Studies of decarboxylation in photolysis of α-carboxy-2-nitrobenzyl (CNB) caged compounds†
John E. T.
Corrie
*a,
V. Ranjit N.
Munasinghe
a,
David R.
Trentham
a and
Andreas
Barth
b
aMRC National Institute for Medical Research, The Ridgeway, Mill Hill, London, UK NW7 1AA. E-mail: jcorrie@nimr.mrc.ac.uk
bDepartment of Biochemistry and Biophysics, Arrhenius Laboratories for Natural Sciences, Stockholm University, SE-106 91, Stockholm, Sweden
First published on 5th November 2007
Abstract
Photolysis of α-carboxy-2-nitrobenzyl (CNB) caged compounds, studied here by time-resolved IR and UV spectroscopy, involves at least two pathways. In one, a conventional 2-nitrobenzyl type rearrangement takes place to release the photoprotected species via rapid decay of an aci-nitro intermediate. The α-carboxylate moiety of the CNB group is retained and the final by-product from this pathway is 2-nitrosophenylglyoxylate. Direct measurements of product formation confirmed that release via this pathway is faster for CNB-caged compounds than for related caged compounds without an α-carboxylate substituent and a rationale for the faster release rate is proposed. In a second pathway, photodecarboxylation of the starting material occurs: this pathway leads only to a slow, minor release of the photoprotected species. The extent to which the latter pathway contributes is affected by the nature of buffer salts in the irradiated solution. It was more prominent in an amine-based buffer (MOPS) than in phosphate buffer.
Introduction
The photofragmentation of 2-nitrobenzyl (NB) and 1-(2-nitrophenyl)ethyl (NPE) esters or ethers of a wide range of compounds, including phosphates, carbamates and alcohols, has been widely applied as the most common photochemistry among the available range of photolabile protecting groups for rapid release of biologically-active species at or near their site of action in biological systems.1 Commonly, the technique is used to enable time-resolved study of fast (often sub-millisecond) biological processes. The photocleaved nitrobenzyl substituent is colloquially termed a caging group and the derivatives are known as caged compounds.2 One variant of the usual NB or NPE groups has a carboxy substituent on the benzylic carbon, giving the α-carboxy-2-nitrobenzyl (CNB) cage group 1 (Scheme 1). This modification was introduced by Walker and Hess, principally to improve the pharmacological properties of a caged acetylcholine analogue.3 The CNB cage has since been applied to a range of compounds, either because the ionised side chain confers improved water solubility or because of indications of more rapid release of the bioactive species upon flash photolysis than occurs from corresponding NB or NPE cages.4–9 | ||
| Scheme 1 Usual reaction scheme for photolysis of CNB-caged compounds. | ||
We became interested in these compounds because all previous studies of CNB cages make the implicit assumption that the carboxy substituent is an inert bystander that does not participate in the photocleavage reaction. Reaction schemes are typically shown as in Scheme 1, leading via a putative aci-nitro compound 2 and subsequent bicyclic species 3 to release of the product (shown generically as X in Scheme 1) and formation of 2-nitrosophenylglyoxylate 4 as the by-product of photolysis.3–9 We note that Scheme 1 incorporates recent results on the equilibria between the aci-nitro species 2 and its isomeric nitronic acids.10 The assumption that the carboxylate group is retained overlooks well-known data for simple nitrophenylacetates, where it has been shown that all three isomers undergo photodecarboxylation with varying degrees of efficiency (m- ≈ p- > o-).11 The possibility of photodecarboxylation has been considered in only one previous study of CNB-caged compounds,5b where there was no attempt at direct detection of CO2 and decarboxylation was ruled out on the sole basis that irradiated solutions of the glycine derivative 5 did not contain the decarboxylated product 6 (Scheme 2).
 | ||
| Scheme 2 | ||
However, if photodecarboxylation were operative for CNB-caged compounds, the basis of reliance on product release rates estimated from decay of the UV-visible transient ascribed to the aci-nitro compound 2 could be compromised to some extent. Unlike NPE-cages, where the aci-nitro decay rates are closely proportional to the proton concentration of the solution over the pH range 4–9 (e.g. ref. 12,13), reported rate profiles for CNB-caged compounds as a function of pH generally show little systematic relationship to pH.3a,5b–d,8a The decay transients are often biphasic5a–e,8a and different UV-visible absorption spectra have been reported for the two components of such transients.5a–c,8a These considerations suggest that the photochemical reactions of CNB-caged compounds are probably more complex than generally supposed. To obtain further insight on these processes, we now describe time-resolved infrared (IR) spectroscopic and other studies of representative CNB and related caged compounds. IR spectroscopy is particularly appropriate in this context as it enables simple detection of CO2 release by observation of a strong band at 2343 cm–1 that is diagnostic for CO2 in aqueous solution.14 Other workers have used time-resolved IR spectroscopy to investigate photolysis of CNB-caged compounds9,15 but their experiments did not cover the spectral range that would have enabled observation of photoreleased CO2.
Results
Synthesis of compounds
In this work we focused on the CNB-caged monomethyl phosphate ester 7 and the glycine carbamate 8. These specific compounds were chosen for two principal reasons. First, they are thermally stable over a wide pH range, so spectroscopic studies would not be compromised by background hydrolysis. Second, they had favourable properties with respect to IR spectroscopy. For example, release of monomethyl phosphate upon photolysis of 7 was expected to be observable in the IR spectra at pH ≥ 7 because of the change of ionisation of the phosphate group, from a monoanion in 7 to a largely dianionic form in the released monomethyl phosphate (pK 6.2).16 The glycine carbamate 8 was expected to allow direct observation of the decarboxylation of the photoreleased carbamate salt: this rate corresponds to the rate of glycine release and is relevant to previous work on the analogous glutamate conjugate.6a Compounds 7 and 8 were also prepared as 13C isotopomers, labelled in the carboxylate of the CNB group (marked * in the structures of these compounds) for positive identification of CO2 formation in the IR spectra. In the case of compound 8, the isotopomer enables the possibility of separately observing formation of 12CO2 from the carbamate anion released by photolysis and 13CO2 from the benzylic carboxylate, if the latter process does take place as discussed above. We also made the amide 9 to probe whether the carboxylate group exerted its effect on the rate of product release simply via an inductive mechanism or by other means.
Synthesis of these compounds followed minor modifications of known methodology (see Experimental section). In addition to the compounds for photochemical study, we attempted to prepare an authentic sample of 2-nitrosophenylglyoxylic acid 4 for use as a reference standard in analysis of photolysed reaction mixtures. We had previously found that treatment of 2-nitrobenzyl alcohol with triflic anhydride and 2,6-di-t-butylpyridine gave 2-nitrosobenzaldehyde,17 and the same procedure applied to the 2-nitromandelate esters 10 or 11 (Scheme 3) readily gave the respective methyl and t-butyl nitrosophenylglyoxylates 12 and 13. The methyl ester 12 has been reported18 in matrix isolation photolysis of methyl α-diazo-2-nitrophenylacetate at 10 K, but was obtained here as a stable, crystalline compound. The 4-nitro-substituted congener of 12 has also been described.19 Attempted conversion of the esters to the free acid 4, either by mild basic hydrolysis of 12 or TFA treatment of 13 immediately gave very dark reaction mixtures from which no recognisable product could be isolated. A triflic anhydride-mediated rearrangement of 2-nitromandelic acid itself failed, not least because of inadequate solubility of the acid in a suitable solvent. Thus we were unable to obtain a sample of 4 as an isolated pure compound and did not pursue this attempt.
 | ||
| Scheme 3 Reagents: (a) (CF3SO2)2O–2,6-di-t-butylpyridine–CH2Cl2. | ||
Spectroscopic studies of (7)–(9)
At the outset of our investigations of these compounds, we wished to determine whether decarboxylation did indeed occur when they were photolysed. IR spectra for photolysis of each isotopomer of 7 and 8 were initially recorded in MOPS buffer, pH 7 at 1 °C, and focused particularly on formation of CO2. Bands observed for 12CO2 and/or 13CO2 at 2343 and 2278 cm–1 respectively were integrated with respect to time (see Experimental section) to determine the kinetics of their formation and decay (see ref. 20 for previous reports of the IR absorption of 13CO2 in aqueous solution). For 7, the formation of CO2 was biphasic, with a fast phase complete within the first 60 ms and a second phase in which CO2 formation continued with a rate constant of 0.82 s–1 (see Fig. 1 for these data, recorded for the 13C isotopomer). The amplitude ratio of the fast and slower phases of formation was ∼2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Formation of 13CO2 confirms its origin from the isotopic compound itself and not from atmospheric CO2 or other environmental source. Slow decay of the band, expected because of hydration21 of the evolved CO2, took place with a rate constant of 0.14 min–1. This decay was two orders of magnitude slower than the slow phase of formation discussed above and did not perturb the observed rates or amplitudes of CO2 formation for either of compounds 7 or 8. When the experiment was repeated in the presence of excess dithiothreitol (DTT), the slow phase of CO2 formation was abolished and the amplitude of the CO2 band was the same as that of the fast phase of CO2 formation in the absence of the thiol (Fig. 1). As noted above, these spectra were recorded in MOPS buffer: for experiments in phosphate-buffered solution (also at pH 7) the rate constants and relative amplitudes of the two phases of CO2 formation were somewhat different, with the slow phase release being only about half its amplitude in MOPS (data not shown), but the qualitative results were unaltered.
:1. Formation of 13CO2 confirms its origin from the isotopic compound itself and not from atmospheric CO2 or other environmental source. Slow decay of the band, expected because of hydration21 of the evolved CO2, took place with a rate constant of 0.14 min–1. This decay was two orders of magnitude slower than the slow phase of formation discussed above and did not perturb the observed rates or amplitudes of CO2 formation for either of compounds 7 or 8. When the experiment was repeated in the presence of excess dithiothreitol (DTT), the slow phase of CO2 formation was abolished and the amplitude of the CO2 band was the same as that of the fast phase of CO2 formation in the absence of the thiol (Fig. 1). As noted above, these spectra were recorded in MOPS buffer: for experiments in phosphate-buffered solution (also at pH 7) the rate constants and relative amplitudes of the two phases of CO2 formation were somewhat different, with the slow phase release being only about half its amplitude in MOPS (data not shown), but the qualitative results were unaltered.
![Kinetics of CO2 release from [13C]CNB-caged monomethyl phosphate 7 (78 mM; H2O solution with 200 mM MOPS, pH 7, 1 °C). Traces are for the intensities of the 13CO2 band at 2278 cm–1 in the absence (●) or (○) presence of 100 mM dithiothreitol, respectively. The inset is on a longer time scale to show the slow hydration of CO2. Lines through the data points are double (●) or single (○) exponential fits to the relevant points. Except for the 2278 cm–1 band, spectra recorded with or without DTT were of very similar intensity, so no normalisation was applied to the CO2 integrations.](/image/article/2008/PP/b711398f/b711398f-f1.gif) | ||
| Fig. 1 Kinetics of CO2 release from [13C]CNB-caged monomethyl phosphate 7 (78 mM; H2O solution with 200 mM MOPS, pH 7, 1 °C). Traces are for the intensities of the 13CO2 band at 2278 cm–1 in the absence (●) or (○) presence of 100 mM dithiothreitol, respectively. The inset is on a longer time scale to show the slow hydration of CO2. Lines through the data points are double (●) or single (○) exponential fits to the relevant points. Except for the 2278 cm–1 band, spectra recorded with or without DTT were of very similar intensity, so no normalisation was applied to the CO2 integrations. | ||
Photolysis spectra recorded similarly for 8, using its 13C isotopomer to enable distinction between CO2 formed from the benzylic carboxylate group and from thermal decomposition of the carbamate salt that results from photolytic cleavage, also showed that CO2 was lost from the benzylic carboxylate. However, there were significant differences from the results for 7 and these are discussed later in the text. At this point of our study it was evident that decarboxylation was a feature of the photolysis of these compounds, albeit that the extent of its contribution to the overall reaction flux was not yet determined.
With the fact of some level of photodecarboxylation established, we next sought to obtain evidence for or against operation of the usual photolysis pathway (Scheme 1), specifically whether or not there was involvement of an aci-nitro intermediate that retained the benzylic carboxylate group. As a lead into this search, we began with the amide 9, where the possibility of CO2 loss is blocked and a relatively straightforward photolysis course was anticipated. Some support for this expectation came from laser flash irradiation of 9 to record the aci-nitro transient kinetics (at 406 nm, where the transient from NPE-caged compounds is at its maximum amplitude22). This transient had a single-exponential decay with a rate constant of 30 s–1 (20 °C, pH 7, data not shown) and this slow decay rate readily enabled recording of the IR spectrum of the aci-nitro intermediate (at pH 7.5, 1 °C), shown as the full line in Fig. 2. IR difference spectra of photolysis were recorded as described previously,23,24 with a full spectrum (2500–900 cm–1) recorded in each 60 ms time interval after the photolysis flash. In the difference spectra shown throughout, negative bands represent species that disappear upon photolysis and positive bands represent species that are formed in the reaction(s).
 | ||
Fig. 2 Infrared difference spectra for photolysis of 9 (104 mM; H2O solution in 200 mM MOPS) at pH 7.5, 1 °C, averaged over time intervals 1–673 ms (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ) and 3.4–30.7 s (⋯) after the photolysis flash. ) and 3.4–30.7 s (⋯) after the photolysis flash. | ||
Spectra averaged over the time band 1–673 ms after the light flash support the formation an aci-nitro intermediate, since they show medium to strong positive bands at 1458, 1382, 1322 and 1243 cm–1 that are reminiscent of bands at similar positions in the aci-nitro spectrum of NPE-caged ATP23 and NPE-caged monomethyl phosphate (see ESI, Fig. S1†). We note that there are additional bands for 9 at 1434, 1405, and 1269 cm–1, which are not observed for the NPE caged-compounds and evidently arise from the presence of the α-substituent. All the bands listed above decay in the transition to the final spectrum and a broad positive envelope develops in the region of 1100 cm–1. This envelope is characteristic of the released monomethyl phosphate anion and a similar feature is present in the final spectrum for photolysis of NPE-caged monomethyl phosphate (see ESI, Fig. S1†).
The aci-nitro spectrum also shows strong bands at 1633 cm–1 (negative) and 1602 cm–1 (positive). These can be assigned to the amide carbonyl in the starting material and aci-nitro intermediate respectively. The shift to lower frequency is consistent with reduced bond order of the amide carbonyl upon conversion of 9 to the highly conjugated system present in an aci-nitro intermediate equivalent to 2 (i.e. with the structure of 2 appropriately modified to incorporate the amide group). Over time, the 1602 cm–1 band decays and is replaced by a positive band at 1649 cm–1. Concomitantly the negative band at 1633 cm–1 is reduced in intensity, as would be expected from its partial overlap with the adjacent new positive band. The final species formed is expected to be the amide 14 and should show an additional band for a second carbonyl group. Other arylglyoxylamides have an amide band at 1640–1650 cm–1 and a second carbonyl band in the range 1670–1715 cm–1.25 The final spectrum for photolysis of 9 does show a weak band at 1690 cm–1 and its low intensity may arise from substantial hydration of the ketone group of the α-keto amide in aqueous solution. Support for this comes from data for pyruvic acid, which is strongly hydrated (60–80%) in its neutral form, although its anion is only slightly hydrated.26 The amide 14 would be expected to show behaviour more like that of the neutral form.
Integration of the bands at 1633, 1322, 1243 and the broad envelope near 1100 cm–1 with respect to time gave a rate constant for the aci-nitro decay at pH 7.5, 1 °C with a mean (±S.D.) value of 1.3 ± 0.2 s–1. This is compatible with the rate constant of 30 s–1 measured above at pH 7.0, 20 °C for the 406 nm transient. As expected, no absorption band at 2343 cm–1 was present in any of the spectra for this compound (data not shown), consistent with its inability to release CO2.
Before attempting to record IR spectra of aci-nitro species formed from 7 and 8, we determined the kinetics of their UV-visible transients. Inter alia, this information was required to plan appropriate conditions for the IR experiments. UV-visible transients following laser flash irradiation at pH 7, 20 °C were recorded for 7 and 8. At 406 nm the decay traces were at least biphasic, in contrast with the monophasic decay described above for 9. Compound 7 showed a fast phase at ∼3000 s–1 and a slower phase of similar amplitude at 16 s–1. For compound 8, the fastest phase was at ∼6600 s–1, with a much slower phase of similar amplitude at 12 s–1. These transients were similar whether or not DTT was present in the solution. At progressively longer wavelengths, the relative amplitude of the fast phase increased for each compound and by 480 nm the decay traces were well fitted by single exponentials. An example of such a trace is shown in Fig. 3 for compound 8.
 | ||
| Fig. 3 Transient 480 nm absorbance following 308 nm laser flash photolysis of an aqueous solution of 8 (0.5 mM) in 25 mM MOPS, 150 mM NaCl at pH 7, 20 °C. The grey line superimposed on the experimental points is a single exponential fit to the data (kobs = 7000 s–1). | ||
The pH dependence of the fast phase at this wavelength was examined for both 7 and 8, and the data are shown in Table 1. Single exponential fits to these data were highly robust, unlike double exponential fits to the 406 nm data for the same compounds. Notably, the decay rates at 480 nm (at least over the pH range 6.5–8) are quite closely proportional to [H+], apparently consistent with that of “conventional” aci-nitro behaviour.12 A red shift of the transient absorbance to a maximum of ∼440 nm has been noted previously for some CNB-caged compounds5c,8a but the wavelength-dependence of the decay kinetics of this transient appear not to have been examined. Our data suggest that more than one species contributes to the transient absorbance in the 400–440 nm range, and single-exponential decay of the longer wavelength component can be observed by recording in the red tail of the composite band. The red shift is indicative of a more extended conjugated system, consistent with an aci-nitro intermediate that retains a carboxylate group as shown in Scheme 1. Taken together with the IR data described below, it is reasonable to infer that the red-shifted transient corresponds to the aci-nitro intermediate (2 in Scheme 1) and that the slower-decaying phase(s) at shorter wavelength correspond to species with less-extended π-system(s), probably related to product(s) of the decarboxylation pathway(s). In contrast to the data for 7 and 8, the transient for 9 was a single-exponential decay with the same rate constant at all wavelengths measured (406, 420 and 440 nm).
| pH | Rate constant/s–1 | |
|---|---|---|
| 7 | 8 | |
| 6.5 | 9100 | 15200 |
| 7.0 | 3500 | 7000 |
| 8.0 | 510 | 1540 |
| 9.0 | 125 | 380 |
| 10.0 | 80 | 220 |
The time scale for acquisition of our IR difference spectra does not allow us to observe processes that occur on a time scale faster than ∼10 ms. Thus, for compounds 7 and 8, IR spectra recorded at pH 7, 1 °C were not appropriate to monitor processes that might be related to the fast UV-vis transients described above. We thus chose to record spectra at pH 8.5 and –10 °C in the hope of being able to observe processes that could give evidence for or against the usual reaction pathway of Scheme 1. We have previously studied photolysis of NPE-caged ATP at pH 8.5, 1 °C and obtained spectra consistent with aci-nitro formation and decay.23
Spectra recorded for photolysis of 7 in EPPS buffer (pH 8.5, –10 °C, D2O) are shown in Fig. 4 for the 13C isotopomer, for which the spectrum obtained in the time range 2–19 ms after the light flash can be interpreted in terms of an almost pure aci-nitro species 2 (Scheme 1, X = OPO2OMe–). Spectra for the 12C isotopomer had comparable features but decay of the aci-nitro species was less well separated, probably because of slight differences in timing, temperature or pH. Evidently our capacity to capture a clean aci-nitro spectrum for this rapidly-decaying species is quite finely balanced, and for clarity we have chosen to show only the 13C data, while referring in the text to band shifts for the 12C isotopomer.
![Infrared difference spectra for photolysis of [13C]CNB-caged monomethyl phosphate 7 (78 mM; D2O solution in 200 mM EPPS, pH 8.5) at –10 °C, averaged over time intervals of 2–19 ms () and 2.3–20.7 s (⋯) after the photolysis flash.](/image/article/2008/PP/b711398f/b711398f-f4.gif) | ||
| Fig. 4 Infrared difference spectra for photolysis of [13C]CNB-caged monomethyl phosphate 7 (78 mM; D2O solution in 200 mM EPPS, pH 8.5) at –10 °C, averaged over time intervals of 2–19 ms () and 2.3–20.7 s (⋯) after the photolysis flash. | ||
The spectrum in Fig. 4 recorded immediately after the light flash shows, inter alia, positive bands at 1458, 1326, 1239 and 1176 cm–1. These bands were scarcely affected by the isotopic substitution and are at very similar positions to bands previously assigned to vibrations associated with the aci-nitro group in the intermediate formed upon photolysis of NPE-caged ATP.23 There is also a positive band at ∼1515 cm–1 and shoulder at ∼1343 cm–1 (12C at 1568 and 1374 cm–1 respectively, the latter as a well-resolved band). In view of their isotopic shifts, these bands can reasonably be assigned to the antisymmetric and symmetric vibrations of a carboxylate group (see ref. 27 for data on isotopic shifts of carboxylate groups and Table 2 for a compilation of carboxylate band positions studied in this work). Note that overlap with adjacent bands distorts the position of the true maxima of these carboxylate bands in several cases. Nevertheless the values are consistent with a conjugated carboxylate group and, together with the four other positive bands described above, are consistent with an aci-nitro species bearing a carboxylate group, i.e. structure 2 as shown in Scheme 1 (X = OPO2OMe–). Elsewhere in the spectrum, the sharp positive band at 1081 cm–1 and the broad negative band at 1209 cm–1, that are scarcely perturbed from their positions in the 12C isotopomer, are very similar to corresponding bands seen in the photolysis spectrum of NPE-caged monomethyl phosphate (see ESI, Fig. S1†) and are likely to be phosphate vibrations.
| Carboxylate | Starting compound | aci-Nitro intermediate | Final product |
|---|---|---|---|
| a Values in parentheses are determined after subtraction of the aci-nitro spectrum from the spectrum of the final products (recorded in EPPS buffer, pH 8.5). This procedure enables more accurate determination of the true band maxima. b 2-Nitrosophenylglyoxylate by-product 4. c Glycine. | |||
| α-Carboxylate of 7: 12C, 13C | 1604, 1564 | 1568 (1576a), ∼1515 (∼1530a) | 1634, 1592b |
| α-Carboxylate of 8: 12C, 13C | 1611, ∼1568 | n.d., 1515 | ∼1640 (shoulder)b, not identified for 13C isotopomer |
| Carbamate group of 8 | 1692 | 1726 | — |
| Glycine moiety of 8 | ∼1590 | n.d. | 1621c |
A negative band at 1564 cm–1 (1604 cm–1 in the 12C isotopomer) is assigned to the antisymmetric vibration of the α-carboxylate group that disappears upon photolysis. The disappearance is a two-fold process, leading both to the observed biphasic release of CO2 (Fig. 1), and to formation of the positive carboxylate bands of the aci-nitro intermediate, as discussed above. The position of the negative band for the antisymmetric vibration of the disappearing nitro group at 1538 cm–1 is distorted by overlap with the adjacent negative 1564 cm–1 band but it appears at a more normal position (1531 cm–1) in the 12C isotopomer. The symmetric vibration of the disappearing carboxylate appears as a composite with the symmetric vibration of the disappearing nitro group in the negative band at 1368 cm–1 [the 12C isotopomer has two resolved negative bands at 1394 and 1355 cm–1 (carboxylate and nitro respectively)].
The aci-nitro intermediate is present at a time when rapid CO2 release is complete and its decay is faster than the slow phase of CO2 release. The CO2 release data shown in Fig. 1 were recorded at pH 7, but the biphasic release was also seen at pH 8.5 (data not shown). We considered the possibility that the rapid decarboxylation process might lead to the same aci-nitro intermediate as is formed on photolysis of 2-nitrobenzyl monomethyl phosphate, i.e. by the route shown in Scheme 4. In principle, this seems a feasible pathway as it is the same as the first step in photodecarboxylation of simple nitrophenylacetates.11,28 However, the aci-nitro intermediate formed on photolysis of 2-nitrobenzyl monomethyl phosphate exhibits a number of prominent bands, including two at 1398 and 1289 cm–1, that all decay on a time scale of minutes at pH 8.5 and seconds at pH 7.0 (see ESI, Fig. S2† for a spectrum at pH 7, 1 °C), i.e. much slower than decay of the aci-nitro bands formed by irradiation of 7. Although some of the bands in the aci-nitro spectrum for this compound are close to the positions of similar bands for the aci-nitro species formed from 7, those at 1398 and 1289 cm–1 are not present in the photolysis spectra of 7. This indicates that the rapid loss of CO2 upon irradiation of 7 does not follow the process postulated in Scheme 4, and the fate of the decarboxylation product remains unclear. Nevertheless, we conclude that formation and decay steps of the aci-nitro intermediate in photolysis of 7 are not associated with either of the CO2 release processes.
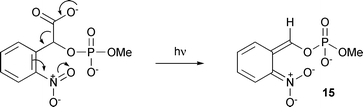 | ||
| Scheme 4 Putative photodecarboxylation of 7 to generate an aci-nitro intermediate. | ||
In the transition to the final spectrum for photolysis of [13C]7, all the positive bands assigned above to the aci-nitro intermediate undergo decay and are replaced by four principal positive bands at 1694, 1592, 1104 and 981 cm–1. Only the band at 1592 cm–1 is isotopically sensitive (1634 cm–1 in the 12C isotopomer) and is consistent with the presence of a carboxylate group in the final product, the antisymmetric vibration of which is upshifted by ∼30 cm–1 with respect to that of the starting compound. The positive band at 1694 cm–1 is in a similar position to the weak 1690 cm–1 band in the final spectrum for photolysis of 9 (Fig. 1), assigned there to the keto group of 14. In the present case, it would be consistent with the keto group of the nitrosophenylglyoxylate by-product 4. In pyruvate anion this band is at 1710 cm–1,29 and it would be downshifted in 4 by conjugation with the adjacent aromatic ring. The band at 1592 cm–1 (1634 cm–1 in the 12C isotopomer) can reasonably be assigned to the carboxylate group of 4. The frequencies for this band are almost identical to those reported for the carboxylate of [12C] and [13C]pyruvate.29b Lastly, the broad band at 1104 cm–1 and the sharp one at 981 cm–1 are similar to corresponding bands in the final spectrum from photolysis of 9, and are characteristic of the monomethyl phosphate product. The spectrum recorded immediately after the photolysis flash does not show the product band at 1104 cm–1, so the fast phase of CO2 release is not associated with product release. Integration with respect to time of the aci-nitro bands at 1515, 1458 and 1239 cm–1, the by-product band at 1592 cm–1 and the product band at 1104 cm–1, and exponential fits to these data points showed that all bands decayed or grew (as appropriate) at similar rate constants (range 23–34 s–1, mean 30 ± 4.5 s–1), i.e. that the product and by-product were formed as the aci-nitro intermediate decayed.
These data show that part of the reaction flux follows the usual mechanism (Scheme 1), with an aci-nitro intermediate 2 that retains the carboxylate group. Decay of 2 leads to product release (monomethyl phosphate in this case) and formation of the nitrosophenylglyoxylate by-product 4. Product release is faster than from the related NPE-caged compound, for which the aci-nitro intermediate can be readily observed even at pH 7.5 (Fig. S1, ESI†) and for much longer times at pH 8.5 [respective half times estimated from time-resolved IR spectra in Bicine buffer are ∼350 ms (at 1 °C)30versus ∼27 ms for 7 at the same pH but at –10 °C].
Spectra of 7 recorded in MOPS or phosphate buffers at pH 7, 1 °C (data not shown) were broadly comparable with the final spectrum at pH 8.5 discussed above, although the spectra in MOPS showed an additional band at 1733 cm–1 that developed on the same time scale as the slow release of CO2 and was insensitive to isotopic substitution. We cannot assign a structure to the species responsible for this band but some further consideration of the effects of MOPS buffer is given below. No aci-nitro intermediate was observed in the pH 7 spectra because of its fast decay under these conditions. In consequence, release of the monomethyl phosphate product was almost complete in the first spectrum after the flash in these spectra. A further small phase (∼10%) of slow product formation could be seen during the slow phase of CO2 release at pH 7 and 8.5, but we could not definitively determine whether the CO2 and the small added amount of monomethyl phosphate product were formed from the same species.
It would be expected that the CO2 released in the slow phase originates from a carboxylate group but no obvious changes could be seen in the difference spectra. However in double difference spectra, where the difference spectrum at the start of the slow CO2 release was subtracted from that at the end of the release, it was possible to detect a band at 1578 cm–1 (1629 cm–1 for the 12C isotopomer) that disappeared on the same time scale as the slow formation of CO2. These bands could be detected in the double difference spectra in either MOPS or phosphate buffer at pH 7. A further subtraction of the double difference spectrum of the 13C isotopomer from that of the 12C isotopomer revealed the isotopically-sensitive bands of these spectra even more clearly, while cancelling those bands that are insensitive to isotopic substitution. This isotope effect spectrum is shown in Fig. S3 of the ESI.† The isotopic shift discussed above shows as a minimum/maximum structure at 1629/1580 cm–1, with a second such structure at 1395/1358 cm–1. These band pairs can be assigned respectively to the antisymmetric and symmetric stretching vibrations of a carboxylate group. The two band pairs in the isotope effect spectrum were not observed for spectra recorded in the presence of DTT. Since the slow phase of CO2 formation was abolished in the presence of DTT (Fig. 1), these isotopically-sensitive bands evidently arise from a carboxylate group that is the source of the slow phase CO2. With the limited information available, it is not possible to propose a complete structure for the species that bears this carboxylate.
In marked contrast to the data for CO2 formation from 7, the kinetics of CO2 release from the 13C isotopomer of carbamate 8 at pH 7, 1 °C showed that evolution of CO2 from the labelled (benzylic) carboxylate was complete within the time span of the first spectrum (i.e. 0–60 ms) (Fig. 5). Unlike for 7, the amplitude of CO2 formation from the benzylic carboxylate was the same in the presence or absence of DTT (data not shown). In the spectra for [13C]8, the band for 12CO2 at 2343 cm–1 increased exponentially, with a rate constant of 4.3 s–1. This process represents decarboxylation of the photolytically-generated N-carboxyglycine salt (Scheme 5) and thus is the rate of glycine release. The data shown were recorded for a solution in potassium phosphate at pH 7, where the amounts of 12CO2 and 13CO2 were virtually identical. A similar experiment for a solution in pH 7 MOPS buffer showed the same kinetic behaviour but formation of approximately twice as much 13CO2 relative to the 12CO2 (data not shown). This result also was the same whether or not DTT was present. The substantial difference between the data recorded in different buffers is a strong indication that the course of this photolysis reaction can be influenced by buffer salts. Furthermore, the formation in MOPS buffer of substantially more CO2 from the benzylic carboxylate than from the carbamate group suggests that not all release of CO2 from the benzylic carboxylate results in release of the caged species, at least in this buffer solution. We return to this point later in the text.
![Kinetics of release of CO2 from [13C]CNB-caged glycine 8 (H2O solution with 200 mM potassium phosphate, pH 7, 1 °C). Data points are for the intensities of the 13CO2 (▲) and 12CO2 (●) bands at 2278 and 2343 cm–1 respectively and the relative intensities of the 12CO2 and 13CO2 have been corrected for differences in the absorption coefficients of the two species (see ESI). The solid line is a double exponential fit to the relevant points to model rise and decay of the 12CO2 band, although decay by hydration of released CO2 was insignificant over the time range shown.](/image/article/2008/PP/b711398f/b711398f-f5.gif) | ||
| Fig. 5 Kinetics of release of CO2 from [13C]CNB-caged glycine 8 (H2O solution with 200 mM potassium phosphate, pH 7, 1 °C). Data points are for the intensities of the 13CO2 (▲) and 12CO2 (●) bands at 2278 and 2343 cm–1 respectively and the relative intensities of the 12CO2 and 13CO2 have been corrected for differences in the absorption coefficients of the two species (see ESI†). The solid line is a double exponential fit to the relevant points to model rise and decay of the 12CO2 band, although decay by hydration of released CO2 was insignificant over the time range shown. | ||
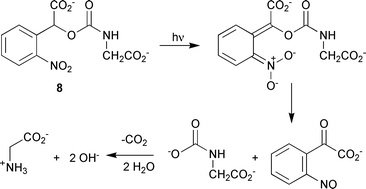 | ||
| Scheme 5 Usual photolysis reaction for 8, showing formation and subsequent decarboxylation of the intermediate N-carboxyglycine salt. | ||
IR spectra of the carbamate 8 were more complex than for 7, as could be expected from the presence of three distinct carbonyl groups in the starting compound (i.e. the carboxylates of the CNB group and of the glycine moiety, plus the carbamate group), all of which are expected to undergo changes (see Scheme 5).
The kinetic plot of CO2 release and spectra that show the formation and decarboxylation of this carbamate salt (Fig. 5 and 6 respectively) were obtained in pH 7 phosphate buffer at 1 °C. The initial spectrum (Fig. 6A) covers a time range of 1–126 ms after the light flash, over which time the carbamate salt had formed and partially decarboxylated, in line with the kinetics of CO2 release (Fig. 5). In both the 12C and 13C spectra, the negative band at 1692 cm–1 can be assigned to the carbonyl of the carbamate group in 8. The N-carboxy moiety of the carbamate salt that forms on photolysis can be identified as a positive band at 1583 cm–1 in double difference spectra (not shown) between the late and early spectra of Fig. 6, a position similar to that reported (∼1590 cm–1) for other carbamate salts.31 However this position is overlapped with a negative band (discussed below) so no definitive bands in this region can be seen in Fig. 6A itself. The strong negative band at 1611 cm–1 (downshifted to ∼1568 cm–1 in the 13C spectrum) must arise from the carboxylate of the CNB caging group and has a similar frequency to the band of the same origin in the caged monomethyl phosphate 7. The negative band at 1596 cm–1, that is resolved in the 13C spectrum but appears as a shoulder in the 12C spectrum, is probably from the carboxylate of the glycine moiety in the starting compound. Published data support assignment of this band to the carboxylate of an N-acylated glycine, e.g.N-acetylglycine is reported to absorb at 1600 cm–1 (D2O solution, pH 7).32 This negative band is overlapped with the putative positive band of the carbamate salt at almost the same frequency. It is notable that a negative feature at this frequency becomes more intense in the final spectra (Fig. 6B) when the expected positive band of the carbamate salt will have decayed. In the final 12C spectrum, reduced overlap with other bands results in the apparent minimum for the lost α-carboxylate of the CNB group shifting from 1611 cm–1 to 1594 cm–1 in the early and late spectra respectively.
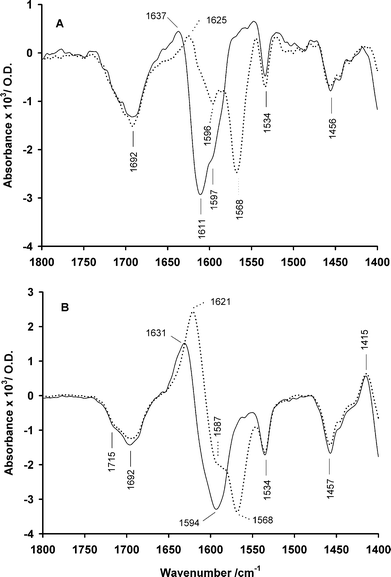 | ||
| Fig. 6
IR difference spectra in the range 1800–1400 cm–1 for photolysis of 8 at pH 7, 1 °C (in 200 mM potassium phosphate, D2O), shown for the 12C () and 13C (⋯) isotopomers (both ∼115 mM). Panels A and B show spectra averaged over the time intervals 0–126 ms and 0.7–3.4 s respectively after the photolysis flash. | ||
Apart from the intensity changes near 1590 cm–1 discussed above, the most prominent feature in the final spectra (Fig. 6B) is the growth of a strong band at 1621 cm–1 for the 13C isotopomer. This band corresponds to the expected carboxylate absorption of free glycine,33i.e. the end product after decarboxylation of the initially-formed carbamate salt. This band appears weaker in the 12C spectrum, evidently because of overlap with the adjacent negative band of the CNB carboxylate at 1611 cm–1 that also distorts its maximum position to 1631 cm–1. A further reason for this shift is an isotopically-sensitive band seen as a shoulder on the high-wavenumber side of the 1631 cm–1 band. This shoulder can be assigned to the carboxylate group of the nitrosophenylpyruvate by-product 4, which is at a similar position in the photolysis spectra of [12C]7.
As noted above in the data for CO2 evolution, photolysis of 8 was significantly influenced by the nature of the buffer salt. Further evidence of this was apparent in spectra recorded in MOPS solution under otherwise identical conditions (data not shown). Although both the initial and final spectra for both isotopomers in this solution had bands that were consistent with the interpretation presented above, the initial spectra showed an additional positive band at 1740 cm–1 that disappeared in the transition to the final spectra. The origin of this band remains obscure but may involve electron-transfer processes from the amine buffer. We have recently described examples of both inter- and intramolecular electron transfer in photolysis of other nitrobenzyl-type compounds.20
As for 7, we sought to verify formation of the usual aci-nitro intermediate from 8, (as shown in Schemes 1 and 5) and in this case the spectra were obtained in Bicine buffer, pH 8.5 in D2O at –10 °C (data not shown). The initial spectrum for the 12C isotopomer, recorded in the time interval of 2–19 ms after the photolysis flash, showed positive bands of medium to strong intensity at frequencies of 1440, 1326, 1238 and 1176 cm–1 that were scarcely affected in the 13C isotopomer (1437, 1327, 1237 and 1174 cm–1). These are all similar to bands discussed above for the aci-nitro intermediate formed from 7. The spectrum of the 13C isotopomer also had positive bands at 1515 and ∼1340 cm–1, similar to those of the unsaturated carboxylate in the aci-nitro intermediate formed from the 13C isotopomer of 7. The ∼1340 cm–1 band was shifted to 1366 cm–1 in the 12C isotopomer, whereas the band corresponding to that at 1515 cm–1 was not visible for the 12C isotopomer because of overlap with a negative band at 1568 cm–1 arising from protonation of the Bicine buffer. Both isotopomers showed a positive band at 1726 cm–1 in the initial spectra, that would be consistent with the carbonyl of the enol carbamate moiety that is present in the aci-nitro intermediate (see Scheme 5). The evidence of these spectra for 8 strongly suggests that an aci-nitro intermediate retaining the carboxylate of the CNB group is formed, as shown in Scheme 5. As already noted, the carboxylate bands in these spectra are summarised in Table 2.
As noted in the results of the photolysis spectra for 7, the photodecarboxylation pathway of these compounds does not result in significant product release and we have measured both the extent of this decarboxylation and, where feasible, the yield of photolysis product. Details for measurement of the extent of decarboxylation are given in the ESI,† and were based on calibration of the intensity of the CO2 band produced by photolysis of a calculated amount of 4-nitrophenylacetate. This calibration was used to determine the amount of CO2 formed upon photolysis of the CNB-caged compounds. Interestingly, both for 7 and 8, the amount of CO2 formed from the benzylic carboxylate was substantially greater in MOPS buffer than in phosphate buffer (both pH 7), which may be relevant to the differences in IR spectra recorded in the different buffers. For 7, the calculations indicate that 57% of photolysed molecules produced CO2 in MOPS, but only 35% in phosphate (combined total for the fast and slow phases of formation, as in Fig. 1). Results for [13C]8 were quite similar, with 47% of photolysed molecules yielding 13CO2 in MOPS, but only 35% in phosphate. Formation of 12CO2 from [13C]8 by decarboxylation of the photogenerated carbamate salt (as shown in Fig. 5 and Scheme 5), was 24% of photolysed 8 in MOPS and 33% in phosphate. The relative amounts of 12CO2 and 13CO2 measured from photolysis of 8 in these quantitative experiments are in line with the qualitative ratios discussed above in the two buffer solutions (Fig. 5 and associated text). Although these relative ratios determined within one set of spectra are accurate, the absolute level of CO2 measured here has significant error (for example, the error in the value of 57% measured for 7 in MOPS is estimated as ± 23%. See details in ESI† for error calculations). Thus the values given above, while indicative of the magnitude of the decarboxylation, cannot be taken as exact measures. They nevertheless confirm that a substantial proportion of the reaction flux involves photodecarboxylation.
We had no convenient means to measure the yield of monomethyl phosphate released by photolysis of 7, but for 8 it was possible to make quantitative measurements of the yield of glycine, as we have done previously for release of amino acids from other caged precursors.34 For a solution of 8 photolysed in phosphate buffer, the measured yields of glycine were 12.5 and 19% at conversions of 22 and 35% respectively. Thus under these conditions, only just over 50% of the molecules undergoing photolysis released glycine. We did not do a comparable experiment in MOPS buffer, but the CO2 results discussed above imply that the glycine yield in that case would only be in the region of 33% of that expected for a stoichiometric reaction. Qualitative support for this prediction was provided by comparison of the IR spectra for photolysis of 8 in phosphate and MOPS buffers. The band at 1622 cm–1 that arises from the released glycine was approximately twice as intense for the spectrum recorded in phosphate buffer as it was in MOPS (data not shown).
Discussion
The results presented here comprise the most detailed study to date of the photochemical reactions of CNB-caged compounds. While the data provide substantial new information on these compounds, they certainly do not give a complete picture of their photolysis: both the positive findings and the remaining areas of uncertainty are considered below.One strong result is the confirmation that these compounds do, in part, photolyse via a usual 2-nitrobenzyl type rearrangement, as shown in Scheme 1. The data are clearer for the monomethyl phosphate 7, where decay of the aci-nitro intermediate is ∼2-fold slower than for the carbamate 8 and there are fewer carbonyl groups that undergo changes. Evidence for the usual pathway comes from bands in the IR spectra that are very similar to bands previously assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, N–O and CC vibrations in the aci-nitro species formed from NPE-caged ATP.23b Isotopically-sensitive bands that can be assigned to the carboxylate group in the aci-nitro intermediate 2 and the by-product 4 give additional confirmation for operation of the usual pathway. The extended conjugation provided by the carboxylate substituent in the aci-nitro system is also consistent with the red shift of the UV-visible absorption band for this intermediate, as described in the Results. The IR data also give unequivocal evidence that the dominant pathway for release of the caged species, monomethyl phosphate in the case of 7, necessarily involves formation and decay of the aci-nitro intermediate and also formation of the 2-nitrosophenylglyoxylate by-product 4. This is shown by the fact that development of the characteristic absorption of the MeOPO32– product near 1100 cm–1 correlates with decay of the aci-nitro bands and not with the fast phase of CO2 release during photolysis of 7. There does appear to be a small amount of product release during the slow CO2 phase for 7, but the chemistry involved in this minor pathway is not clear. There is no evidence for a comparable slow phase of product release from 8.
N, N–O and CC vibrations in the aci-nitro species formed from NPE-caged ATP.23b Isotopically-sensitive bands that can be assigned to the carboxylate group in the aci-nitro intermediate 2 and the by-product 4 give additional confirmation for operation of the usual pathway. The extended conjugation provided by the carboxylate substituent in the aci-nitro system is also consistent with the red shift of the UV-visible absorption band for this intermediate, as described in the Results. The IR data also give unequivocal evidence that the dominant pathway for release of the caged species, monomethyl phosphate in the case of 7, necessarily involves formation and decay of the aci-nitro intermediate and also formation of the 2-nitrosophenylglyoxylate by-product 4. This is shown by the fact that development of the characteristic absorption of the MeOPO32– product near 1100 cm–1 correlates with decay of the aci-nitro bands and not with the fast phase of CO2 release during photolysis of 7. There does appear to be a small amount of product release during the slow CO2 phase for 7, but the chemistry involved in this minor pathway is not clear. There is no evidence for a comparable slow phase of product release from 8.
As would be expected from previous experience the amide compound 9, for which the possibility of photodecarboxylation is blocked, also photolyses via the usual pathway, where product release is again concurrent with decay of the aci-nitro intermediate. However, the striking difference between 9 and its analogue 7 is in the relative rates of aci-nitro decay and hence of product release. From the UV-visible transients, this rate constant for 9 is over 100-fold slower than for 7, an effect related solely to the change between a carboxamide and a free carboxylate as the α-substituent of the caging group. Since the fast rate of product release is a key feature of CNB-caged compounds, this effect deserves consideration. In practice, substantial differences in aci-nitro decay rates consequent on structural changes have also been described for NPE-caged compounds. For example, the rate for NPE-caged phosphate is fast (2.4 × 104 s–1 at pH 7.1, 16 °C)35 whereas, when the phosphate group is present as an anhydride (e.g. in NPE-caged ATP) or a diester (e.g. in NPE-caged monomethyl phosphate), the product release rate is several hundred-fold slower: aci-nitro decay rates measured from UV-visible transients for NPE-caged ATP and NPE-caged monomethyl phosphate under similar conditions are 86 s–1 (pH 7.1, 21 °C)12 and 140 s–1 (pH 7.0, 21 °C).36 The latter value is ∼5-fold faster than for the caged monomethyl phosphate 9 and 25-fold slower than for the CNB-caged monomethyl phosphate 7.
We suggest that these rate differences can, at least in part, be explained on the basis of recent experimental and computational studies.10 These showed that decomposition of aci-nitro intermediates such as 2via the bicyclic species 3 required uptake of a proton by 2, since cyclisation to the deprotonated form of 3 was highly endothermic. Since the conjugate acid of 2 is a moderately strong acid (for example, pKa = 3.7 for the nitronic acid formed from 2-nitrotoluene37) it will be almost fully ionised at neutral pH values, and flux through the bicyclic intermediate 3 will be influenced by the position of the ionisation equilibrium (Scheme 1). While this may not be the only influence on the overall reaction rate, the presence of other negative charge in the caging group or the caged moiety can reasonably be expected to decrease the acidity of the nitronic acid, i.e. a higher proportion of the aci-nitro intermediate will be present as its conjugate acid at neutral pH. Such effects can be substantial: in the extreme, the two pKa values for maleic acid differ by >4 pH units, while even for the rotationally-mobile succinic acid there is a difference of ∼1.5 units.38 Thus the presence of the ionised carboxylate in all CNB cages should promote the rate of aci-nitro decay. In line with this argument, the aci-nitro decay rates (Table 1) of the CNB-caged compounds 7 and 8 are within a 10-fold factor of that for NPE-caged phosphate. All three compounds have two negative charges. Conversely, it is reasonable to suppose that the aci-nitro species formed from 9 will be a weaker base than that formed from NPE-caged monomethyl phosphate because the differing inductive effects of the α-carboxamide or methyl substituents would result in relative stabilisation of the anion formed from 9, hence to a lower equilibrium proportion of its conjugate acid and a slower aci-nitro decay rate.
Importantly, our results confirm that product release from CNB-caged compounds is faster than for the generality of NPE-caged compounds, in line with much previous data (see ref. 3–9 for data on a range of CNB-caged compounds and ref. 39 for a survey of rates for a range of NPE-caged compounds). While this has previously been inferred from aci-nitro decay rates and results of biological experiments, the present data provide the first direct observation of product release.
In the case of carbamate 8, release of the end product (glycine) is limited by thermal decarboxylation of the initially-formed carbamate salt (Scheme 5), which is much slower than the photolytic release rate. We have previously reported40 a rate constant of 150 s–1 for decarboxylation of N-carboxysarcosine at 21 °C, pH 7 but the present value for N-carboxyglycine (4.3 s–1, 1 °C) is slower than that expected at the lower temperature. Differences in the pK of the different amines and in the solution conditions are likely to account for the discrepancy. Previous studies of carbamate decarboxylation have discussed at length the effects of ionic strength, buffer concentration and pK of the amine on the rate of this decarboxylation.41
In the context of product release rate measurements, we note the previous work of Cheng et al.,9 who used the faster time resolution of step-scan IR to monitor aci-nitro decay for a CNB-caged carboxylate. They reported a rate constant of ∼50000 s–1 for product release (at an unspecified temperature) based on decay of a positive band at 1504 cm–1 that was assigned to a stretching vibration of the CN bond in the aci-nitro intermediate. The assignment was based on citation of our previous work,23 in which we had definitively assigned a band at 1465 cm–1 to this vibration. Given the results reported herein, where the CNB-caged compounds show a band consistent with this vibration in the range 1440–1458 cm–1, (i.e. slightly downshifted from the frequency for this vibration in the intermediate from NPE-caged ATP) it is unclear that the same vibration would be upshifted by ∼40 cm–1 in the compound studied by Cheng et al.,9 and their assignment must remain tentative.
Up to this point, our data support the usual scheme for photorelease of products from these compounds, as shown in Scheme 1. However, we have also shown that there is a second, less well-characterised aspect of their reactivity, namely the photodecarboxylation that could be expected from previous findings with simple nitrophenylacetates.11,28 However, the decarboxylation can also be more complex than a rapid photo-induced process, as seen in the slow phase of CO2 release that was observed for 7, and the species that remain after loss of CO2 from the benzylic carboxylate group have not been identified here. To some extent, they may be responsible for the slowly-decaying, shorter wavelength components of UV-visible transients observed with 7 and 8 but this cannot be a full explanation as the IR spectra for photolysis of 8 show no observable slow phase of decarboxylation from the benzylic carboxylate. Attempts to quantify the extent of this decarboxylation showed that up to ∼60 (±24)% of the reaction flux was by the photodecarboxylation path in MOPS buffer, dropping to about half this level in phosphate buffer. As described above, the spectra in MOPS buffer were somewhat more complex than in phosphate or EPPS buffers, with some unexplained bands that cannot be related to the usual pathway. Unfortunately, the decarboxylation pathway(s) do not lead to bands in the IR spectra that can easily be correlated with structures of products thereby formed (apart from CO2 itself and the carboxylate group discussed in the Results section that is associated with slow CO2 release from 7). The data do provide some evidence that different intermediates may be involved for different caged compounds, as shown by the different kinetics of CO2 liberation from compounds 7 and 8. Significantly, the spectra show that the photodecarboxylation process(es) do not result in release of the caged species in these compounds, with the possible exception of the minor slow release from 7. Product release appears to take place solely, or almost so, via the usual aci-nitro pathway, as is shown by the data for 7 and the rapid phase of product release is certainly via the usual pathway. These data are less definitive for 8, although the quantitative data for the latter compound (see below) give some support to this view. A priori, it might have been expected from earlier studies11,28 that decarboxylation would lead simply to an aci-nitro species 15 (see Scheme 4), identical to that formed in a conventional photo-rearrangement of a 2-nitrobenzyl derivative. However, comparative data presented in the Results section for 2-nitrobenzyl monomethyl phosphate rule out this possibility.
Further support for the proposition that the photodecarboxylation does not lead to product release comes from the observation that the yield of glycine from photolysis of 8 (in phosphate buffer) is only about 55% of that expected if the photolysis were stoichiometric. This value is unlikely to be affected by measurement error, since we have previously shown by the same analytical methods (quantitative HPLC and amino acid analysis) that we obtain essentially stoichiometric release of amino acids from other caged precursors.34 However, there is a discrepancy between this value and that indicated by the estimations of CO2 formation, which indicate that the amounts of CO2 released from the CNB side chain and from the carbamate of 8 are equal (∼33% each) in phosphate buffer. While the latter data are consistent in respect of the equal formation of CO2 from the two groups under these conditions, the absolute values are less secure in view of possible errors in the method of CO2 estimation (see ESI†). Nevertheless, the combined IR and amino acid analytical data are consistent with substantial photolytic loss of the carboxylate of the CNB cage. Those molecules undergoing this decarboxylation do not release their caged moiety to any significant extent. As shown in the Results section, the extent of this non-productive photodecarboxylation is influenced by the nature of the buffer salt. We suggest that electron-transfer processes may be operative in amine-based buffers, but other radical intermediates could also be involved. In the absence of data on the identity of product(s) formed in this reaction, it is not feasible to speculate further on the underlying processes. However, we note that a related study of products formed on photolysis of 4-nitrophenylmandelic acid and its derived acetate showed considerable complexity, with product formation influenced by solvent conditions such as pH and the presence or absence of oxygen.42
Finally, we have not commented in detail on the effects of dithiothreitol, other than to note that it blocked the slow phase of CO2 release from 7. This or other thiols are expected to cause rapid reduction of a nitroso group, such as in the by-product 4, but the photolysis spectra showed only small changes when recorded in the presence of DTT. This contrasts with the detailed chemistry and related spectroscopic changes previously reported for the 2-nitrosoacetophenone by-product from NPE-caged compounds,23b and no evidence was observed of products related to those found in that earlier work. We did not attempt to investigate the thiol-related processes further.
Conclusions
The work described has revealed the fact of hitherto unrecognised decarboxylation in each of the model CNB-caged compounds, which is therefore likely to apply to all such compounds. At least for the two compounds studied, the fraction of molecules that undergo this side reaction do not release their caged species. Our data are consistent with product release being rapid and proceeding via the usual aci-nitro intermediate (Scheme 1), and this rate is accurately reported by UV-visible transients measured at 480 nm. However, we emphasise that in cases such as for the carbamate 8, the actual product released by photolysis is an N-carboxy salt, as shown in Scheme 5. Decay of this species requires thermal loss of the N-carboxy group and is rate-limiting when the photocleavage process is rapid, as for the CNB-caged compounds discussed here. In the context of biological applications of these compounds, the fact that at least half the photolysis product is the expected one means that the interpretation of biological experiments with them will not be qualitatively altered by our results here. Importantly, photocleavage takes place, at least to a predominant extent, in a single, fast phase.Given the diversity and complexity of the results discussed above, it was impractical to undertake a detailed identification of the range of photo-products that arise from the compounds studied, and we are unable to comment on the nature or fate of species formed by loss of the benzylic carboxylate. It is possible that other types of CNB-caged compound may show further differences and we do not imply that the specific results obtained here will apply in every case: however the common factor is almost certainly that all CNB-caged compounds undergo significant levels of decarboxylation upon photolysis. We suggest that the results serve as an alert to other workers to consider the detailed nature of the photochemistry from a particular CNB-caged compound that might be in use, notably as to whether any of the effects reported here, such as the reduced stoichiometry of product release, could have an influence upon experimental results.
Experimental
General
1H NMR spectra were recorded on JEOL FX90Q or Varian Unityplus 500 instruments, with internal reference of TMS (for solutions in CDCl3) or acetone (for solutions in D2O). J values are given in Hz. Analytical anion exchange chromatography was performed on a Whatman Partisphere SAX column (Cat. No. 4621–0505) with mobile phase flow rates 1.5 mL min–1 and detection at 254 nm. Preparative anion exchange chromatography was on a column of DEAE cellulose (2.5 × 40 cm). Triethylammonium bicarbonate (TEAB) buffer was prepared by bubbling CO2 into an ice-cold 1 M solution of redistilled triethylamine in water until the pH stabilised at ∼7.4. Pooled column fractions were evaporated at ∼1 mm Hg and freed from residual triethylamine by repeated evaporation from methanol. For NMR spectroscopy, triethylammonium salts were converted to sodium salts by treatment with Dowex 50 (Na form). Preparative reverse-phase chromatography was performed on a 7.8 × 300 mm column packed with Waters C18 Bulk Packing Material (Cat. No. 20594) at a flow rate of 2 mL min–1. Quantification of aqueous solutions of 7 and 8 was based on the spectrum of 2-nitromandelic acid in aqueous solution at pH 7, λmax/nm 265 (ε/M–1 cm–1 4200).13C-Labelled t-butyl 2-nitromandelate was prepared as described for the non-isotopic compound,17 using [13C]KCN (99% isotopic abundance, Goss Scientific, Essex, UK). Details for the synthesis of the precursors to 9 are reported in the ESI.†
:65)] to give a pale oil (0.4 g) which was dissolved in TFA (5 mL) and kept for 1 h at room temperature. TFA was then evaporated under reduced pressure and the residue was mixed with water (10 mL) and adjusted to pH 7.2 with 1 M NaOH. The material was purified by anion exchange chromatography (linear gradient formed from 10 and 500 mM TEAB, each 1 L) and fractions were analysed by anion-exchange HPLC (mobile phase 100 mM Na phosphate, pH 5.5–MeOH (100:13)], tR 7.6 min. Combined fractions were processed as described above to yield α-carboxy-2-nitrobenzyl phosphate as the Et3NH+ salt (0.67 mmol). 1H NMR (90 MHz, D2O), δ 8.09 (d, J 7, 1H, Ar-H3), 7.30–7.90 (m, 3H, Ar-H), 5.96 (d, JH,P 9.7, 1H, ArCH); LR-MS: calcd for (C8H5NO8P + 2H)–: 276; found: 276. The 13C isotopomer was prepared similarly. In addition to the aromatic proton signals described above, the 1H NMR spectrum (90 MHz, D2O) showed the benzylic proton as a doublet of doublets (JH,P 9.7 and JH,C 5.8).
An aqueous solution of the above phosphate (16.5 mL of 20 mM) was adjusted to pH 5 with dilute HCl, cooled in ice and stirred with an ethereal solution of diazomethane (∼0.3 M, 20 mL) for 30 min. Anion-exchange HPLC analysis [mobile phase 50 mM ammonium phosphate, pH 6–MeOH (10:1)] showed 2 new peaks of similar intensity, tR 1.2 and 2.2 min and a trace of the starting phosphate at 10.0 min. The earlier-eluting peaks were assumed to correspond to 7 (2.2 min peak) and its carboxylate methyl ester. The ether layer was removed and the aqueous solution was adjusted to pH 13 and kept overnight at room temperature. Further HPLC analysis showed that the 1.2 min peak had disappeared and the intensity of the 2.2 min peak had increased, consistent with hydrolysis of the carboxylate ester. The solution was adjusted to pH 7.4 and chromatographed on DEAE cellulose (linear gradient formed from 10 and 400 mM TEAB, each 1 L). Fractions containing the product were processed as described above to yield 7 as the Et3NH+ salt (0.29 mmol, 87%). 1H NMR (90 MHz, D2O), δ 8.06 (d, J 7, 1H, Ar-H3), 7.50–7.80 (m, 3H, ArH), 6.02 (d, JH,P 9.7, 1H, ArCH), 3.39 (d, JH,P 11.0, 3H, OMe); LR-MS: calcd for (C9H8NO8P + Na)–: 312; found: 312. The 13C isotopomer was prepared similarly and, in addition to the aromatic and methyl proton signals described above, its 1H NMR spectrum (90 MHz, D2O) showed the benzylic proton as a doublet of doublets (JH,P 9.7 and JH,C 5.8).
:3)] to give the di-t-butyl ester of 8 as a pale gum (0.27 g). This material was dissolved in TFA (4 mL) and kept at room temperature for 1 h. The TFA was evaporated and the residue was dissolved in water (60 mL) and adjusted to pH 7.2. The solution was chromatographed on DEAE cellulose (linear gradient formed from 10 and 350 mM TEAB, each 1 L) and fractions were analysed by anion-exchange HPLC [mobile phase 50 mM NH4 phosphate, pH 6–MeOH (10:1)], tR 3.2 min. Fractions containing the product were processed as described above to give 8 (Et3NH+ salt; 0.5 mmol, 50%). 1H NMR (90 MHz, D2O), δ 8.03 (d, J 7, 1H, Ar-H3), 7.48–7.84 (m, 3H, ArH), 6.40 (s, 1H, ArCH), 3.70 (s, 2H, CH2CO2–); LR-MS: calcd for (C11H8N2O8 + H)–: 297; found: 297. The 13C isotopomer was prepared similarly and, in addition to the aromatic and methylene proton signals described above, its 1H NMR spectrum (90 MHz, D2O) showed the benzylic proton as a doublet (JH,C 5.7).
:13)] showed a major peak, tR 1.5 min, with a trace amount of E (tR 4.3 min). The aqueous layer was washed with Et2O and adjusted to pH 6.5. The solution was freeze–dried, dissolved in a small volume of water and injected onto the preparative reverse-phase HPLC column equilibrated in 10 mM sodium phosphate, pH 5.5. The column was eluted with the same buffer for 2 h, with water for 75 min, then with H2O–MeCN (3:2). Fractions containing 9 were identified by anion-exchange HPLC (as above), partially evaporated to remove acetonitrile, adjusted to pH 7.2 and chromatographed on DEAE cellulose with a linear gradient formed from 10 and 200 mM TEAB (each 1000 mL). Pure fractions, identified by anion-exchange HPLC were combined, quantified by UV spectroscopy as for E and processed as described above to give 9 as its Et3NH+ salt (195 µmol; 72%). 1H NMR (D2O, 500 MHz) δ 8.21 (dd, J 8.4, 1.2, 1H, 3-H), 7.95 (dd, J 7.9, 1.0, 1H, 6-H), 7.89 (dt, J 7.9, 1.2, 1H, Ar-H), 7.66 (dt, J 8.4, 1.4, 1H, Ar-H), 6.64 (d, JH,P 9.6, 1H, CH), 3.52 (d, JH,P 11.0, 3H, OMe), 3.02 (s, 3H, NMe), 3.39 (s, 3H, NMe). LR-MS: calcd for (C11H14N2O7P)–: 317; found: 317.
:7)] to give a pale green syrup which solidified on standing. Crystallisation from EtOH gave 12 as yellow crystals (0.45 g, 52%), mp 108–109.5 °C; UV [λmax (EtOH)]/nm (ε/M–1 cm–1) 228 (7000), 301 (5700), 318 (5300); 1H NMR (500 MHz, CDCl3) δ 8.14 (dd, J 7.3, 1.8, 1H, H-3′), 7.85–7.91 (m, 2H, Ar-H), 7.72 (m, 1H, Ar-H), 3.83 (s, 3H, OMe); HR-MS: calcd for (C9H7NO4 + H)+: 194.0453: found: 194.0448.
:85)] to give a pale green syrupy solid that crystallised from diethyl ether–light petroleum to give 13 as yellow crystals (0.87 g, 62%), mp 79–81 °C; 1H NMR (500 MHz, CDCl3) δ 7.98–8.02 (m, 1H, Ar-H), 7.83–7.87 (m, 2H, Ar-H), 7.72–7.76 (m, 1H, Ar-H), 1.47 (s, 9H, CMe3). Anal. calcd for C12H13NO4: C, 61.27; H, 5.57; N, 5.95; found: C, 61.40; H, 5.56; N, 5.93.
Infrared spectroscopic measurements
Infrared difference spectra were recorded as described previously,24 in buffers made by adjusting 200 mM solutions of MOPS, KH2PO4 or EPPS with 3 M NaOH (or KOH for the phosphate buffer) to pH values specified in the text. Samples to observe and quantify normal and isotopic CO2 were prepared in H2O solvent. Samples to show the range 1800–900 cm–1 were prepared by evaporation of the test compound and buffer (both in H2O solution) on one of the IR cell windows and reconstituting the dried spots in D2O. pH values specified in the text refer to the pH of the original buffer and are not corrected for an effect of D2O.Evaluation of CO2 bands (Fig. 1 and 5) was performed by integration of peak areas between 2346 and 2340 cm–1 for 12CO2 and between 2280 and 2274 cm–1 for 13CO2 with respect to a baseline drawn between two points at either side of the bands. These were obtained by averaging data points between 2410 and 2380 cm–1 and between 2220 and 2120 cm–1 using method “E” for integration of band areas in the Bruker OPUS software. Details of the quantitative estimation of CO2 are given in the ESI.† For the four bands integrated for compound 9, the integration limits and baselines were as follows: 1646–1625 cm–1 (baseline limits 1738–1668 and 1575–1544 cm–1); 1328–1314 cm–1 (1340–1329 and 1311–1291 cm–1); 1244–1236 cm–1 (1255–1246 and 1232–1219 cm–1) and 1128–1092 cm–1 (1153–1132 and 970–935 cm–1).
Time-resolved UV-visible measurements
Absorption transients in the range 406–480 nm were triggered by a 30 ns, 308 nm pulse from a LambdaPhysik LPX105 XeCl excimer laser directed into a quartz cell with 10 × 10 mm path lengths, that was located in a thermostatted (20 °C) sample holder positioned in a Luzchem LFP-111 transient recorder (Luzchem Research, Quebec, Canada). Solutions contained the relevant compound (0.5 mM) in 25 mM buffers of MES (pH 6.5), MOPS (pH 7.0), EPPS (pH 8.0), CHES (pH 9.0) or CAPS (pH 10.0), all also containing 150 mM NaCl. Data were collected using the Luzchem software and analysed in Microsoft Excel.Acknowledgements
We thank the MRC Biomedical NMR Centre for access to facilities, the Knut och Alice Wallenbergs Stiftelse for funding the infrared spectrometer, and the EPSRC National Mass Spectrometry Centre for negative ion mass spectrometry. This work was supported in part by an ESEP grant from the Royal Society.References
- (a) J. E. T. Corrie, 2-Nitrobenzyl and 7-nitroindoline derivatives, in Dynamic Studies in Biology: Phototriggers, Photoswitches and Caged Biomolecules, ed. M. Goeldner and R. Givens, Wiley VCH, Weinheim, ch. 1.1, 2005 Search PubMed; (b) S. R. Adams and R. Y. Tsien, Controlling cell chemistry with caged compounds, Annu. Rev. Physiol., 1993, 55, 755–784 CrossRef CAS; (c) J. H. Kaplan, Photochemical manipulation of divalent cation levels, Annu. Rev. Physiol., 1990, 52, 897–914 CrossRef CAS; (d) Caged Compounds, Methods in Enzymology, ed. G. Marriott, Academic Press, New York, 1998, vol. 291 Search PubMed; (e) A. Pelliccioli and J. Wirz, Photochemical protecting groups: reaction mechanisms and applications, Photochem. Photobiol. Sci., 2002, 1, 441–458 RSC.
- J. H. Kaplan, B. Forbush and J. F. Hoffman, Rapid photorelease of adenosine 5′-triphosphate from a protected analogue: utilization by the Na:K pump of human red blood cell ghosts, Biochemistry, 1978, 17, 1929–1935 CrossRef CAS.
- (a) J. W. Walker, J. A. McCray and G. P. Hess, Photolabile protecting groups for an acetylcholine-receptor ligand-synthesis and photochemistry of a new class of ortho-nitrobenzyl derivatives and their effects on receptor function, Biochemistry, 1986, 25, 1799–1805 CrossRef CAS; (b) T. Milburn, N. Matsubara, A. P. Billington, J. B. Udgaonkar, J. W. Walker, B. K. Carpenter, W. W. Webb, J. Marque, W. Denk, J. A. McCray and G. P. Hess, Synthesis, photochemistry, and biological activity of a caged photolabile acetylcholine receptor ligand, Biochemistry, 1989, 28, 49–55 CrossRef CAS.
- (a) C. Y. Chang, B. Niblack, B. Walker and H. Bayley, A photogenerated pore-forming protein, Chem. Biol., 1995, 2, 391–400 CrossRef CAS; (b) P. Pan and H. Bayley, Caged cysteine and thiophosphoryl peptides, FEBS Lett., 1997, 405, 81–85 CrossRef CAS.
- (a) R. Wieboldt, K. R. Gee, L. Niu, D. Ramesh, B. K. Carpenter and G. P. Hess, Photolabile precursors of glutamate: synthesis, photochemical properties, and activation of glutamate receptors on a microsecond time scale, Proc. Natl. Acad. Sci. USA, 1994, 91, 8752–8756 CAS; (b) A. P. Billington, K. M. Walstrom, D. Ramesh, A. P. Guzikowski, B. K. Carpenter and G. P. Hess, Synthesis and photochemistry of photolabile N-glycine derivatives and effects of one on the glycine receptor, Biochemistry, 1992, 31, 5500–5507 CrossRef CAS; (c) D. Ramesh, R. Wieboldt, A. P. Billington, B. K. Carpenter and G. P. Hess, Photolabile precursors of biological amides: synthesis and characterization of caged o-nitrobenzyl derivatives of glutamine, asparagines, glycinamide and g-aminobutyrate, J. Org. Chem., 1993, 58, 4599–4605 CrossRef CAS; (d) K. R. Gee, L. Niu, K. Schaper and G. P. Hess, Caged bioactive carboxylates. Synthesis, photolysis studies, and biological characterization of a new caged N-methyl-D-aspartic acid, J. Org. Chem., 1995, 60, 4260–4263 CrossRef CAS; (e) R. Wieboldt, D. Ramesh, B. K. Carpenter and G. P. Hess, Synthesis and photochemistry of photolabile derivatives of g-aminobutyric acid for chemical kinetic investigations of the g-aminobutyric acid receptor in the millisecond time region, Biochemistry, 1994, 33, 1526–1533 CrossRef CAS.
- (a) F. M. Rossi, M. Margulis, C. M. Tang and J. P. Y. Kao, N-Nmoc-L-glutamate, a new caged glutamate with high chemical stability and low pre-photolysis activity, J. Biol. Chem., 1997, 272, 32933–32939 CrossRef CAS; (b) F. M. Rossi and J. P. Y. Kao, N-Nmoc-DBHQ, a new caged molecule for modulating sarcoplasmic/endoplasmic reticulum Ca2+ ATPase activity with light flashes, J. Biol. Chem., 1997, 272, 3266–3271 CrossRef CAS.
- (a) J. W. Walker, H. Martin, F. R. Schmidt and R. J. Barsotti, Rapid release of an α-adrenergic receptor ligand from photolabile analogues, Biochemistry, 1993, 32, 1338–1345 CrossRef CAS; (b) J. W. Walker, Z. Lu and R. L. Moss, Effects of Ca2+ on the kinetics of phosphate release in skeletal muscle, J. Biol. Chem., 1992, 267, 2459–2466 CAS.
- (a) C. Grewer, J. Jäger, B. K. Carpenter and G. P. Hess, A new photolabile precursor of glycine with improved properties: a tool for chemical kinetic investigations of the glycine receptor, Biochemistry, 2000, 39, 2063–2070 CrossRef CAS; (b) K. Schaper, S. A. M. Mobarekeh and C. Grewer, Synthesis and photophysical characterization of a new, highly hydrophilic caging group, Eur. J. Org. Chem., 2002, 1037–1046 CrossRef CAS.
- Q. Cheng, M. G. Steinmetz and V. Jayaraman, Photolysis of γ-(α-carboxy-2-nitrobenzyl)-L-glutamic acid investigated in the microsecond time scale by time-resolved FTIR, J. Am. Chem. Soc., 2002, 124, 7676–7677 CrossRef CAS.
- (a) M. Schwörer and J. Wirz, Photochemical reaction mechanisms of 2-nitrobenzyl compounds in solution. I. 2-Nitrotoluene: thermodynamic and kinetic parameters of the aci-nitro tautomer, Helv. Chim. Acta, 2001, 84, 1441–1457 CrossRef CAS; (b) Y. V. Il'ichev and J. Wirz, Rearrangements of 2-nitrobenzyl compounds. I. Potential, energy surface of 2-nitrotoluene and its isomers explored with ab initio and density functional theory methods, J. Phys. Chem. A, 2000, 104, 7856–7870 CrossRef CAS.
- J. D. Margerum and C. T. Petrusis, The photodecarboxylation of nitrophenylacetate ions, J. Am. Chem. Soc., 1969, 91, 2467–2472 CrossRef CAS.
- J. W. Walker, G. P. Reid, J. A. McCray and D. R. Trentham, Photolabile 1-(2-nitrophenyl)ethyl phosphate esters of adenine nucleotide analogues. Synthesis and mechanism of photolysis, J. Am. Chem. Soc., 1988, 110, 7170–7177 CrossRef CAS.
- J. E. T. Corrie, A. de Santis, Y. Katayama, K. Khodakhah, J. B. Messenger, D. C. Ogden and D. R. Trentham, Post-synaptic activation at the squid giant synapse by flash photolytic release of glutamate from a “caged” L-glutamate, J. Physiol., 1993, 465, 1–8 CAS.
- F. S. Parker, Applications of Infrared Spectroscopy in Biochemistry, Biology and Medicine, Plenum, New York, 1971, p. 365 Search PubMed.
- V. Jayaraman, S. Thiran and D. R. Madden, Fourier transform infrared spectroscopic characterization of a photolabile precursor of glutamate, FEBS Lett., 2000, 475, 278–282 CrossRef CAS.
- H. Brintzinger and G. G. Hammes, Inner- and outer-sphere complex formation in aqueous solutions of nickel(II)-methyl phosphate, Inorg. Chem., 1966, 5, 1286–1287 CrossRef CAS.
- J. E. T. Corrie, M. J. Gradwell and G. Papageorgiou, Non-photochemical rearrangements of o-nitrobenzyl compounds, J. Chem. Soc., Perkin Trans. 1, 1999, 2977–2982 RSC.
- H. Tomioka, N. Ichikawa and K. Komatsu, Photochemistry of (2-nitrophenyl)diazomethane studied by the matrix isolation technique. (Nitrophenyl)carbene to (carboxypheny)carbene rearrangement by successive reduction of the nitro group with the carbonic center, J. Am. Chem. Soc., 1992, 114, 8045–8053 CrossRef CAS.
- R. Sreekumar, Y. Q. Ping, X. P. Huang and J. W. Walker, Stereospecific protein kinase C activation by photolabile diglycerides, Bioorg. Med. Chem. Lett., 1997, 7, 341–346 CrossRef CAS.
- A. Barth, S. R. Martin and J. E. T. Corrie, Decarboxylation is a significant reaction pathway for photolabile calcium chelators and related compounds, Photochem. Photobiol. Sci., 2006, 5, 107–115 RSC.
- C. Ho and J. M. Sturtevant, The kinetics of hydration of carbon dioxide at 25°, J. Biol. Chem., 1963, 238, 3499–3501 CAS.
- J. E. T. Corrie, Y. Katayama, G. P. Reid, M. Anson and D. R. Trentham, The development and application of photosensitive caged compounds to aid time-resolved structure determination of macromolecules, Philos. Trans. R. Soc. London, Ser. A, 1992, 340, 233–244 CAS.
- (a) A. Barth, K. Hauser, W. Mäntele, J. E. T. Corrie and D. R. Trentham, The photochemical release of ATP from “caged ATP” studied by time-resolved infrared spectroscopy, J. Am. Chem. Soc., 1995, 117, 10311–10316 CrossRef CAS; (b) A. Barth, J. E. T. Corrie, M. J. Gradwell, Y. Maeda, W. Mäntele, T. Meier and D. R. Trentham, Time-resolved infrared spectroscopy of intermediates and products from photolysis of 1-(2-nitrophenyl)ethyl phosphates: reaction of the 2-nitrosoacetophenone by-product with thiols, J. Am. Chem. Soc., 1997, 119, 4149–4159 CrossRef CAS.
- G. Papageorgiou, A. Barth and J. E. T. Corrie, Flash photolytic release of alcohols from photolabile carbamates or carbonates is rate-limited by decarboxylation of the photoproduct, Photochem. Photobiol. Sci., 2005, 4, 216–220 RSC.
- (a) E. Campaigne, G. Skwronski and R. B. Rogers, Synthesis of β-dimethylaminoethyl and α-hydroxy-β-dimethylaminoethyl derivatives from organolithium reagents and tetramethoxamide, Synth. Commun., 1973, 3, 325–332 CrossRef CAS; (b) T. Kolasa and M. J. Miller, Reactions of α-hydroxy carbonyl compounds with azodicarboxylates and triphenylphosphine: synthesis of α-N-hydroxy amino acid derivatives, J. Org. Chem., 1987, 52, 4978–4984 CrossRef CAS; (c) J. Chen and R. F. Cunico, Synthesis of α-ketoamides from a carbamoylsilane and acid chlorides, J. Org. Chem., 2004, 69, 5509–5511 CrossRef CAS; (d) H. Bredereck, G. Simchen, G. Kapaun and R. Wahl, Über die Umsetzung von p-Tolyl-und Anisaldehyd mit einem Aminal-tert-butyl ester zu [1.2-Bis-dimethylamino-vinyl]-arylketonen, Chem. Ber., 1970, 103, 2980–2983 CrossRef CAS.
- Y. Pocker, J. E. Meany, B. J. Nist and C. Zadorojny, The reversible hydration of pyruvic acid. I. Equilibrium, studies, J. Phys. Chem., 1969, 73, 2979–2882.
- S. Pinchas and I. Laulicht, Infrared Spectra of Labelled Compounds, Academic Press, London, 1971, p. 212 Search PubMed.
- P. Wan and S. Muralidharan, Structure and mechanism in the photo-retro-aldol type reactions of nitrobenzyl derivatives. Photochemical heterolytic cleavage of C–C bonds, J. Am. Chem. Soc., 1988, 110, 4336–4365 CrossRef CAS.
- (a) W. P. Jencks and J. Carriuolo, Structure of pyruvate in aqueous solution, Nature, 1958, 182, 598–599 CrossRef CAS; (b) M. Kakihana and M. Okamoto, Vibrational analysis of pyruvate ion molecules and estimation of equilibrium constants for their hydrogen isotopic exchange reactions, J. Phys. Chem., 1984, 88, 1797–1804 CrossRef CAS.
- A. Barth and J. E. T. Corrie, unpublished data.
- M. Aresta, A. Dibenedetto and E. Quaranta, Reaction of alkali-metal tetraphenylborates with amines in the presence of CO2; a new easy way to aliphatic and aromatic alkali-metal carbamates, J. Chem. Soc., Dalton Trans., 1995, 3359–3363 RSC.
- J. Chouteau, Sur certaines anomalies du spectre infrarouge des acylglycines, C. R. Hebd. Seances Acad. Sci., 1953, 237, 891–893 Search PubMed.
- W. W. Wright and J. M. Vanderkooi, Use of IR absorption of the carbonyl group of amino acids and their metabolites to determine pKs, to study proteins and to monitor enzyme activity, Biospectroscopy, 1997, 3, 457–467 CrossRef CAS.
- (a) G. Papageorgiou, D. C. Ogden, A. Barth and J. E. T. Corrie, Photorelease of carboxylic acids from 1-acyl-7-nitroindolines in aqueous solution: rapid and efficient photorelease of L-glutamate, J. Am. Chem. Soc., 1999, 121, 6503–6504 CrossRef CAS; (b) G. Papageorgiou and J. E. T. Corrie, Effects of aromatic substitution on the photocleavage of 1-acyl-7-nitroindolines, Tetrahedron, 2000, 56, 8197–8205 CrossRef CAS.
- J. A. Dantzig, Y. E. Goldman, N. C. Millar, J. Lacktis and E. Homsher, Reversal of the cross-bridge force-generating transition by photogeneration of phosphate in rabbit psoas muscle fibres, J. Physiol., 1992, 451, 247–278 CAS.
- D. R. Trentham and J. E. T. Corrie, unpublished result.
- G. Wettermark, E. Black and L. Dogliotti, Reactions of photochemically formed transients from 2-nitrotoluene, Photochem. Photobiol., 1965, 4, 229–239 CrossRef CAS.
- (a) G. Kortüm, W. Vogel and K. Andrusson, Dissociation Constants of Organic Acids in Aqueous Solution, Butterworths, London, 1961, p. 267 Search PubMed; (b) E. P. Sergeant and B. Dempsey, Ionisation Constants of Organic Acids in Aqueous Solution, Pergamon, Oxford, 1979, p. 75 Search PubMed.
- J. E. T. Corrie and D. R. Trentham, in Biological Applications of Photochemical Switches, ed. H. Morrison, Wiley, New York, 1993, pp. 267–271 Search PubMed.
- G. Papageorgiou and J. E. T. Corrie, Synthesis and properties of carbamoyl derivatives of photolabile benzoins, Tetrahedron, 1997, 53, 3917–3932 CrossRef CAS.
- (a) M. Caplow, Kinetics of carbamate formation and breakdown, J. Am. Chem. Soc., 1968, 90, 6795–6803 CrossRef CAS; (b) S. P. Ewing, D. Lockshon and W. P. Jencks, Mechanism of cleavage of carbamate anions, J. Am. Chem. Soc., 1980, 102, 3072–3084 CrossRef CAS.
- J. Morrison, P. Wan, J. E. T. Corrie and V. R. N. Munasinghe, Chemistry of photogenerated α-hydroxy-p-nitrobenzyl carbanions in aqueous solution: protonation vs. disproportionation, Can. J. Chem., 2003, 81, 586–597 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Photolysis spectra of NPE-caged monomethyl phosphate (Fig. S1) and of 2-nitrobenzyl monomethyl phosphate (Fig. S2); isotope effect spectra for photolysis of the two isotopomers of 7 (Fig. S3); details of the synthesis of the precursor compounds for synthesis of 9 together with the method used for quantitative measurement of CO2 formation in the IR spectroscopic cells. See DOI: 10.1039/b711398f |
| This journal is © The Royal Society of Chemistry and Owner Societies 2008 |