, 1269–1286 RSC.
- A. N. Lukyanov and V. P. Torchilin, Adv. Drug Delivery Rev., 2004, 56, 1273–1289 CrossRef CAS.
- M. Malmsten, Soft Matter, 2006, 2, 760–769 RSC.
- V. Torchilin, Drug Discovery Today, 2003, 6, 259–265 CrossRef.
- Th M. Allen and P. Cullis, Science, 2004, 303, 1818–1822 CrossRef CAS.
- O. V. Gerasimov, J. A. Boomer, M. M. Qualls and D. H. Thompson, Adv. Drug Delivery Rev., 1999, 38, 317–338 CrossRef CAS.
- M. A. Yessine and J. C. Leroux, Adv. Drug Delivery Rev., 2004, 56, 999–1021 CrossRef CAS.
- V. Nikolov, R. Lipowsky and R. Dimova, Biophys. J., 2007, 92, 4356–4368 CrossRef CAS.
- C. Hiergeist and R. Lipowsky, J. Phys. II, 1996, 6, 1465–1481 CrossRef.
- D. Marsh, Biophys. J., 2001, 81, 2154–2162 CrossRef CAS.
- E. Helfer, S. Harlepp, L. Bourdieu, J. Robert, F. C. MacKintosh and D. Chatenay, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2001, 6302 Search PubMed , 021904.
- J. Simon, M. Kuhner, H. Ringsdorf and E. Sackmann, Chem. Phys. Lipids, 1995, 76, 241–258 CrossRef CAS.
- M. A. Mader, V. Vitkova, M. Abkarian, A. Viallat and T. Podgorski, Eur. Phys. J. E, 2006, 19, 389–397 CrossRef CAS.
- S. L. Li and A. F. Palmer, Langmuir, 2004, 20, 7917–7925 CrossRef CAS.
- A. F. Palmer, P. Wingert and J. Nickels, Biophys. J., 2003, 85, 1233–1247 CrossRef CAS.
- G. Krishna, J. Schulte, B. A. Cornell, R. J. Pace and P. D. Osman, Langmuir, 2003, 19, 2294–2305 CrossRef CAS.
- W. H. Binder, V. Barragan and F. M. Menger, Angew. Chem., Int. Ed., 2003, 42, 5802–5827 CrossRef CAS.
- A. A. Yaroslavov, N. S. Melik-Nubarov and F. M. Menger, Acc. Chem. Res., 2006, 39, 702–710 CrossRef CAS.
- J. L. Thomas and D. A. Tirrell, J. Controlled Release, 2000, 67, 203–209 CrossRef CAS.
- F. Vial, A. G. Oukhaled, L. Auvray and C. Tribet, Soft Matter, 2007, 1, 75–78 RSC.
- H. Ringsdorf, B. Schlarb and J. Venzmer, Angew. Chem., Int. Ed. Engl., 1988, 27, 113–158 CrossRef.
- H. M. Chen, V. Torchilin and R. Langer, J. Controlled Release, 1996, 42, 263–272 CrossRef CAS.
- V. P. Torchilin and V. S. Trubetskoy, Adv. Drug Delivery Rev., 1995, 16, 141–155 CrossRef CAS.
- A. Mecke, C. Dittrich and W. Meier, Soft Matter, 2006, 2, 751–759 RSC.
- L. Bromberg, in Handbook of Surfaces and Interfaces of Materials, ed. H.S. Nalwa, Academic Press, New York, 2001, vol. 4, ch. 7, pp. 369–404 Search PubMed.
- G. J. Fleer, M. A. Cohen Stuart, J. M. Scheutjens, T. Cosgrove and B. Vincent, Polymers at interfaces, Chapman & Hall, London, 1993 Search PubMed.
- J. Y. Wong, J. Majewski, M. Seitz, C. K. Park, J. N. Israelachvili and G. S. Smith, Biophys. J., 1999, 77, 1445–1457 CrossRef CAS.
- E. T. Castellana and P. S. Cremer, Surf. Sci. Rep., 2006, 61, 429–444 CrossRef CAS.
- M. Merzlyakov, E. Li, I. Gitsov and K. Hristova, Langmuir, 2006, 22, 10145–10151 CrossRef CAS.
- F. Vial, S. Rahbi and C. Tribet, Langmuir, 2005, 21, 853–862 CrossRef CAS.
- D. Marsh, R. Bartucci and L. Sportelli, Biochim. Biophys. Acta, 2003, 1615, 33–59 CAS.
- J. M. Sanderson, Org. Biomol. Chem., 2005, 3, 201–212 RSC.
- J. M. Boon and B. D. Smith, J. Am. Chem. Soc., 2001, 123, 6221–6226 CrossRef CAS.
- A. A. Yaroslavov, E. G. Yaroslavova, A. A. Rakhnyanskaya, F. M. Menger and V. A. Kabanov, Colloids Surf., B, 1999, 16, 29–43 CrossRef CAS.
- I. Porcar, R. Garcia, V. Soria and A. Campos, Polymer, 1997, 38, 3545–3552 CrossRef CAS.
- C. Gourier, F. Pincet, E. Perez, Y. M. Zhang, Z. Y. Zhu, J. M. Mallet and P. Sinay, Angew. Chem., Int. Ed., 2005, 44, 1683–1687 CrossRef CAS.
- M. Mizusaki, Y. Morishima and F. M. Winnik, Polymer, 2001, 42, 5615–5624 CrossRef CAS.
- A. Polozova and F. M. Winnik, Biochim. Biophys. Acta, 1997, 1326, 213–224 CAS.
- J. N. Israelachvili, Intermolecular and Surface Forces, Academic Press, New York, 1992 Search PubMed.
- J. L. Thomas, K. A. Borden and D. A. Tirrell, Macromolecules, 1996, 29, 2570–2576 CrossRef CAS.
- B. Ceh, M. Winterhalter, P. M. Frederik, J. J. Vallner and D. Lasic, Adv. Drug Delivery Rev., 1997, 24, 165–177 CrossRef CAS.
- D. T. Auguste, R. K. Prud'homme, P. L. Ahl, P. Meers and J. Kohn, Biochim. Biophys. Acta, 2003, 1616, 184–195 CAS.
- S. Beugin-Deroo, M. Ollivon and S. Lesieur, Colloids Surf. Sci., 1998, 202(2), 324–333 Search PubMed.
- A. Polozova and F. M. Winnik, Langmuir, 1999, 15, 4222–4229 CrossRef CAS.
- K. Kawakami, Y. Nishihara and K. Hirano, J. Phys. Chem. B, 2001, 105, 2374–2385 CrossRef CAS.
- J. M. Sanderson and E. J. Whelan, Phys. Chem. Chem. Phys., 2004, 6, 1012–1017 RSC.
- L. Zhang, T. Peng, S. X. Cheng and R. X. Zhuo, J. Phys. Chem. B, 2004, 108, 7763–7770 CrossRef CAS.
- N. O. Kozlova, I. B. Bruskovskaya, I. B. Okuneva, N. S. Melik-Nubarov, A. A. Yaroslavov, V. A. Kabanov and F. M. Menger, Biochim. Biophys. Acta, 2001, 1514, 139–151 CAS.
- T. K. Bronich, S. V. Solomatin, A. A. Yaroslavov, A. Eisenberg, V. A. Kabanov and A. V. Kabanov, Langmuir, 2000, 16, 4877–4881 CrossRef CAS.
- M. C. Woodle and P. Scaria, Curr. Opin. Colloid Interface Sci., 2001, 6, 78–84 CrossRef CAS.
- O. Boussif, F. Lezoualch, M. A. Zanta, M. D. Mergny, D. Scherman, B. Demeneix and J. P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7297–7301 CAS.
- W. T. Godbey and A. G. Mikos, J. Controlled Release, 2001, 72, 115–125 CrossRef CAS.
- J. F. Tan, T. A. Hatton, K. C. Tam and H. P. Too, Biomacromolecules, 2007, 8, 448–454 CrossRef CAS.
- M. I. Angelova, N. Hristova and I. Tsoneva, Eur. Biophys. J. Biophys. Lett., 1999, 28, 142–150 Search PubMed.
- A. Mecke, I. J. Majoros, A. K. Patri, J. R. Baker, M. M. B. Holl and B. G. Orr, Langmuir, 2005, 21, 10348–10354 CrossRef CAS.
- M. Baciu, S. C. Sebai, O. Ces, X. Mulet, J. A. Clarke, G. C. Shearman, R. V. Law, R. H. Templer, C. Plisson, C. A. Parker and A. Gee, Philos. Trans. R. Soc. London, Ser. A: Math., Phys. Eng. Sci., 2006, 364, 2597–2614 Search PubMed.
- D. Murray, A. Arbuzova, G. Hangyas-Mihalyne, A. Gambhir, N. Ben-Tal, B. Honig and S. McLaughlin, Biophys. J., 1999, 77, 3176–3188 CrossRef CAS.
- N. Ben-Tal, B. Honig, C. Miller and S. McLaughlin, Biophys. J., 1997, 73, 1717–1727 CrossRef CAS.
- V. A. Kabanov and A. A. Yaroslavov, J. Controlled Release, 2002, 78, 267–271 CrossRef CAS.
- C. Delajon, T. Gutberlet, R. Steitz, H. Mohwald and R. Krastev, langmuir, 2005, 21, 8509–8514 CrossRef CAS.
- S. Moya, E. Donath, G. B. Sukhorukov, M. Auch, H. Baumler, H. Lichtenfeld and H. Mohwald, Macromolecules, 2000, 33, 4538–4544 CrossRef CAS.
- L. Y. Wang, M. Schonhoff and H. Mohwald, J. Phys. Chem. B, 2002, 106, 9135–9142 CrossRef CAS.
- L. Ramos, M. Schonhoff, Y. Luan and H. Mohwald, Colloids Surf., A, 2007, 303, 79–88 CrossRef CAS.
- D. Huster and K. Arnold, Biophys. J., 1998, 75, 909–916 CrossRef CAS.
- O. Zschornig, W. Richter, G. Paasche and K. Arnold, Colloid Polym. Sci., 2000, 278, 637–646 Search PubMed.
- N. Sakai and S. Matile, J. Am. Chem. Soc., 2003, 125, 14348–14356 CrossRef CAS.
- E. P. Holowka, V. Z. Sun, D. T. Kamei and T. J. Deming, Nat. Mater., 2007, 6, 52–57 CrossRef CAS.
- S. Das, U. D. Lengweiler, D. Seebach and R. N. Reusch, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 9075–9079 CrossRef CAS.
- S. Das, P. Kurcok, Z. Jedlinski and R. N. Reusch, Macromolecules, 1999, 32, 8781–8785 CrossRef CAS.
- S. Das and R. N. Reusch, Biochemistry, 2001, 40, 2075–2079 CrossRef CAS.
- T. Demina, I. Grozdova, O. Krylova, A. Zhirnov, V. Istratov, H. Frey, H. Kautz and N. Melik-Nubarov, Biochemistry, 2005, 44, 4042–4054 CrossRef CAS.
- P. Chandaroy, A. Sen, P. Alexandridis and S. W. Hui, Biochim. Biophys. Acta, 2002, 1559, 32–42 CrossRef CAS.
- M. Tanaka and E. Sackmann, Nature, 2005, 437, 656–663 CrossRef CAS.
- D. Anrather, M. Smetazko, M. Saba, Y. Alguel and T. Schalkhammer, J. Nanosci. Nanotechnol., 2004, 4, 1–22 CrossRef CAS.
- V. L. B. Braach Maksvytis, B. A. Cornell, L. G. King, P. D. J. Osman, R. J. Pace, B. Raguse and L. Wieczorek, Biophys. J., 1997, 72 TH288-TH288 B. A. Cornell, V. L. B. Braach Maksvytis, L. G. King, P. D. J. Osman, B. Raguse, L. Wieczorek and R. J. Pace, Nature, 1997, 387, 580–583 Search PubMed.
- O. Purrucker, A. Fortig, R. Jordan and M. Tanaka, ChemPhysChem, 2004, 5, 327–335 CrossRef CAS.
- T. Baumgart and A. Offenhausser, Langmuir, 2003, 19, 1730–1737 CrossRef CAS.
- P. Theato and R. Zentel, Langmuir, 2000, 16, 1801–1805 CrossRef CAS.
- M. L. Wagner and L. K. Tamm, Biophys. J., 2000, 79(3), 1400–1414 CrossRef CAS.
- F. Albertorio, A. J. Diaz, T. L. Yang, V. A. Chapa, S. Kataoka, E. T. Castellana and P. S. Cremer, Langmuir, 2005, 21, 7476–7482 CrossRef CAS.
- N. Fang, W. J. Tan, K. W. Leong, H. Q. Mao and V. Chan, Colloids Surf., B, 2005, 42, 245–252 CrossRef CAS.
- P. Theato and R. Zentel, J. Colloid Interface Sci., 2003, 268, 258–262 CrossRef CAS.
- J. J. Benkoski, A. Jesorka, M. Edvardsson and F. Hook, Soft Matter, 2006, 2, 710–715 RSC.
- M. A. Deverall, E. Gindl, E. K. Sinner, H. Besir, J. Ruehe, M. J. Saxton and C. A. Naumann, Biophys. J., 2005, 88, 1875–1886 CAS.
- E. A. Smith, J. W. Coym, S. M. Cowell, T. Tokimoto, V. J. Hruby, H. I. Yamamura and M. J. Wirth, Langmuir, 2005, 21, 9644–9650 CrossRef CAS.
- A. Fortig, R. Jordan, K. Graf, G. Schiavon, O. Purrucker and M. Tanaka, Macromol. Symp., 2004, 210, 329–338 CrossRef.
- S. Granick, J. Zhao, A. F. Xie and S. C. Bae, Macromol. Symp., 2003, 201, 89–94 CrossRef CAS.
- M. A. Deverall, E. Gindl, E. K. Sinner, H. Besir, J. Ruehe, M. J. Saxton and C. A. Naumann, Biophys. J., 2005, 88, 1875–1886 CAS.
- H. Hayashi, K. Kono and T. Takagishi, Bioconjugate Chem., 1998, 9, 382–389 CrossRef CAS.
- E. Ruel-Gariepy, G. Leclair, P. Hildgen, A. Gupta and J. C. Leroux, J. Controlled Release, 2002, 82, 373–383 CrossRef CAS.
- H. D. Han, T. W. Kim, B. C. Shin and H. S. Choi, Macromol. Res., 2005, 13, 54–61 Search PubMed.
- F. E. Antunes, E. F. Marques, R. Gomes, K. Thuresson, B. Lindman and M. G. Miguel, langmuir, 2004, 20, 4645–4656.
- H. S. Ashbaugh, K. Boon and R. K. Prud'homme, Colloid Polym. Sci., 2002, 280, 783–788 Search PubMed.
- K. Loyen, I. Iliopoulos, R. Audebert and U. Olsson, Langmuir, 1995, 11, 1053–1056 CrossRef CAS.
- J.-H. Lee, J. P. Gustin, T. Chen, G. F. Payne and S. R. Raghavan, Langmuir, 2005, 21, 26–33 CrossRef CAS.
- Y. Okumura, H. Mizushima and J. Sunamoto, J. Colloid Interface Sci., 2007, 307, 296–299 CrossRef CAS.
- M. A. Khaleque, Y. Okumura, S. Yabushita and M. Mitani, Chem. Lett., 2003, 32, 416–417 CrossRef CAS; M. A. Khaleque, Y. Okumura, S. Yabushita and M. Mitani, Colloids Surf., B, 2004, 37, 35–42 CrossRef CAS.
- M. Pantusa, R. Bartucci, D. Marsh and L. Sportelli, Biochim. Biophys. Acta, 2003, 1614, 165–170 CAS.
- D. Needham and D. H. Kim, Colloids Surf., B, 2000, 18, 183–195 CrossRef CAS.
- T. Auth and G. Gompper, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2003, 68, 051801 Search PubMed.
- T. Auth and G. Gompper, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2005, 72, 031904 Search PubMed.
- F. Castro-Roman, G. Porte and C. Ligoure, Langmuir, 2001, 17, 5045–5058 CrossRef CAS.
- P. L. Hansen, J. A. Cohen and R. Podgornik, V.A. Parsegian, Biophys. J., 2003, 84, 350–355 Search PubMed.
- F. F. Rossetti, I. Reviakine, G. Csucs, F. Assi, J. Voros and M. Textor, Biophys. J., 2004, 87, 1711–1721 CrossRef CAS.
- D. W. Pack, A. S. Hoffman, S. Pun and P. S. Stayton, Nat. Rev. Drug Discovery, 2005, 4, 581–593 CrossRef CAS.
- A. S. Hoffman, P. S. Stayton, O. Press, N. Murthy, C. A. Lackey, C. Cheung, F. Black, J. Campbell, N. Fausto, T. R. Kyriakides and P. Bornstein, Polym. Adv. Technol., 2002, 13, 992–999 CrossRef CAS.
- E. Roux, M. Lafleur, E. Lataste, P. Moreau and J. C. Leroux, Biomacromolecules, 2003, 4, 240–248 CrossRef CAS.
- T. Chen, D. McIntosh, Y. H. He, J. Kim, D. A. Tirrell, P. Scherrer, D. B. Fenske, A. P. Sandhu and P. R. Cullis, Mol. Membr. Biol., 2004, 21, 385–393 CrossRef CAS.
- H. Ringsdorf, E. Sackmann, J. Simon and F. M. Winnik, Biochim. Biophys. Acta, 1993, 1153, 335–344 CAS.
- J. C. Kim and J. D. Kim, Colloids Surf., B, 2002, 24, 45–52 CrossRef CAS.
- M. A. Firestone and S. Seifert, Biomacromolecules, 2005, 6, 2678–2687 CrossRef CAS.
- A. C. Couffin-Hoarau and J. C. Leroux, Biomacromolecules, 2004, 5, 2082–2087 CrossRef CAS.
- M. F. Francis, G. Dhara, F. M. Winnik and J. C. Leroux, Biomacromolecules, 2001, 2, 741–749 CrossRef CAS.
- J. C. Leroux, E. Roux, D. Le Garrec, K. L. Hong and D. C. Drummond, J. Controlled Release, 2001, 72, 71–84 CrossRef CAS.
- V. Bulmus, M. Woodward, L. Lin, N. Murthy, P. Stayton and A. Hoffman, J. Controlled Release, 2003, 93, 105–120 CrossRef CAS.
- S. M. Henry, M. E. H. El-Sayed, C. M. Pirie, A. S. Hoffman and P. S. Stayton, Biomacromolecules, 2006, 7, 2407–2414 CrossRef CAS.
- C. A. Lackey, N. Murthy, O. W. Press, D. A. Tirrell, A. S. Hoffman and P. S. Stayton, Bioconjugate Chem., 1999, 10, 401–405 CrossRef CAS.
- M. E. Eccleston, M. Kuiper, F. M. Gilchrist and N. K. H. Slater, J. Controlled Release, 2000, 69, 297–307 CrossRef CAS.
- C. Kusonwiriyawong, P. van de Wetering, J. A. Hubbell, H. P. Merkle and E. Walter, Eur. J. Pharm. Biopharm., 2003, 56, 237–246 CrossRef CAS.
- N. Bergstrand and K. Edwards, J. Colloid Interface Sci., 2004, 276, 400–407 CrossRef CAS.
- K. Kostarelos, T. F. Tadros and P. F. Luckham, Langmuir, 1999, 15, 369–376 CrossRef CAS.
- J. K. Ferri, R. Miller and A. V. Makievski, Colloids Surf., A, 2005, 261, 39–48 CrossRef CAS.
- C. Ladaviere, M. Toustou, T. Gulik-Krzywicki and C. Tribet, J. Colloid Interface Sci., 2001, 241, 178–187 CrossRef CAS.
- J. L. Thomas and D. A. Tirrell, Acc. Chem. Res., 1992, 25, 336–342 CrossRef CAS.
- A. Krabi and M. A. Cohen Stuart, Macromolecules, 1998, 31, 1285–1291 CrossRef CAS.
- J. G. Gobel, N. A. M. Besseling, M. A. Cohen Stuart and C. Poncet, J. Colloid Interface Sci., 1999, 209, 129–135 CrossRef CAS.
- J. Simon, M. Kuhner, H. Ringsdorf and E. Sackmann, Chem. Phys. Lipids, 1995, 76, 241–258 CrossRef CAS.
- H. Miyata and K. Kinosita, Biophys. J., 1994, 67, 922–928 CrossRef CAS.
- H. Miyata, S. Nishiyama, K. Akashi and K. Kinosita, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 2048–2053 CrossRef CAS.
- I. Tsafrir, Y. Caspi, M. A. Guedeau-Boudeville, T. Arzi and J. Stavans, Phys. Rev. Lett., 2003, 91, 138102 CrossRef.
- C. Ladaviere, C. Tribet and S. Cribier, Langmuir, 2002, 18, 7320–7327 CrossRef CAS.
- I. Tsafrir, D. Sagi, T. Arzi, M. A. Guedeau-Boudeville, V. Frette, D. Kandel and J. Stavans, Phys. Rev. Lett., 2001, 86, 1138–1141 CrossRef CAS.
- V. Frette, I. Tsafrir, M. A. Guedeau-Boudeville, L. Jullien, D. Kandel and J. Stavans, Phys. Rev. Lett., 1999, 83(12), 2465–2468 CrossRef CAS.
- Y. W. Kim and W. Sung, Europhys. Lett., 1999, 47, 292–297 CrossRef CAS.
- J. Stavans, Physica A, 2002, 306, 368–375 CrossRef CAS.
- T. Bickel, C. Marques and C. Jeppesen, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Interdiscip. Top., 2000, 62, 1124–1127 Search PubMed.
- M. Breidenich, R. R. Netz and R. Lipowsky, Europhysics Letters, 2000, 49, 431–437 Search PubMed.
- T. Bickel, C. Jeppesen and C. M. Marques, European Physical Journal E, 2001, 4, 33–43 Search PubMed.
- A. Nicolas and B. Fourcade, European Physical Journal E, 2003, 10, 355–367 Search PubMed.
- R. Lipowsky, Europhys Letters, 1995, 30, 197–202 Search PubMed.
- J. Wang, K. Guo, F. Qiu, H. Zhang and Y. Yang, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2005, 71(4), 041908 Search PubMed.
- M. Rovira-Bru, D. H. Thompson and I. Szleifer, Biophys. J., 2002, 83, 2419–2439 CrossRef CAS.
- R. Joannic, L. Auvray and D. D. Lasic, Phys. Rev. Lett., 1997, 78(17), 3402–3405 CrossRef CAS.
- N. I. Hristova, M. I. Angelova and I. Tsoneva, Bioelectrochemistry, 2002, 58, 65–73 CrossRef CAS.
- O. Krylova, N. Melik-Nubarov, G. Badun, A. Ksenofontov, F. Menger and A. Yaroslavov, Chem.–Eur. J., 2003, 9, 3930–3936 CrossRef CAS.
- S. Bhattacharya, R. A. Moss, H. Ringsdorf and J. Simon, Langmuir, 1997, 13, 1869–1872 CrossRef CAS.
- N. Rodriguez, J. Heuvingha, F. Pincet and S. Cribier, Biochim. Biophys. Acta, 2005, 1724, 281–287 CrossRef CAS.
- T. Tolpekina, W. den Otter and W. Briels, J. Chem. Phys., 2004, 121(16), 8014–8020 CrossRef CAS.
- O. Sandre, L. Moreaux and F. Brochard-Wyart, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 10591–10596 CrossRef CAS.
- M. Fosnaric, V. Kralj-Iglic, K. Bohinc, A. Iglic and S. May, J. Phys. Chem. B, 2003, 107, 12519–12526 CrossRef CAS.
- M. D. Betterton and M. P. Brenner, Phys. Rev. Lett., 1999, 82, 1598–1601 CrossRef CAS.
- M. T. Lee, F. Y. Chen and H. W. Huang, Biochemistry, 2004, 43, 3590–3599 CrossRef CAS.
- Y. Gohon, F. Giusti, C. Prata, D. Charvolin, P. Timmins, C. Ebel, C. Tribet and J.-L. Popot, Langmuir, 2006, 22, 1281–1290 CrossRef CAS.
- C. A. Buser, J. Kim, S. McLaughlin and R. M. Peitzsch, Mol. Membr. Biol., 1995, 12, 69–75 CAS.
- S. McLaughlin and D. Murray, Nature, 2005, 438, 605–611 CrossRef CAS.
- W. Hartmann and H.-J. Galla, Biochim. Biophys. Acta, 1978, 509, 474–490 CAS.
- F. E. Antunes, R. O. Brito, E. F. Marques, B. Lindman and M. Miguel, J. Phys. Chem. B, 2007, 111, 116–123 CrossRef CAS.
- O. Regev, E. F. Marques and A. Khan, Langmuir, 1999, 15, 642–645 CrossRef CAS.
- A. A. Yaroslavov, O. Y. Kuchenkova, I. B. Okuneva, N. S. Melik-Nubarov, N. O. Kozlova, V. I. Lobyshev, F. M. Menger and V. A. Kabanov, Biochim. Biophys. Acta, 2003, 1611, 44–54 CrossRef CAS.
- A. A. Pashkovskaya, E. P. Lukashev, P. E. Antonov, O. A. Finogenova, Y. A. Ermakov, N. S. Melik-Nubarov and Y. N. Antonenko, Biochim. Biophys. Acta, 2006, 1758, 1685–1695 CAS.
- C. R. Safinya, Curr. Opin. Struct. Biol., 2001, 11, 440–448 CrossRef CAS.
- I. Koltover, T. Salditt, J. O. Radler and C. R. Safinya, Science, 1998, 281, 78–81 CrossRef CAS.
- E. Vivares and R. L. Ramos, Langmuir, 2005, 21, 2185–2191 CrossRef CAS.
- Z. V. Feng, S. Granick and A. A. Gewirth, Langmuir, 2004, 20, 8796–8804 CrossRef CAS.
- U. K. Schroeder and D. A. Tirrell, Macromolecules, 1989, 22, 765–769 CrossRef CAS.
- F. Vial, Master Thesis, University Paris 6, 2003.
- A. Mecke, D. K. Lee, A. Ramamoorthy, B. G. Orr and M. M. B. Holl, Langmuir, 2005, 21, 8588–8590 CrossRef CAS.
- S. A. Safran, T. L. Kuhl and J. N. Israelachvili, Biophys. J., 2001, 81, 659–666 CrossRef CAS.
- R. Lipowsky and R. Dimova, J. Phys.: Condens. Matter, 2003, 15, S31–S45 CrossRef CAS.
- L. A. Bagatolli and E. Gratton, Biophys. J., 2000, 78, 290–305 CrossRef CAS.
- S. Garg, J. Ruhe, K. Ludtke, R. Jordan and C. A. Naumann, Biophys. J., 2007, 92, 1263–1270 CAS.
- J. C. Chung, J. L. Thomas, D. A. Tirrell and L. R. Opsahl-Ong, Macromolecules, 1996, 29, 4636–4641 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2008 |