Photophysical properties of non-homoconjugated 1,2-dihydro, 1,2,3,4-tetrahydro and 1,2,3,4,5,6-hexahydro-C60 derivatives
Khin K.
Chin
a,
Shih-Ching
Chuang
a,
Billy
Hernandez
b,
Luis M.
Campos
a,
Matthias
Selke
b,
Christopher S.
Foote†
a and
Miguel A.
Garcia-Garibay
*a
aDepartment of Chemistry and Biochemistry, University of California, Los Angeles, CA 90095-1569, USA. E-mail: mgg@chem.ucla.edu
bDepartment of Chemistry and Biochemistry, California State University, Los Angeles, CA 90032, USA
First published on 28th November 2007
Abstract
The photophysical properties of a novel series of non-homoconjugated 1,2-di-, 1,2,3,4-tetra-, and 1,2,3,4,5,6-hexasubstituted fullerenes (compounds 1, 2, and 3, respectively) have been systematically investigated. In this report, we examine the effect of substitution pattern of non-homoconjugated derivatized fullerenes on the ground state UV-Vis absorption, triplet state properties (lifetime, quantum yield, extinction coefficient), and singlet oxygen quantum yield. The non-homoconjugated fullerene derivatives 1–3 exhibit higher singlet oxygen quantum yield than analogous homoconjugated Bingel adducts with the same number of saturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds and exhibit decreasing quantum yield of singlet oxygen generation upon increasing the degree of functionalization on a single six member ring on the fullerene cage. This trend is similar for triplet quantum yield and triplet lifetime. The triplet extinction coefficient increases with functionalization. A detailed discussion comparing 1, 2, and 3 with functionalized homoconjugated systems and with other non-homoconjugated derivatives is presented.
C bonds and exhibit decreasing quantum yield of singlet oxygen generation upon increasing the degree of functionalization on a single six member ring on the fullerene cage. This trend is similar for triplet quantum yield and triplet lifetime. The triplet extinction coefficient increases with functionalization. A detailed discussion comparing 1, 2, and 3 with functionalized homoconjugated systems and with other non-homoconjugated derivatives is presented.
Introduction
The synthesis of multiply functionalized fullerene derivatives possessing a specific addition pattern has been one of the most challenging areas of fullerene chemistry.1 Elucidating the physical and photophysical properties of these isomers has been challenging due to difficult synthesis and rigorous chromatographic processes needed to obtain pure regioisomers. The separation of regioisomers synthesized by double additions of Bingel,2 Diels–Alder,3 Prato,4 [2+2],5nitrene,6 and carbene adducts of C607 requires extensive purification prior to the characterization of their physical properties.Our laboratories have been interested in understanding how the photophysical properties of fullerene derivatives are affected by their electronic structure. Although the photophysical properties of many fullerene derivatives have been reported,8 there are several structures that have not been synthetically available. The absence of these regioisomers has thus limited a thorough elucidation of the relationship between photophysical properties and electronic structure. For example, the C3v trisadduct derivative (all-cis-1) has only recently been synthetically available (Fig. 1)9,11 for its photophysical properties to be documented.10
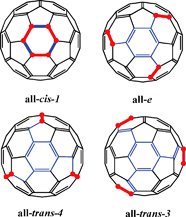 | ||
| Fig. 1 Trisadducts of C60 with C3 rotational axis. The sites of addition are indicated with a red bond and a dot. Different addition patterns carve out different π-chromophores. | ||
Recently, Rubin and coworkers demonstrated a stepwise synthetic approach to saturate three CC bonds within a single six member ring of C60 by a double Diels–Alder reaction followed by an intramolecular radical addition.11 Naturally, the perturbation caused by the saturation of three CC bonds of a single six member ring should affect the extent of π-conjugation and electronic properties of C60 (Fig. 2).
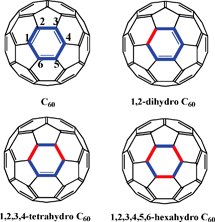 | ||
| Fig. 2 Saturation of a single six-member ring on C60. Derivatization at sequential positions on the six-member ring should affect the π-conjugation of C60 resulting in changes in photophysical properties. | ||
To date, most studies of fullerene derivatives aimed at elucidating the relation between electronic structure and photophysical properties have used homoconjugated Bingel adducts.
However, the dependence of structural topology on the photophysical properties for non-homoconjugated systems has not been well studied due to the lack of synthetically available examples that can be systematically correlated.12,18 Herein, we report triplet state properties and singlet oxygen photosensitization of a series of non-homoconjugated C60 derivatives with substitution at two, four, and all six carbons of a single six member ring.
Experimental
General methods
All reactions were performed under an argon atmosphere unless otherwise noted. Chemicals were used as purchased without further purification. Distilled solvents were dried as follows: THF from Na/benzophenone, and ODCB from CaH2. 1H and 13C NMR spectra were recorded using Avance 500 MHz spectrometers. FTIR spectra were recorded using a Perkin-Elmer Paragon 1000 spectrometer. UV-Vis spectra were recorded using HP 8452 and 8453 diode-array single beam spectrometers. HRMS of non-C60 products were obtained after ionizing through EI. HRMS and LRMS of C60 derivatives were obtained after ionizing through MALDI. Column chromatography was carried out on silica gel 40–63 µm from EMD; thin layer chromatography was carried on glass plates coated with silica gel 250 µm from Sorbent Technologies. HOMO/LUMO and HOMO–1/LUMO+1 orbitals were computed using AM1 method using MacSpartan. Compounds 1, 2, and 3 are synthesized according to a reported procedure.11Materials
2,3-Benzanthrancene (purity >99%), toluene (spectrophotometric grade), anthracene (purity >98%) and rubrene (98%) were purchased from Aldrich Chemical Company. Deuterated toluene was obtained from Cambridge Isotope Laboratories. C60 (>99.5%) was obtained from MER Corporation.13Perylene (98%) was obtained from Sigma Chemical Company. Benzo[a]pyrene (98%) was obtained from Fluka. All chemicals were used as received except for anthracene which was recrystallized from acetone prior to use.Measurements
Samples of 1, 2, or 3 were prepared in spectrophotometric grade toluene with an absorbance at the excitation wavelength of A355 ≈ 0.3. Solutions were purged with argon for 30 min. There was negligible change in the concentration of the solution after purging and was verified by noting only a slight (∼0.02) increase in absorbance at the excitation wavelength. UV-Vis absorption spectra were obtained using Hewlett-Packard 8453 and Beckman DU-650 spectrophotometers.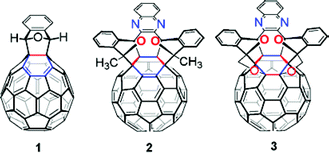
Time resolved measurements: triplet decay kinetics, triplet–triplet spectra, triplet–triplet extinction coefficients, triplet quantum yields, and triplet energies were obtained by transient absorption methods described previously.8f Briefly speaking, measurements were obtained by excitation of argon purged solutions at 355 nm using a frequency tripled Quanta Ray DCR-2 Nd:YAG laser (3–20 mJ pulse–1). Transient absorption was monitored using a probe beam from a Hanovia 100-W xenon lamp passed through a Jarrell-Ash 82-410 monochromator (250 µm slits) and detected using a Hamamatsu R928 PMT. Data output from the PMT was collected using a LeCroy 9350 oscilloscope coupled to a Macintosh G4 computer using Labview software. Kinetic curves were averaged over 30–70 laser pulses.
Triplet energies and triplet extinction coefficients were determined by energy transfer experiments. Energy transfer was observed by quenching the triplet state of 1, 2, and 3 and monitoring the decay kinetics using transient absorption. Solutions of adduct 1, 2, and 3 in toluene (A355 ≈ 0.3) were placed in 1 cm quartz cuvettes with a selected quencher at increasing concentrations. The solutions were purged under argon for 30 min and were measured under identical experimental conditions and laser power. The quenching rate constant was determined using a Stern–Volmer analysis where the transient absorption traces were curve fitted to a monoexponential decay using Igor Pro 3.1 software. Compound 1, 2, or 3 were excited at 355 nm in the presence of quenchers rubrene, benzo[a]pyrene, perylene, and 2,3-benzanthracene. The quenching rate of the triplet of 1, 2, or 3 were monitored at a local maximum in the triplet–triplet absorption at 365 nm. The absorbance (ΔOD) of the triplet of rubrene was measured at 490 nm (ET = 26.0 kcal mol–1, ΔεT-T (490) = 26000 M–1 cm–1) and at 670 nm for 1, 2, or 3 in experiments to determine the triplet molar absorption coefficient.
Singlet oxygen quantum yield was determined by measuring the singlet oxygen near infrared luminescence using a cryogenic germanium photodiode detector (model 403 HS; Applied Detector Corp., USA) cooled by liquid nitrogen.23 Air saturated samples of 1, 2, or 3 were dissolved in toluene-d8 and excited at either 355 nm or 532 nm using anNd:YAG laser (model MiniLaseII/10 Hz; New Wave Research Inc., USA), with pulse energies of 2–5 mJ pulse–1. Scattered laser light and other undesired radiation was eliminated using a series of filters, namely a Schott color glass filter (model RG850; cut-on 850 nm; Newport, USA) taped to the sapphire entrance of the detector. The port opening to the detector contains the remaining filters, a long wave pass filter (silicon filter model 10LWF∼1000; Newport, USA) which transmits in the range of 1100–2220 nm and blocks at 800–954 nm, and a band pass filter (model BP-1270-080-B*; CWL 1270 nm; Spectrogon, USA) with maximum transmission of 60% at 1270 nm. Singlet oxygen luminescence was measured orthogonal to laser excitation. The initial 1O2 intensity was extrapolated to t= 0. The data points of the initial 0–5 µs were not used due to electronic interference signals from the detector. Signals were digitized on a LeCroy 9350 CM 500 MHz oscilloscope and analyzed using Origin software. Decay traces were averaged over 10–40 laser pulses.
Results and discussion
UV-Vis spectroscopy
The ground state absorption spectra of derivatives 1, 2, and 3 reveal significant changes as compared to pristine C60. The intensity of the characteristic UV absorption peak at ca. 330 nm decreases and becomes more red-shifted with increasing substitution (Fig. 3). The three isobenzofuran derivatives 1, 2, and 3 also display very different absorption features between 420–650 nm when compared to that of C60. Increasing substitution from two to six carbons on a single six member ring also results in the appearance of a small absorption band at ∼430 nm. The non-homoconjugated derivatives 1, 2, and 3 show absorption peaks at 434, 428 and 429 nm, respectively.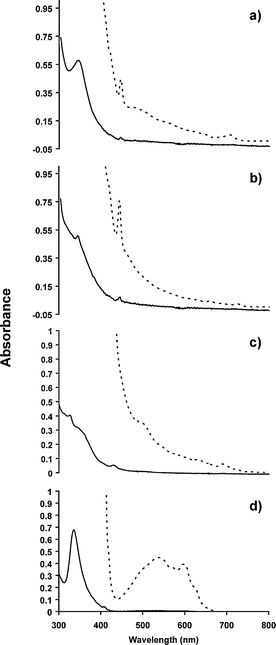 | ||
| Fig. 3 UV-Vis absorption spectra of C60 derivatives a) 1,2-dihydro- (1), b) 1,2,3,4-tetrahydro- (2), c) 1,2,3,4,5,6-hexahydro-isobenzofuran (3), and d) C60 recorded in toluene. | ||
The broad absorption band of C60 spanning from 430 to 660 nm becomes featureless upon derivatization. The overall spectral differences in compounds 1, 2 and 3 may result both from the progressive changes in the conjugation of the fullerene chromophore and the presence of a rigid and strained 1,4-epoxy naphthalene moiety.11,14 The absorption changes in 1, 2, 3 are similar to those of previously reported for non-homoconjugated pyrrolidinofullerene derivatives.12b In addition, derivatives, 1, 2, and 3 exhibit slight absorbance in the red portion of the spectrum. Since these compounds do not show any observable emission up to 900 nm at room temperature, or in glassy matrices at 77 K, the current study is restricted to the characterization of their triplet state photophysical properties by nanosecond laser flash photolysis.
Triplet state properties of compounds 1, 2, 3
Triplet–triplet absorption spectra
The triplet–triplet absorption spectra of the three fullerene derivatives recorded in toluene are shown in Fig. 4. The triplet–triplet absorption spectra were acquired with the laser flash photolysis setup described previously.8f The observed transients are attributed to the triplet state because they are readily quenched by triplet quenchers in argon purged solutions (vide infra) and by oxygen in air-saturated and oxygen-purged solutions.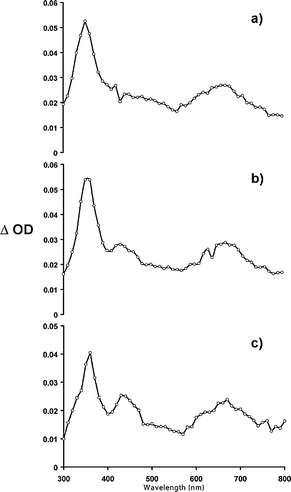 | ||
| Fig. 4 Triplet–triplet absorption spectrum of a) 1,2-dihydro-, b) 1,2,3,4-tetrahydro-, c) 1,2,3,4,5,6-hexahydro-isobenzofuran C60 derivatives (1, 2, and 3 respectively) recorded in toluene (λexc = 355 nm). | ||
The triplet–triplet absorption of all three derivatives show a broad absorption band near 670 nm that is blue shifted by about 50 nm with respect to that of C60. Non-homoconjugated derivatives 1, 2, and 3 also possess a weak absorption band at 430 nm. This band appears as a small shoulder for the 1,2-dihydro compound 1, but increases in intensity for the 1,2,3,4-tetrasubstituted derivative 2 and shows more pronounced absorption with subsequent functionalization exhibited by the 1,2,3,4,5,6-hexasubstituted 3. A much more intense peak which is a common absorption for the triplet state of C60, compounds 1–3, and other multiply functionalized fullerenes, appears at ca. 360 nm.
Triplet extinction coefficient and triplet quantum yield
The triplet extinction coefficient of 1, 2, and 3 were determined by energy transfer experiments15 using rubrene (Rub0, Scheme 1) as an energy acceptor.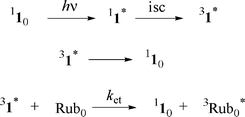 | ||
| Scheme 1 | ||
The rate of energy transfer was measured by monitoring the triplet–triplet absorption of a donor in the presence of various concentrations of quencher. The quenching rate was determined by measuring the changes in the rate of decay of the triplet state of the donor. Energy transfer was confirmed by observation of the triplet absorption of the quencher. As energy transfer is monitored by triplet–triplet absorption, the donor and acceptor must not have significant overlapping transient absorption at the observed wavelengths. Laser excitation at 355 nm of argon-purged toluene solutions of fullerene derivative 1 (A355 ∼ 0.3) results in the formation of the singlet excited state (11*), which efficiently undergoes intersystem crossing to form the triplet state (31*). Energy transfer from the triplet state of the fullerene derivative to rubrene, which has lower triplet energy, occurs by collisional quenching (Scheme 1).
The fullerene triplet (31*) was monitored at 670 nm and the rubrene triplet (3Rub*) was followed at 490 nm where 31* does not have significant triplet absorbance. Since rubrene has only negligible absorbance at the 355 nm excitation wavelength under the experimental conditions, the rubrene triplet (3Rub*) observed at 490 nm (ET = 26.0 kcal mol–1, ΔεT-T (490) = 26000 M–1 cm–1)16 can be solely attributed to energy transfer from each fullerene derivative. The triplet molar absorption coefficient of the fullerene derivatives is given by eqn (1) where ΔOD is the signal intensity of the triplet at the monitoring wavelengths. The triplet coefficient was corrected for incomplete energy transfer. The probability of energy transfer (Ptr) from 31* to rubrene (Rub0) was calculated using eqn (2), where [Rub] is the rubrene concentration, kD is the intrinsic decay rate constant of 1, and ket is the rate constant of energy transfer from 31* to rubrene.
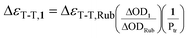 | (1) |
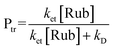 | (2) |
The triplet extinction coefficients of the non-homoconjugated systems increase with increasing saturation of the fullerene chromophore. The triplet coefficient of 1,2-dihydro compound 1, 14000 M–1 cm–1, is much higher than that of a previously studied homoconjugated 1,2-dihydrofullerene, whose coefficient is 4200 M–1 cm–1.17 Subsequent functionalizations gave a triplet coefficient of 19800 M–1 cm–1 for the 1,2,3,4-tetrahydro-fullerene 2 and an even higher value of 21600 M–1 cm–1 for the 1,2,3,4,5,6-hexaadduct 3.
Previous examples of homoconjugated and multiply functionalized fullerene derivatives have shown that the reported triplet extinction coefficients can vary widely, ranging from 4800 M–1 cm–1 to as high as 40042 M–1 cm–1. The triplet extinction coefficient is dependent on the number of substituents, substitution pattern, and type of substituents.18 The 1,2,3,4,5,6-hexaadduct 3 has a similar extinction coefficient as that of a tetrakis Bingel adduct, D2h-all-e-C60[C(COOEt)2]4 (20914 M–1 cm–1) and a hexakis Bingel adduct, Th-all-e-C60[C(COOEt)2]6 (20871 M–1 cm–1). In addition, the coefficient of 1,2-dihydrofullerene 1 is similar to that of a tris-substituted derivative, e-face, e-edge, trans-1-C60[C(COOEt)2]3 (15085 M–1 cm–1). This suggests that substitution by a non-homoconjugated addend has a greater effect on the triplet extinction coefficient of the fullerene chromophore than that of a homoconjugated addend (Bingel adduct). The results of the current study also indicate that for non-homoconjugated systems, the triplet extinction coefficient increases with sequential disruption of the fullerene core, in analogy to trends reported previously for homoconjugated systems.12a,17
The triplet quantum yields of fullerene derivatives 1, 2, and 3 were determined by the comparative method using C60 as a standard.19 Under identical experimental conditions and with optically matched solutions (A355 ∼ 0.3) of each derivative and C60, the triplet quantum yield (φT) was calculated (eqn (3)). ΔOD is the initial intensity of the triplet absorbance and ΔεT-T is the triplet extinction coefficient determined above, at 670 nm for the fullerene derivatives and 740 nm for C60.
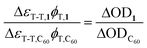 | (3) |
The triplet quantum yields of fullerene derivatives 1, 2, and 3 are shown in Table 1 and shows progressive changes with increasing disturbance in the conjugation of the fullerene chromophore. The quantum yield decreases with increasing saturation of the CC bonds in a six member ring. This is consistent with studies of homoconjugated fullerene derivative systems.17 The quantum yield of monoadduct 1 is similar to that of a mono-Bingel adduct C60[C(COOEt)2] (ΦT = 0.95). The 1,2,3,4-tetrahydro adduct 2 displays a higher triplet quantum yield than a bis-Bingel adduct e-C60[C(COOEt)2]2 (ΦT = 0.79). Hexaadduct 3 exhibits a similar quantum yield as a tris Bingel e-face, e-edge, trans-1-C60[C(COOEt)2]3 adduct (ΦT = 0.69). Overall, the increased perturbation in the π conjugation of a single six-membered ring in a non-homoconjugated system results in a consistent decrease in triplet quantum yield and is similar to previous reports for homoconjugated systems.12a,17
| Propertya | Compound 1 | Compound 2 | Compound 3i |
|---|---|---|---|
| a All experiments were conducted in spectrophotometric grade toluene or toluene-d8 at room temperature and excited at 355 nm or 532 nm with a concentration of 1, 2, or 3 where absorbance at the excitation wavelength is Aexc ≈ 0.3. Reported values are averages of 3 or more measurements. b Determined using the energy transfer method with rubrene as a reference.15 c In argon purged solutions and void of any quenchers. d Calculated using the comparison method with C60 as reference. e Determined in air saturated conditions with sample concentrations from absorbance (OD) = 0.1–0.5 using C60 as standard. f Determined in air saturated conditions with sample concentrations from absorbance (OD) = 0.05–0.2 using C60 as standard. g S Δ = ΦΔ/ΦT. SΔ is the fraction of the triplet state of 1, 2, or 3 that forms singlet oxygen by energy transfer (see text). h No observed quenching of singlet oxygen by 1, 2, or 3 up to 2.98 × 10–5 M. i Data taken from previous report.10 | |||
| E T/kcal mol–1 | 33 ≤ ET ≤ 38 | ||
| ε T /M–1 cm–1b | 14000 ± 1000 | 19800 ± 1000 | 21600 ± 1000 |
| τ T /µsc | 25 ± 1 | 22 ± 1 | 18 ± 1 |
| k O /M–1 s–1 | (1.7 ± 0.09) × 109 | (2.3 ± 0.2) × 109 | (1.56 ± 0.15) × 109 |
| Φ T d | 0.96 ± 0.02 | 0.89 ± 0.03 | 0.67 ± 0.03 |
| Φ 1O2 (355 nm)e | 0.95 ± 0.02 | 0.87 ± 0.03 | 0.65 ± 0.03 |
| Φ 1O2 (532 nm)f | 0.82 ± 0.02 | 0.70 ± 0.03 | 0.55 ± 0.03 |
| S Δ (355 nm)g | 0.99 | 0.98 | 0.97 |
| k q(1O2) | Not measurableh |
Triplet energies
The triplet energy of the series of fullerene derivatives was estimated using bracketing experiments by the energy transfer method detailed in the Experimental section. The relative triplet energy of each fullerene derivative can be estimated by using quenchers of different triplet energies and measuring the rates of energy transfer.15 The rate constants of quenching by various quenchers of the triplet state of each fullerene derivative are shown in Table 2. The triplet state of each derivative 1–3 is quenched at diffusion controlled rates by oxygen (ET = 22 kcal mol–1)15 in air and oxygen saturated solutions of toluene. Rubrene (ET = 26 kcal mol–1)15 and 2,3-benzanthracene (ET = 29 kcal mol–1)15 quench at slightly slower than diffusion controlled rates. Perylene (ET = 34 kcal mol–1)15 quenches the triplet of all three compounds 1–3 at significantly lower than diffusion controlled rates. This suggests that the approximate energies are close (∼0–2 kcal mol–1) to that of perylene. Benzo[a]pyrene (ET = 41.8 kcal mol–1)15 and anthracene (ET = 42.5 kcal mol–1)15 showed no quenching of any of the fullerene triplets. Reverse quenching experiments using each fullerene derivative as a quencher of the triplet of benzo[a]pyrene were conducted and gave results consistent with estimated triplet energies that were much lower than that of benzo[a]pyrene. From energy transfer experiments, we estimate that the triplet energies of the fullerene derivatives are similar to each other and close to that of perylene; i.e. 33 kcal mol–1 ≤ ET (fullerene derivatives 1–3) ≤ 38 kcal mol–1.| Quencher | E T/kcal mol–1 | 1 a | 2 a | 3 a |
|---|---|---|---|---|
| a k q (×1010 M–1 s–1). | ||||
| O2 | 22 | 1 | 1 | 1 |
| Rubrene | 26 | 0.46 | 0.51 | 0.58 |
| 2,3-Benzanthracene | 29 | 0.26 | 0.23 | 0.25 |
| Perylene | 35 | 0.006 | 0.027 | 0.05 |
| Benzo[a]pyrene | 42 | No quenching observed | ||
| Anthracene | 42 | No quenching observed | ||
The triplet energies of 1, 2, and 3 were also calculated by density functional theory at the B3LYP/6-31G*20 and B3LYP/6-31G**//B3LYP/3-21G level of theory (Table 3).21 The triplet energy values show a slight decrease upon consecutive saturation of double bonds on a single six-membered ring, from 36.2 kcal mol–1 to 35.2 kcal mol–1 and 33.6 kcal mol–1 for 1, 2, and 3, respectively.
| Level of theory | 1 | 2 | 3 |
|---|---|---|---|
| B3LYP/6-31G*//B3LYP/6-31G* | 36.2 | 35.2 | 33.6 |
| B3LYP/6-31G**//B3LYP/3-21G | 38.8 | 35.2 | 34.8 |
These values are comparable to the calculated triplet energy of C60 of 36.2 kcal mol–1, which has been experimentally determined to be 36.0 kcal mol–1.22 The calculated triplet energy values are consistent with those estimated in the energy transfer experiments, confirming that the triplet energies of fullerene derivatives 1–3 are within ca. 0–2 kcal mol–1 of the triplet energy of perylene and the parent C60.
Singlet oxygen quantum yield
This series of fullerene derivatives 1–3 gives us the opportunity to examine the effect of substitution pattern on the singlet oxygen quantum yield. Singlet oxygen is generated by energy transfer from the triplet state of each fullerene derivative to ground state molecular oxygen. The quantum yield of 1O2 generation (ΦΔ) can be expressed using the following equation.| ΦΔ = ΦTPT,O2fT,Δ | (4) |
C bond by a non-homoconjugated addend (compound 1, Φ1O2 (355 nm) = 0.95) is comparable to that of homoconjugated analogs.17 The second addition of a non-homoconjugated addend at the adjacent CC bond (cis-1 position) preserves more π-conjugation than the second addition of a homoconjugated addend at the e-position in a second aromatic ring, correlating with a larger quantum yield with compound 2 (Φ1O2 (355 nm) = 0.87) than with the homoconjugated derivative e-C60[C(CO2Me)2]2 (Φ1O2 (355 nm) = 0.79). This trend also applies to a third addition as compound 3 has a higher quantum yield for singlet oxygen generation than a tris-Bingel adduct e-face, e-edge, trans-1-C60[C(COOEt)2]3, which perturbs conjugation in three different benzene rings.17
In agreement with the systematic decrease in the triplet quantum yields as a function of π-conjugation detailed previously, consecutive disruption in the local conjugation of a single six-membered ring by one, two, and three double bonds on the fullerene cage consistently shows a small, but progressive decrease in singlet oxygen quantum yield. However, consecutive addition to a single isolated benzene ring produces a smaller effect than consecutive addition of homoconjugated adducts to separated benzene rings around the fullerene core.
Conclusions
We have reported a systematic study on the photophysics of the non-homoconjugated 1,2-dihydro, 1,2,3,4,-tetrahydro, 1,2,3,4,5,6-hexahydro fullerene derivatives. The synthetic availability for this series of adducts offers us the opportunity to carry out an insightful investigation of the photophysical properties of non-homoconjugated systems. Saturation of CC bonds by a non-homoconjugated addend at adjacent positions preserves more π-conjugation than the derivatives with delocalized addends. This is reflected in measurements of triplet energy, triplet quantum yield, and singlet oxygen quantum yield. Progress toward the understanding of the effects of electronic structure on the photophysical properties of non-homoconjugated systems is made in the current study.
Acknowledgements
This work was supported by NSF grants CHE0551938 and DMR0605688. B.H. and M.S. also gratefully acknowledge support from the NSF-PREM program (Grant number 03-564).Notes and references
- (a) S. Anderson, H. L. Anderson and J. K. M. Sanders, Expanding roles for templates in synthesis, Acc. Chem. Res., 1993, 26, 469–475 CrossRef CAS; (b) L. Isaacs, R. F. Haldimann and F. Diederich, Tether-directed remote functionalization of buckminsterfullerene: regiospecific hexaadduct formation, Angew. Chem., Int. Ed. Engl., 1994, 33, 2339–2342 CrossRef; (c) S. Sergeyev and F. Diederich, Regio- and stereoselective tether-directed remote functionalization of C60 with derivatives of the Tröger base, Angew. Chem., Int. Ed. Engl., 2004, 43, 1738–1740 CrossRef CAS; (d) I. Lamparth, C. Maichle-Mössner and A. Hirsch, Reversible template-directed activation of equatorial double bonds of the fullerene framework: regioselective direct synthesis, crystal structure, and aromatic properties of Th–C66(COOEt)12, Angew. Chem., Int. Ed. Engl., 1995, 34, 1607–1609 CrossRef CAS; (e) F. Diederich and M. Gomez-Lopez, Supramolecular fullerene chemistry, Chem. Soc. Rev., 1999, 28, 263–277 RSC; (f) U. Reuther, T. Brandmüller, W. Donaubauer, F. Hampel and A. Hirsch, A highly regioselective approach to multiple adducts of C60 governed by strain minimization of macrocyclic malonate addends, Chem.–Eur. J., 2002, 8, 2261–2273 CrossRef CAS; (g) W. Qian and Y. Rubin, Convergent, regioselective synthesis of tetrakisfulleroids from C60, J. Org. Chem., 2002, 67, 7683–7687 CrossRef CAS; (h) C. Thilgen and F. Diederich, Tether-directed remote functionalization of fullerenes C60 and C70, C. R. Chim., 2006, 9, 868–880 CrossRef CAS.
- (a) A. Hirsch, I. Lamparth and H. R. Karfunkel, Fullerene chemistry in three dimensions: isolation of seven regioisomeric bisadducts and chiral trisadducts of C60and di(ethoxycarbonyl)methylene, Angew. Chem., Int. Ed. Engl., 1994, 33, 437–438 CrossRef; (b) C. Bingel, Cyclopropanierung von fullerenen, Chem. Ber., 1993, 126, 1957–1959 CrossRef CAS.
- (a) S. R. Wilson and Q. Lu, 2,6-Dimethoxyanthracene - a directing group for regioselective bisaddition to C60, Tetrahedron Lett., 1995, 36, 5707–5710 CrossRef CAS; (b) A. Duarte-Ruiz, T. Müller, K. Wurst and B. Kräutler, The bis-adducts of the [5,6]-fullerene C60 and anthracene, Tetrahedron, 2001, 57, 3709–3714 CrossRef CAS.
- (a) M. Maggini, G. Scorrano and M. Prato, Addition of azomethine ylides to C60: synthesis, characterization, and functionalization of fullerene pyrrolidines, J. Am. Chem. Soc., 1993, 115, 9798–9799 CrossRef CAS; (b) S. R. Wilson, Y. Wang, J. Cao and X. Tan, Amino acids as precursors for N-unsubstituted fulleropyrrolidine derivatives, Tetrahedron Lett., 1996, 37, 775–778 CrossRef CAS; (c) Q. Lu, D. I. Schuster and S. R. Wilson, Preparation and characterization of six bis(N-methylpyrrolidine)-C60 isomers: magnetic deshielding in isomeric bisadducts of C60, J. Org. Chem., 1996, 61, 4764–4768 CrossRef.
- (a) Y. Nakamura, N. Takano, T. Nishimura, E. Yashima, M. Sato, T. Kudo and N. Nishimura, First isolation and characterization of eight regioisomers for [60]fullerene-benzyne bisadducts, Org. Lett., 2001, 3, 1193–1196 CrossRef CAS; (b) F. Diederich and R. Kessinger, Templated regioselective and stereoselective synthesis in fullerene chemistry, Acc. Chem. Res., 1999, 32, 537–545 CrossRef CAS.
- (a) F. Djojo, A. Herzog, I. Lamparth, F. Hampel and A. Hirsch, Regiochemistry of two-fold additions to [6,6] bonds in C60: influence of the addend-independent cage distortion in 1,2-monoadducts, Chem.–Eur. J., 1996, 2, 1537–1547 CrossRef CAS; (b) G. Shick, A. Hirsch, H. Mauser and T. Clark, Opening and closure of the fullerene cage in cis-bisimino adducts of C60: The influence of the addition pattern and the addend, Chem.–Eur. J., 1996, 2, 935–943 CrossRef CAS.
- M. W. J. Beulen, J. A. Rivera, M. A. Herranz, B. Illescas, N. Martín and L. Echegoyen, Reductive electrochemistry of spiromethanofullerenes, J. Org. Chem., 2001, 66, 4393–4398 CrossRef CAS.
- (a) T. Hamano, K. Okuda, T. Mashino, M. Hirobe, K. Arakane, A. Ryu, S. Mashiko and T. Nagano, Singlet oxygen production from fullerene derivatives: effect of sequential functionalization of the fullerene core, Chem. Commun., 1997,(1), 21–22 RSC; (b) F. Prat, R. Stackow, R. Bernstein, W. Y. Qian, Y. Rubin and C. S. Foote, Triplet-state properties and singlet oxygen generation in a homologous series of functionalized fullerene derivatives, J. Phys. Chem. A, 1999, 103, 7230–7235 CrossRef CAS; (c) Y. Nakamura, M. Taki, S. Tobita, H. Shizuka, H. Yokoi, K. Ishiguro, Y. Sawaki and J. Nishimura, Photophysical properties of various regioisomers of [60]fullerene-o-quinodimethane bisadducts, J. Chem. Soc., Perkin Trans. 2, 1999, 1, 127–130 Search PubMed; (d) P. F. Coheur, D. A. dos Santos, J. Cornil, P. R. Birkett, J. Liévin, J. L. Brédas, D. R. M. Walton, R. Taylor, H. W. Kroto and R. Colin, Photophysical properties of hexa-functionalized C60 derivatives: Spectroscopic and quantum-chemical investigations, J. Chem. Phys., 2000, 112, 8555–8566 CrossRef CAS; (e) S. Foley, S. Bosi, C. Larroque, M. Prato, J. M. Janot and P. Seta, Photophysical properties of novel water soluble fullerene derivatives, Chem. Phys. Lett., 2001, 350, 198–205 CrossRef CAS; (f) J. W. Arbogast and C. S. Foote, Photophysical properties of C70, J. Am. Chem. Soc., 1991, 113, 8886–8889 CrossRef CAS.
- U. Reuther, T. Brandmüller, W. Donaubauer, F. Hampel and A. Hirsch, A highly regioselective approach to multiple adducts of C60 governed by strain minimization of macrocyclic malonate addends, Chem.–Eur. J., 2002, 8, 2261–2273 CrossRef CAS.
- K. K. Chin, S.-C. Chuang, B. Hernandez, M. Selke, C. S. Foote and M. A. Garcia-Garibay, Photophysical properties of a 1,2,3,4,5,6-hexasubstituted fullerene derivative, J. Phys. Chem. A, 2006, 110, 13662–13666 CrossRef CAS.
- (a) S.-C. Chuang, M. Sander, T. Jarrosson, S. James, E. Rozumov, S. I. Khan and Y. Rubin, Approaches to open fullerenes: synthesis and kinetic stability of Diels–Alder adducts of substituted isobenzofurans and C60, J. Org. Chem., 2007, 72, 2716–2723 CrossRef CAS; (b) M. Sander, T. Jarrosson, S.-C. Chuang, S. I. Khan and Y. Rubin, Approaches to open fullerenes: synthesis and thermal stability of cis-1 bis(isobenzofuran) Diels–Alder adducts of C60, J. Org. Chem., 2007, 72, 2724–2731 CrossRef CAS; (c) S.-C. Chuang, F. R. Clemente, S. I. Khan, K. N. Houk and Y. Rubin, Approaches to open fullerenes: a 1,2,3,4,5,6-hexaadduct of C60, Org. Lett., 2006, 8, 4525–4528 CrossRef CAS.
- (a) D. M. Guldi and K.-D. Asmus, Photophysical properties of mono- and multiply-functionalized fullerene derivatives, J. Phys. Chem. A, 1997, 101, 1472–1481 CrossRef CAS; (b) K. George Thoma, V. Biju and M. V. George, Excited-state interactions in pyrrolidinofullerenes, J. Phys. Chem. A, 1998, 102, 5341–5348 CrossRef CAS.
- The MER Corp., 7960 S. Kolb Road, Tucson, AZ 85706.
- (a) R. N. Warrener, Isolation of isobenzofuran, a stable but highly reactive molecule, J. Am. Chem. Soc., 1971, 93, 2346–2348 CrossRef CAS; (b) W. Friedrichsen, Adv. Heterocycl. Chem., 1980, 26, 135–241 CAS; (c) R. Rodrigo, Progress in the chemistry of isobenzofurans: Applications to the synthesis of natural products and polyaromatic hydrocarbons, Tetrahedron, 1988, 44, 2093–2135 CrossRef CAS; (d) B. Rickborn, in Advances in Theoretically Interesting Molecules, ed. R. P. Thummel, JAI Press Inc., Greenwich, CT, 1989, vol. I, pp. 1–134 Search PubMed; (e) C. W. Bird, Prog. Heterocycl. Chem., 1990, 2, 87–101 Search PubMed; (f) O. Peters and W. Friedrichsen, Trends Heterocycl. Chem., 1995, 4, 217–259 Search PubMed.
- I. Carmichael and G. L. Hug, Triplet-triplet absorption spectra of organic molecules in condensed phases, J. Phys. Chem. Ref. Data, 1986, 15, 1–250.
- S. L. Murov, I. Carmichael and G. L. Hug, Handbook of Photochemistry, Marcel Dekker, New York, 1993 Search PubMed.
- J. L. Anderson, Y.-Z. An, Y. Rubin and C. S. Foote, Photophysical characterization and singlet oxygen yield of a dihydrofullerene, J. Am. Chem. Soc., 1994, 116, 9763–9764 CrossRef CAS.
- F. Prat, R. Stackow, R. Bernstein, W. Y. Qian, Y. Rubin and C. S. Foote, Triplet-state properties and singlet oxygen generation in a homologous series of functionalized fullerene derivatives, J. Phys. Chem. A, 1999, 103, 7230–7235 CrossRef CAS.
- R. V. Bensasson, E. Bienvenue, J. M. Janot, S. Leach, P. Seta, D. I. Schuster, S. R. Wilson and H. Zhao, Photophysical properties of three hydrofullerenes, Chem. Phys. Lett., 1995, 245, 566–570 CrossRef CAS.
- (a) C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS; (b) A. D. Becke, Density-functional thermochemistry. III. The, role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision C.02), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- W. Qian, M. D. Bartberger, S. J. Pastor, K. N. Houk, C. L. Wilkins and Y. Rubin, C62, a non-classical fullerene incorporating a four-membered ring, J. Am. Chem. Soc., 2000, 122, 8333–8334 CrossRef CAS , and references therein.
- C. Schweitzer and R. Schmidt, Physical, mechanisms of generation and deactivation of singlet oxygen, Chem. Rev., 2003, 103, 1685–1757 CrossRef CAS.
- O. L. Gijzeman, F. Kaufman and G. Porter, Oxygen, quenching of aromatic triplet states in solution, J. Chem. Soc. Faraday Trans. 2, 1973, 69, 708–720 RSC.
Footnote |
| † Deceased June 13, 2005. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2008 |