A cholic acid-based fluorescent chemosenor for the detection of ATP†
Hao
Wang
and
Wing-Hong
Chan
*
Department of Chemistry, Hong Kong Baptist University, Kowloon Tong, Hong Kong SAR, China. E-mail: whchan@hkbu.edu.hk; Fax: +852 34117348; Tel: +852 34117076
First published on 21st November 2007
Abstract
A novel ditopic cholic acid-based fluorescent chemosensor for ATP, 1a, was designed and synthesized. Its interactions with phosphates, AMP, ADP, ATP, CTP, GTP, and TTP have been investigated. When ATP was added to a 1 : 1 aqueous CH3CN solution of the sensor at pH 7.4, a significant decrease in fluorescence of 1a was observed, whereas other guest molecules showed a much smaller effect. The complex between 1a and ATP was confirmed through combined UV, 1H, 13C and 31P NMR spectroscopic methods. The uniqueness of the new sensor is that it binds with ATP 33–124 times more selectively than other nucleotides , as evidenced from the respective binding constants. 1a is a highly sensitive sensing probe; as little as 30 nM ATP can cause 15% fluorescence quenching of the sensor.
Introduction
The development of abiotic molecular systems with tailored binding properties for biologically relevant species in aqueous media remains a challenge for supramolecular chemistry.1 Due to the significance of molecular recognition, numerous efforts have been devoted to designing synthetic luminescent and spectrophotometric sensors capable of reporting recognition events in real time.2 Among various biologically important anions, phosphate derivatives, encompassing nucleotides and inorganic pyrophosphate, have long been significant targets because of their pivotal roles in biological systems. For instance, ATP is known not only as a universal “energy currency”3a but also as an extracellular signaling mediator in many biological processes.3b,c It is therefore not surprising that we have recently witnessed much research into the development of chemosensors for phosphate derivatives in the literature.4 However, in most reported cases, these chemosensors are selective for ATP over the less charged ADP and AMP.4a–c,g,j,l,p On the other hand, sensors bearing two zinc metal centers as the binding sites and exhibiting high selectivity for pyrophosphate over other nucleotides have been documented.4e,h,n In contrast, only two chemosensor developments in the literature are claimed to be able to selectively sense ATP and GTP, respectively, over other nucleotides .4f,kInspired by the seminal work of Davis and coworkers employing cholic acid derivatives as versatile synthetic hosts,5 for the past few years we have been engaged in using cholic acid as the molecular platform to construct fluorescent chemosensors for cations, anions and neutral molecules (i.e. mercuric ion, halides, carboxylates, dicarboxylates and amino acids).6 To augment the binding capacity of the tripodal cholic acid, we have uncovered for the first time that the C17 flexible side chain of the host can be exploited as an additional receptive site in supramolecular chemistry.6c To continue our effort in this significant field of research, we herein report the development of a cholic acid-based fluorescent chemosensor for ATP. This ATP sensor, operating at pH 7.4 in aqueous media, exhibits outstanding selectivity not only over phosphate derivatives but also other nucleotides .
Results and discussion
Design of the chemosensor
At the outset of the study, we wanted to introduce multi-receptive sites for binding to the nucleobase and the triphosphate terminus of ATP. Working on the cholic acid scaffold, our strategy was (1) to transform the C17 side chain carboxylic group into an amidothiourea functionality which can serve as a selective phosphate receptor; and (2) to append a vicinal diamino moiety onto the C7 and C12 hydroxyl groups which can interact cooperatively with nucleobase(s) through hydrogen bonding. To confer the host with fluorescence signal display ability, an anthracene moiety was introduced to the thiourea moiety, with a methylene spacer.2bSynthesis of the chemosensor
To realize the aforementioned strategy, ditopic probe 1a was first constructed by the synthetic route outlined in Scheme 1. Condensation of cholic acid derivative 27 with bromoacetyl bromide afforded dibromide 3 in 85% yield. In the presence of NaOH, treatment of 3 with the (R,R)-1,2-diammoniumcyclohexane mono-(+)-tartrate salt8 in CH2Cl2 led to the formation of cyclic diamine 4 in 86% yield. The chiral 1,2-diaminocyclohexane was used in order to avoid the formation of a diastereomeric mixture of products. Through this transformation, the first receptive site of the sensor, comprising two vicinal amino groups capable of forming hydrogen bonds with guest molecules, was incorporated onto the cholic acid framework. Hydrolysis of acetate 4 with K2CO3 in methanol gave rise to the control compound 1b. Both the amino groups and the ester groups of 1b can potentially act as binding sites for nucleic acid in aqueous solutions. To endow the sensor with a second binding site and to incorporate a fluorophore capable of detecting the binding event, 1b was treated with hydrazine in methanol at 50 °C, affording the corresponding hydrazide 5 in 85% yield. It is worthy of note that the C7 and C12 ester functionalities, due to their steric environment, survived very well under the reaction conditions; to our delight, no trace of the hydrolyzed product was obtained. To complete the synthesis, 5 was allowed to react with 9-(thioisocyanomethyl)anthracene to give rise to sensor 1a.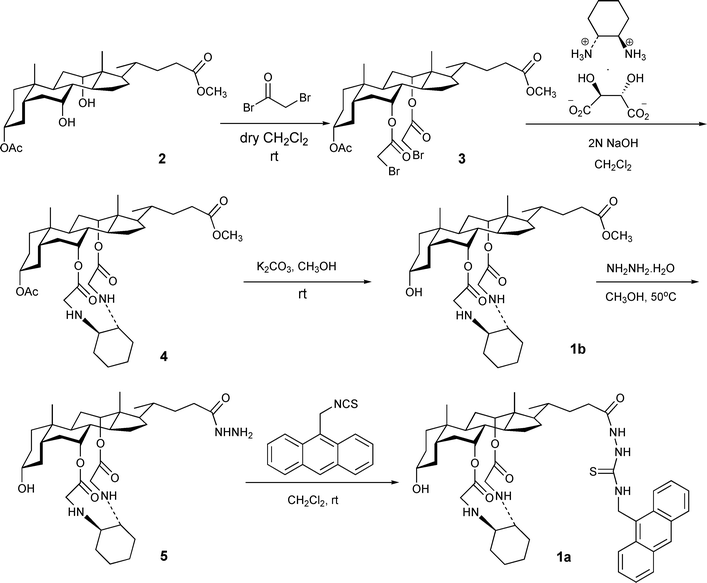
We envisage that the ditopic nature of the sensor should confer it with binding affinity to bifunctional guest molecules such as nucleotides . Notably, the flexible C24 side chain bearing the amidothiourea group should bind strongly with phosphate, while the cyclic vicinal amino groups attached to the C7 and C12 positions of cholic acid could work cooperatively to provide an additional binding site for hydrogen bond donors. The former binding event can modulate the fluorescence of anthracenevia a photo-induced electron transfer (PET) process such that quantitative work can be conducted.6c
Complexation of nucleic acids with 1b
To shed light on the viability of the vicinal diamino groups in complexing nucleobases, a 1 : 1 aqueous CH3CN solution of each of the four nucleobases (i.e.adenine, A; thymine, T; guanine, G; and cytosine, C) was titrated with the control compound 1b in pH 7.4 10 mM HEPES buffer solutions. The extent of interaction between the host and the guest was monitored qualitatively by UV spectroscopy. In all four cases, the addition of the host to the nucleobase solutions induces a bathochromic shift and an enhancement of the absorbance of the bases. Judging from the extent of the shift, it is quite evident that a stronger host–guest interaction took place between adenine and 1b (Fig. 1). By complexing with the host, adenine underwent the greatest red-shift of 8 nm while the other three nucleic bases recorded a bathochromic shift of about 5 nm. In addition, the magnitude of interactions between various nucleobases to the host was reflected by the extent of change in absorbance. An increase of absorbance by 38.7%, 27.3%, 16.7%, and 11.5% was observed for the binding of adenine, cytosine, guanine, and thymine, respectively (Fig. S1† ). The hydrogen bond(s) between the vicinal diamino groups of the host and the amino group of adenine and cytosine is conceivably responsible for triggering the change in the UV spectra of the nucleobases. Pertaining to our sensor design for ATP, the binding ability of the cyclic vicinal diamine moieties towards nucleobases, particularly to adenine, has been firmly established.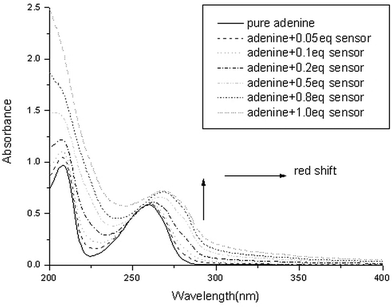 | ||
| Fig. 1 UV titration of adenine (50 µM) with increasing amounts of compound 1b in CH3CN–H2O (1 : 1, v/v), HEPES = 10 mM, pH = 7.4. | ||
Fluorescence titrations of sensor 1a with various phosphate derivatives
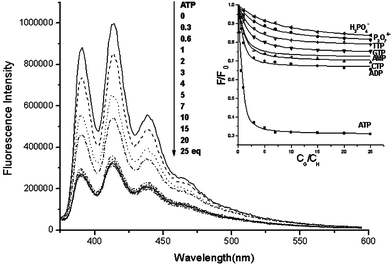
Having set the stage for investigating the fluorescent chemosensing capacity of sensor 1a compared to the control compound 1b, we next initiated the full study of the fluorescence of 1a upon titration with the sodium salts of all four nucleotides , AMP, ADP, tetrabutylammonium dihydrogen phosphate and sodium pyrophosphate. This was carried out by adding incremental amounts of the guests to a 0.1 µM solution of 1a in 10 mM HEPES pH 7.4 aqueous CH3CN (1 : 1, v/v). To explore the potential applications of the sensor, a higher content of water in the aqueous measuring solutions seemed to be desirable. However, such an approach was constrained by the limited solubility of the sensor in water. As a reasonable compromise, a 1 : 1 aqueous CH3CN solution was used in the subsequent study. In all titrations, significant fluorescence emission was observed upon excitation at 366 nm. A typical anthracene emission was observed, with emission peaks at 443 nm, 419 nm, and 397 nm, and a shoulder at 473 nm. Upon the addition of 20 equiv. of the guests, as shown in Fig. 2, the intensity of the anthracene emission bands gradually decreased, with no other spectral changes being detected. Such a phenomenon of fluorescence quench upon anion recognition, due to enhanced efficiency of electron transfer quenching from the receptor to the excited state of the anthracene moiety, has widely documented.9 However, the ability of each of the biologically relevant phosphate derivatives to quench the fluorescence of the sensor is interestingly different, indicating the variation of binding affinity of the host to the guests. ATP displays the greatest effect on quenching the fluorescence of the sensor, reducing the fluorescence by as much as 70%.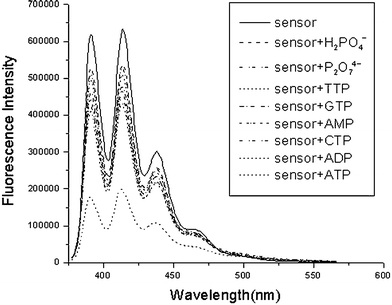 | ||
| Fig. 2 Changes in the fluorescence emission spectra of 1a (0.1 µM) upon addition of 20 equiv. of the sodium salts of AMP, ADP, ATP, CTP, GTP, and TTP, sodium pyrophosphate and tetrabutylammonium dihydrogen phosphate at pH 7.4 (10 mM HEPES) in 1 : 1 aqueous CH3CN solution. | ||
It is also apparent from Fig. 2 that the ability of various guests to quench the sensors via a PET process can be arranged in the following descending order: ATP >> ADP > CTP > GTP > TTP > H2PO4− ∼ P2O74−. The binding stoichiometry of the complex of 1a and ATP was confirmed to be 1 : 1 by a Job plot (Fig. S2† ). Careful and systematic fluorescent titrations of the sensor with individual guests allow the evaluation of the binding constant of each of the complexes (inset of Fig. 3). The sensor is extremely sensitive to ATP, as exemplified in the titration graphs shown in Fig. 3 (i.e. the second curve from the top). 30 nM of ATP can induce 15% fluorescent change of the sensor. The binding constants of 1a and ATP in different compositions of aqueous CH3CN were also evaluated, and the results are compiled in (Table S1† ).
 | ||
| Fig. 3 Changes in the fluorescence emission spectra of 1a (0.1 µM) upon addition of ATP anion (0–2.5 µM) in aqueous CH3CN (1 : 1) buffer solution at pH 7.4. Inset: The dependence of the fluorescence of the sensor on the concentration of different guests. | ||
Using non-linear least-square fitting, the respective binding constants of the studied sensor–guest pairs are reported in Table 1.10 By careful examination of the relative values of the binding constants (which are in agreement with many literature reports), the gradual decrease in binding constant in the ATP, ADP and AMP series is readily explainable. The enhanced electron density accumulating in the phosphate terminus on moving from AMP to ADP to ATP means that they exhibit different binding with the thiourea receptor of the sensor. In contrast to other analogs, it is understandable that dihydrogen phosphate and pyrophosphate, which bear only one effective binding functionality, should display the weakest interaction with the sensor. However, the selectivity exhibited by the sensor, ranging over more than two orders of magnitude (i.e. 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 950000 for ATPversus 56200 for TTP) among the four nucleotides , is most interesting and intriguing. The second receptor, comprising vicinal diamine groups lying in the core of the sensor, must contribute crucially to this discriminative power.
950000 for ATPversus 56200 for TTP) among the four nucleotides , is most interesting and intriguing. The second receptor, comprising vicinal diamine groups lying in the core of the sensor, must contribute crucially to this discriminative power.
| Analyte | Binding constant Kass/M−1 | R | No. of H-bonding sites for 1a |
|---|---|---|---|
| a The value is too small to be calculated. | |||
| ATP | (6.95 ± 0.11) × 106 | 0.9962 | 4 |
| ADP | (5.79 ± 0.18) × 105 | 0.9921 | 4 |
| CTP | (2.08 ± 0.17) × 105 | 0.9914 | 4 |
| AMP | (1.71 ± 0.12) × 105 | 0.9927 | 4 |
| GTP | (6.81 ± 0.46) × 104 | 0.9972 | 3 |
| TTP | (5.62 ± 0.49) × 104 | 0.9938 | 3 |
| H2PO4− | (8.17 ± 0.10) × 103 | 0.9962 | 2 |
| P2O74− | (2.80 ± 0.25) × 104 | 0.9982 | 2 |
| Glutamate | (2.83 ± 0.38) × 105 | 0.9914 | — |
| Acetate | (1.37 ± 0.20) × 103 | 0.9915 | 2 |
| Alanine | 527 ± 17 | 0.9978 | 2 |
| Chloride | —a | — | — |
To rationalize the selectivity of the sensor towards different nucleotides and phosphate, the complementary relationships between the host and the guests in terms of electronic and spatial characteristics were scrutinized. Cursory examination of molecular models revealed that ATP and GTP bearing a purine ring can stretch apart far enough to enable on the one hand the triphosphate terminus to reach the C17 thiourea receptive site, while on the other hand to allow the nucleobase terminus to interact with the diamino groups via hydrogen bonding (Fig. 4). Therefore, on the basis of the spatial complementary factor, the sensor 1a can bind better with ATP and GTP than with CTP and TTP. However, ATP and CTP, possessing an amino group situated in the correct place, can conceivably interact with the vicinal amino groups via two hydrogen bonds, as shown in Fig. 4. In contrast, GTP and TTP can only form one hydrogen bonding interaction with the second receptive site of the host. The additional hydrogen bonding capacity of ATP and CTP makes them better guests than GTP and TTP, which are in turn better than the mono-functionalized phosphates.
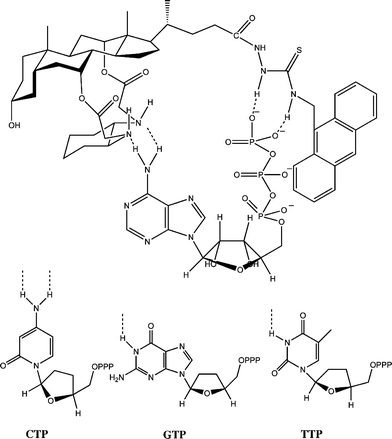 | ||
| Fig. 4 The proposed binding mode of 1a and ATP (and other nucleotides ) by multiple hydrogen bonding. For simplicity, the hydroxyl groups in the ribose units of CTP, GTP and TTP are not shown. | ||
To define the full scope of the selectivity of the sensor, the fluorescence response of 1a to other common anions was also determined, and their association constants are included in Table 1. As expected, the spherical chloride ion did not bind with either receptive site, while trigonal planar carboxylates such as acetate and alanine can complex the amidourea group with two hydrogen bonds. Being a stronger acid than acetate, alanine binds with 1a in a much weaker manner. It is interesting to note that glutamate, being a trifunctional amino acid, binds to the sensor with a fairly high binding constant. Systematic studies of the interaction between various amino acids and 1a to elucidate their binding mode are underway, and the results will be reported in due course.
Investigating the ATP recognition by the sensor using 1H, 13C and 31P NMR
To clarify the structure of the complex, 1H, 13C and 31P NMR experiments were conducted. A mixture of D2O and CH3CN (1 : 1, v/v) and millimolar concentrations of the sensor were used to avoid precipitation. Due to the presence of a large number of cholestic protons, the 1H NMR spectrum of the sensor is very complicated, particularly in the upfield region. Also, in D2O solvent, all amino protons and thiourea N–H protons were too broad to locate. Nevertheless, upon binding, substantial complexation-induced shifts (CISs) of certain critical protons of the sensor and the guest can be clearly identified. As shown in Fig. 5 and Table 2, significant upfield shifts were observed for the two purine protons (H2 and H8) of ATP as well as all aromatic protons of the anthracene moiety of the sensor. The two aromatic protons of the adenine unit, originally at 8.22 and 8.38 ppm, respectively, were significantly shifted to 7.62 and 7.88 ppm, respectively, when an equimolar mixture of ATP and 1a was measured. These considerable CIS values (i.e. −0.6 and −0.5 ppm) strongly suggest that strong interactions are taking place between the host and the guest. In contrast, the CIS of the anthracene aromatic protons of 1a, ranging from −0.08 to −0.30 ppm as a result of the binding to the phosphate group of ATP, has many literature precedents.2d The upfield shifts are common for guest binding, and in our case it denotes an effect of a microenvironment of high electron density, due to the lone electron pairs of vicinal diamino nitrogens and the hydrogen bonded dithiourea group.11 13C NMR data allowed us to completely rule out the participation of the two pendent ester groups in binding ATP, as the chemical shifts of these two carboxyl carbons of the host were 169.2 and 171.1 ppm, remaining unchanged after binding with the guest. On the other hand, clear upfield shifts of all five purine carbons in ATP were observed as a result of the complexation (Fig. S3 and Table S2† ).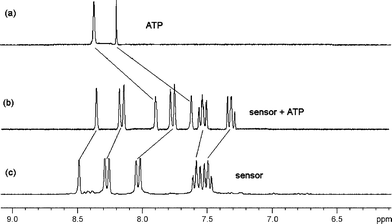 | ||
| Fig. 5 Partial 1H NMR spectrum of (a) ATP, (b) sensor + ATP (1 equiv.) and (c) sensor in CD3CN–D2O (1 : 1). | ||
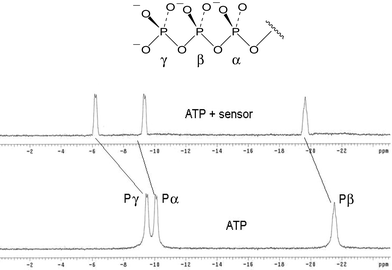
Finally, the 31P NMR study of the complex of the sensor and ATP revealed that, after binding, the α-, β- and γ-phosphorus atoms of ATP recorded different extents of upfield shift, according to their distance from the receptor (Fig. 6). The relatively large upfield shift observable for the 31P signals (4.2 and 1.8 ppm for γ- and β-phosphorus atoms) of the complex suggested the formation of very strong hydrogen bonds between the host and guest.
In summary, on the basis of NMR observations and the fluorescence data of the complex, together with the aforementioned UV spectroscopic results on the interaction of nucleobases with the control compound 1b, a plausible binding mode (as described in Fig. 4) was adequately established.
Conclusions
We have described the design and synthesis of a novel cholic acid-based fluorescent chemosensor for ATP. We have demonstrated that the ditopic sensor 1a, bearing thiourea and vicinal diamine receptive sites, forms complexes with nucleotides via multiple hydrogen bonding interactions. The binding can be conveniently examined by fluorescence titrations. The impressive selectivity of the sensor towards ATP over other biologically related nucleotides was rationalized on the basis of the combined effect of electronic and spatial factors. A plausible binding model was proposed and was substantiated by combined spectroscopic methods. The sensing can be conducted in aqueous solutions with sensitivity on the sub-micromolar level.Experimental
1. General methods
Melting points were determined with an MEL-TEMPII melting-point apparatus (uncorrected). 1H, 13C and 31P NMR spectra were recorded in CDCl3, with Me4Si as the internal standard, on a JEOL EX270 spectrometer. High-resolution mass spectra were recorded on a Bruker Autoflex mass spectrometer (MALDI-TOF). Absorption spectra were recorded on a Hewlett Packard 8453 UV-vis spectrophotometer. Fluorescence emission spectra were collected on a Photon Technology International (PTI) spectrometer at 293 K. CH2Cl2 was dried and distilled from CaH2. All other commercially available reagents were used as received.2. Synthesis of 1a, 1b, and 3–5
Acknowledgements
The project was supported by grants from the Science Faculty and the Research Committee of Hong Kong Baptist University (FRG-05/06-II51).References
- For a recent review: G. V. Oshovsky, D. N. Reinhoudt and W. Verboom, Angew. Chem., Int. Ed., 2007, 46, 2366 Search PubMed.
- (a) Fluorescent Chemosensors for Ion and Molecular Recognition, ed. A. W. Czarnik, American Chemical Society, Washington, DC, 1993 Search PubMed; (b) A. P. de Silva, H. Q. N. Gunaratne, T. A. Gunnlaugsson, T. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515 CrossRef; (c) R. Martinez-Manez and F. Sancanon, Chem. Rev., 2003, 103, 4419 CrossRef CAS; (d) T. Gunnlaugsson, M. Glynn, G. M. Tocci, P. E. Kruger and F. M. Pfeffer, Coord. Chem. Rev., 2006, 250, 3094 CrossRef CAS.
- (a) W. N. Lipscomb and N. Strater, Chem. Rev., 1996, 96, 2375 CrossRef CAS; (b) R. D. Field and B. Stevens, Trends Neurosci., 2000, 23, 625 CrossRef CAS; (c) A. V. Gourine, E. Llaudet, N. Dale and K. M. Spyer, Nature, 2005, 436, 108 CrossRef CAS.
- (a) M. T. Albelda, M. A. Bernardo, E. Garcia-España, M. L. Godino-Salido, S. V. Luis, M. J. Melo, F. Pina and C. Soriano, J. Chem. Soc., Perkin Trans. 2, 1999, 2545 RSC; (b) A. Ojida, S.-K. Park, Y. Mito-oka and I. Hamachi, Tetrahedron Lett., 2002, 43, 6193 CrossRef CAS; (c) S. Atilgan and E. U. Akkaya, Tetrahedron Lett., 2004, 45, 9269 CrossRef CAS; (d) M. S. Vickers, K. S. Martindale and P. D. Beer, J. Mater. Chem., 2005, 15, 2784 RSC; (e) D. H. Lee, S. Y. Kim and J.-I. Hong, Angew. Chem., Int. Ed., 2004, 43, 4777 CrossRef CAS; (f) J. Y. Kwon, N. J. Singh, H. N. Kim, S. K. Kim, K. S. Kim and J. Yoon, J. Am. Chem. Soc., 2004, 126, 8892 CrossRef CAS; (g) C. Li, M. Numata, M. Takeuchi and S. Shinkai, Angew. Chem., Int. Ed., 2005, 44, 6371 CrossRef CAS; (h) H. K. Cho, D. H. Lee and J.-I. Hong, Chem. Commun., 2005, 1690 RSC; (i) A. M. Costero, J. Sanchis, S. Gil, V. Sanz and J. A. G. Williams, J. Mater. Chem., 2005, 15, 2848 RSC; (j) Z. Kejik, K. Zaruba, D. Michalik, J. Sebek, J. Dian, S. Pataridis, K. Volka and V. Kral, Chem. Commun., 2006, 1533 RSC; (k) C. Bazzzicalupi, S. Biagini, A. Bencini, E. Faggi, C. Giorgi, I. Matera and B. Valtancoli, Chem. Commun., 2006, 4087 RSC; (l) A. Ojida, H. Nonaka, Y. Miyahara, S.-I. Tamaru, K. Sada and I. Hamachi, Angew. Chem., Int. Ed., 2006, 45, 5518 CrossRef CAS; (m) A. Ojida, Y. Miyahara, J. Wongkongkatep, S.-I. Tamaru, K. Sada and I. Hamachi, Chem.–Asian J., 2006, 1, 555 CrossRef CAS; (n) H. N. Lee, Z. Xu, S. K. Kim, K. M. K. Swamy, Y. Kim, S.-J. Kim and J. Yoon, J. Am. Chem. Soc., 2007, 129, 3828 CrossRef CAS; (o) L. Vial and P. Dumy, J. Am. Chem. Soc., 2007, 129, 4884 CrossRef CAS; (p) P. P. Neelakandan, M. Hariharan and D. Ramaiah, Org. Lett., 2005, 7, 5765 CrossRef CAS.
- (a) A. P. Davis and J.-B. Joos, Coord. Chem. Rev., 2003, 240, 143 CAS; (b) A. P. Davis, Coord. Chem. Rev., 2006, 250, 2939 CrossRef CAS.
- (a) S. M. Cheung and W. H. Chan, Tetrahedron, 2006, 62, 8379 CrossRef CAS; (b) H. Wang and W. H. Chan, Tetrahedron, 2007, 63, 8825 CrossRef CAS; (c) L. Fang, W. H. Chan, Y. B. He, D. W. J. Kwong and A. W. M. Lee, J. Org. Chem., 2005, 70, 7640 CrossRef CAS; (d) S. Y. Liu, Y. B. He, W. H. Chan and A. W. M. Lee, Tetrahedron, 2006, 62, 11687 CrossRef CAS; (e) S. Y. Liu, L. Fang, Y. B. He, W. H. Chan, K. T. Yeung, Y. K. Cheng and R. H. Yang, Org. Lett., 2005, 7, 5825 CrossRef CAS; (f) S.-Y. Liu, K. Y. Law, Y.-B. He and W. H. Chan, Tetrahedron Lett., 2006, 47, 7857 CrossRef CAS.
- A. P. Davis and M. N. Perez-Payan, Synlett, 1999, S1, 991.
- J. F. Larrow and E. N. Jacobsen, J. Org. Chem., 1994, 59, 1939 CrossRef CAS.
- T. Gunnlaugsson, A. P. Davis, G. M. Hussey, J. Tierney and M. Glynn, Org. Biomol. Chem., 2004, 2, 1856 RSC and references therein.
- H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703 CrossRef CAS.
- M. Dukh, D. Saman, K. Lang, V. Pouzar, I. Cerny, P. Drasar and V. Kral, Org. Biomol. Chem., 2003, 1, 3458 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: UV titration of 1b with four nucleobases, solvent effect on the binding of 1a with ATP, Job plot of 1a with ATP, and 13C NMR data for the binding experiments. See DOI: 10.1039/b715086e |
| This journal is © The Royal Society of Chemistry 2008 |