Synthesis of gem-difluorinated nucleoside analogues of the liposidomycins and evaluation as MraY inhibitors†
Xiu-Hua
Xu
a,
Amy E.
Trunkfield
b,
Timothy D. H.
Bugg
b and
Feng-Ling
Qing
*a
aKey Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Science, 354 Fenglin Lu, Shanghai, 200032, China. E-mail: flq@mail.sioc.ac.cn; Fax: +86-21-64166128
bDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK
First published on 16th November 2007
Abstract
Two gem-difluoromethylenated nucleoside moieties of liposidomycins, 3 and 4, were designed and synthesized. Compound 3 was assembled from lactol 5 and gem-difluoromethylenated nucleoside 6. In the synthesis of target molecule 4, the coupling of the trichloroacetimidate derivative of gem-difluoromethylenated furanose 7 with nucleoside 8 in the presence of TMSOTf gave the unexpected compound 16 when CH3CN was used as solvent. This results from acetonitrile acting as a nucleophile and participating in the glycosylation reaction. This unusual process may be correlated with the presence of the electron-withdrawing gem-difluoro substituents at the C-2 position of furanose . Compound 3 demonstrated 29% inhibition of MraY at 11.4 mM.
Introduction
Phospho-MurNAc-pentapeptide translocase (MraY)1 is an essential enzyme for bacteria.2 It catalyzes a key step during the synthesis of precursors of peptidoglycan , which provides much of the strength and rigidity to withstand the high internal osmotic pressure within the cell. Consequently, MraY is a target of choice for the discovery of new antibacterial agents in view of bringing a solution to today's problem of antibiotic resistance.3 Liposidomycins (LPMs), isolated from the fermentation broth of Streptomyces griseosporeus in 1985 by Kimura and Isono et al.,4 are a family of fatty acyl nucleoside natural products containing uracil as the nucleoside , a ribofuranoside, a diazepine, and a lipid region (Fig. 1). LPMs were found to be potent and selective inhibitors of MraY,5 and selectively inhibited the biosynthesis of undecaprenol pyrophosphate N-acetylmuramyl pentapeptide. In 2000, Dini and co-workers synthesized a simplified analogue 16 of LPMs based on the structure–activity relationship (SAR) analysis between LPMs and tunicamycins (TCMs).7 Compound 1 displayed a moderate inhibitory activity against MraY (IC50 = 50 µM) and was identified as the key fragment responsible for preserving a reasonable inhibitory activity of this family of naturally occurring inhibitors of MraY. Later, a number of analogues of 1 were synthesized and tested against MraY.8 These studies showed that an unmodified uracil moiety, a hydroxy group in the 3″-position and a primary amine group in the 5″-position were crucial for the inhibition of MraY. Conversely, the absence of the 3′-hydroxyl gave rise to an inhibitor 2, which was five times more potent (IC50 = 10 µM). When the 2′-hydroxyl of 2 was removed, the activity decreased significantly (IC50 = 120 µM), and so, in our opinion, the 2′-hydroxyl may also be important for the inhibition of MraY.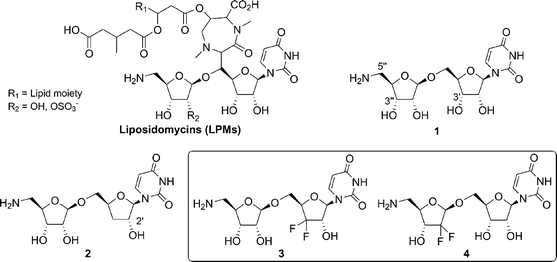 | ||
| Fig. 1 Design of gem-difluoronucleoside analogues of the liposidomycins. | ||
It is well known that introduction of fluorine atom(s) or fluorine-containing groups into an organic compound can bring about remarkable changes in the physical, chemical and biological properties.9 Fluorine has a small van der Waals radius (1.35 Å) which closely resembles that of hydrogen (1.20 Å).10 Therefore, replacement of a hydrogen by fluorine in a bioactive molecule is expected to cause minimal steric perturbations with respect to the molecule's mode of binding to receptors or enzymes. The substitution of fluorine for hydrogen also can profoundly affect chemical reactivity, because of the powerful electron-withdrawing properties of fluorine relative to hydrogen and the increased stability of the carbon–fluorine bond relative to the carbon–hydrogen bond. Moreover, Gemcitabine11 (2′-deoxy-2′,2′-difluorocytidine) has been approved by FDA for treatment of inoperable pancreatic cancer and of 5-fluorouracil-resistant pancreatic cancer. The high antiviral and antineoplastic activities of Gemcitabine reveals that the replacement of the gem-difluoromethylene group (CF2) at the C-2 position of the furanose may bring about special influences of biological activities of nucleoside analogues. Based on the above consideration and our ongoing efforts to develop biologically interesting fluorine-containing compounds, we designed target molecule 3, with a CF2 group replacing the methylene group (CH2) at the 3′-position of 2, and target molecule 4, with a CF2 group replacement at the 2″-position (the 2′-hydroxyl is crucial for the inhibition of MraY, as described above). Herein, we describe the synthesis and inhibition activities of the gem-difluoromethylenated compounds 3 and 4.
Results and discussion
Based on retrosynthetic analysis (Scheme 1), the stereocontrolled construction of the glycosidic linkage turns out to be the key for the synthesis of 3. The stereoselective formation of the 1,2-trans-β-furanoside linkage could be performed by glycosylation utilizing donor substrates containing a neighbouring participating group, and thus furanose 5 has a 2-O-acetyl group. Recently, we have developed a practical route to the gem-difluoromethylenated nucleoside 6.12 The target molecule 4 could be prepared by the coupling of gem-difluoromethylenated lactol 7 with nucleoside 8,13 the gem-difluoromethylenated furanose 7 being a key intermediate in this synthesis. The gem-difluoromethylene group is frequently formed through the difluorination of a carbonyl group using fluorinating reagents such as (diethylamino)sulfur trifluoride (DAST).14 However, very few sterically hindered five-membered cyclic ketones have been difluorinated by DAST. Furthermore, in the example of difluorination of an α,α′-disubstituted five-membered cyclic ketone , the carbonyl group is relatively unhindered, yet the yield is low (25%).15 Recently, the application of fluorine-containing building blocks for synthesis of gem-difluoromethylenated furanoses has been reported.11a,12,16 Thus, the gem-difluoromethylenated furanose 7 will be prepared from the corresponding fluorine-containing building block.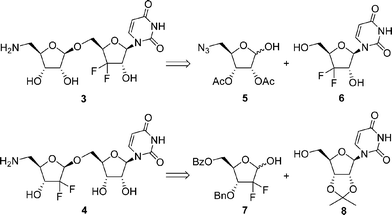
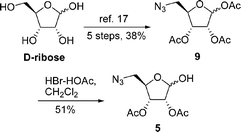
The lactol 5 was synthesized starting from D-ribose (Scheme 2). D-Ribose was transformed into known 1,2,3-tri-O-acetyl-5-azido-5-deoxy-D-ribofuranose9 in 38% yield over five steps.17 The selective removal of the anomeric acetate of 9 was a key step for the preparation of 5. Although hydrazine acetate was widely used for the selective removal of the anomeric acetate of pyranoses containing an azido group,18 Hui and co-workers reported that the selective removal of the anomeric acetate of tetra-O-acetyl-α-L-arabinofuranose in the presence of hydrazine acetate gave the corresponding hemiacetal in poor yield (∼20%) but the yield was improved to quantitative in the presence of HBr/HOAc.19 Accordingly, treatment of compound 5 with HBr/HOAc in CH2Cl2 provided 2,3-di-O-actyl-5-azido-5-deoxy-D-ribofuranose in 51% yield.
 | ||
| Scheme 2 | ||
With lactol 5 and nucleoside 6 in hand, the coupling reaction of these two compounds was carried out (Scheme 3). Treatment of 5 with CCl3CN–DBU afforded the corresponding trichloroacetimidate derivative in 76% yield, which was treated with nucleoside 6 using TMSOTf as an activator in the presence of 4 Å MS in CH3CN, to give the β-anomer 10. The neighbouring group participation of the acetyl group benefited the formation of our desired β-anomer (the absolute configuration of compound 10 was determined by a NOESY experiment on compound 11). Removal of the acetyl groups of 10 with saturated methanolic ammonia produced 11 in 95% yield. Finally, hydrogenation of 11 with Pd/C in methanol for 30 minutes afforded the target molecule 3 in 82% yield. It was noteworthy that the hydrogenation time was very important for the conversion of the azido group of 11 to the amino group. With a longer recation time (>30 minutes), the hydrogenation gave a complex mixture, and the over-hydrogenated product may have been formed. Boojamra et al. previously also found that hydrogenation of nucleoside produced the dihydronucleoside analogue.20
 | ||
| Scheme 3 Reagents and conditions: (a) (i) CCl3CN, DBU, CH2Cl2, 76%, (ii) 6, 4 Å MS, TMSOTf, CH3CN, 68%; (b) NH3, MeOH, 95%; (c) Pd/C, MeOH, H2, 82%. | ||
The gem-difluoromethylenated furanose 7 was prepared from gem-difluorohomoallyl alcohol 12 (Scheme 4). Compound 12 was prepared from 1-(R)-glyceraldehyde acetonide and 3-bromo-3,3-difluoropropene according to our recent report.16d Utilizing the kinetic resolution method and optimized reaction conditions, the benzylation of 12 with BnBr in the presence of NaH (0.8 equiv.) and catalytic tetrabutylammonium iodide (TBAI) afforded the desired single anti-isomer 13 in 79% yield. Removal of the isopropylidene ketal of 13 with one equivalent of p-toluenesulfonic acid (PTSA) in MeOH gave diol 14 in 91% yield. Selective benzoylation of the primary hydroxyl group in 14 with BzCl afforded the benzoate 15 in 90% yield. The conversion of 15 to furanose 7 was achieved in 86% yield by ozonization and subsequent cyclization. The ratio of the two diastereoisomers in 7 was 56 : 44, as determined by 19F NMR. These two diastereoisomers could not be separated by silica gel chromatography .
 | ||
| Scheme 4 Reagents and conditions: (a) NaH, BnBr, TBAI, THF, 79%; (b) PTSA, MeOH, 91%; (c) BzCl, Py, CH2Cl2, 86%; (d) O3, CH2Cl2, 86%. | ||
With lactol 7 in hand, the procedures of the coupling of 5 and 6 were applied to coupling of 7 and 8. Accordingly, treatment of lactol 7 with CCl3CN–DBU afforded the corresponding trichloroacetimidate derivative in 87% yield (Scheme 5). Unfortunately, the reaction of the trichloroacetimidate derivative with nucleoside 8 in CH3CN, under the promotion of TMSOTf, failed to give our desired product 17, but compound 16 was obtained in 62% yield. The formation of 16 indicated that CH3CN participated in the coupling reaction. Acetonitrile (CH3CN) is usually used as the solvent in conventional glycosylation reactions to favor products with the β-configuration.21 Wong et al. found that in the fluorination–nucleophilic addition reaction of glycols , when the nucleophile was added in stoichiometric amounts with acetonitrile as the solvent, the acetonitrile participates in the reaction by attack on the anomeric position, and consequent addition of the nucleophile at the nitrile carbon to give the fluorinated disaccharide product.22 Thus, a mechanism for the formation of compound 16 is proposed (Scheme 5). As the gem-difluoromethylene is an electron-withdrawing group, the acetonitrile-containing intermediate II predominates over intermediate I (prepared from the trichloroacetimidate derivative of lactol 7 with activation by TMSOTf). The attack of nucleoside 8 upon II results in the formation of 16.
 | ||
| Scheme 5 Reagents and conditions: (a) (i) CCl3CN, DBU, CH2Cl2, 87%; (ii) 8, 4 Å MS, TMSOTf, CH3CN, 62%. | ||
To avoid this undesirable reaction, nitromethane was used as the solvent because it dissolves compound 8 well and remains inert during the reaction (Scheme 6). As a result, the desired product 17 was obtained as a mixture of β- and α-anomers with the 72 : 28 ratio (determined by 19F NMR). The two anomers could not be separated by silica gel chromatography . Treatment of 17 with saturated methanolic ammonia smoothly produced compound 18 with a free hydroxyl in the 5″-position, which was used for introduction of the azido group. Reaction of 18 with trifluoromethanesulfonic anhydride in CH2Cl2 at −30 °C, followed by treatment with sodium azide in acetone at room temperature, gave the desired product 19. Finally, deprotection of 24 with BCl3 gave the target molecule 4 and the α-isomer, which were readily separated by column chromatography .
 | ||
| Scheme 6 Reagents and conditions: (a) (i) CCl3CN, DBU, CH2Cl2; (ii) 8, 4 Å MS, TMSOTf, CH3NO2, 32% over two steps; (b) NH3, MeOH, 88%; (c) (i) Tf2O, pyridine, CH2Cl2; (ii) NaN3, acetone, 58% over two steps; (d) BCl3, CH2Cl2, 63%. | ||
The stereochemistries of products 3 and 4 were established by 2D NMR NOESY experiment of compounds 11 (the precursor of compound 3) and 4, respectively. As shown in Fig. 2, correlations between H1″ (5.15 ppm) and H4″ (4.08–4.13 ppm) were clearly observed in 4, which was identified as having the β-configuration at the C-1″ position. As for compound 11, the chemical shifts of H4″, H3″ and H5′b were overlapped, so the correlations between H1″ and H4″ were not easily identified. However, correlations between H1″ (4.93 ppm) and H4′ (3.68 ppm) were clearly observed in 11, which could thus be identified as having the β-configuration at the C-1″ position.
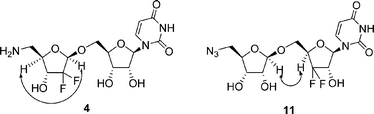 | ||
| Fig. 2 NOE correlation of compounds 4 and 11. | ||
Compounds 3 and 4 were evaluated for activity as an inhibitor of MraY. A coupled MraY–MurG radiochemical assay was utilized, whereby Micrococcus flavus membranes containing high levels of MraY23 were solubilised and used to generate lipid intermediate I in situ, to which was added purified Escherichia coli MurG and UDP-[3H]GlcNAc. The 3H-labelled lipid intermediate II was extracted into n-butanol and analyzed for radioactivity. Compound 3 demonstrated 29% inhibition of MraY at a concentration of 11.4 mM, whereas compound 4 showed no inhibition at 10 mM concentration.
In summary, two gem-difluoromethylenated nucleoside analogues of liposidomycins, 3 and 4, were both synthesized. Compound 3 was assembled from lactol 5 and gem-difluoromethylenated nucleoside 6. The neighbouring group participation of the 2-O-acetyl group in 5 ensured the construction of the 1,2-trans-β-furanoside linkage during the glycosylation reaction, which resulted in the stereocontrolled formation of 3. In assembling 4, the trichloroacetimidate derivative of the gem-difluoromethylenated lactol 7 was coupled with nucleoside 9 with TMSOTf as an activator in CH3CN. However, acetonitrile was found to participate as a nucleophile in the glycosylation reaction, resulting in the production of 16. To avoid this undesirable reaction, nitromethane was used as the solvent, resulting in the desired product 17, from which the target molecule 4 was prepared in a few steps. Compound 3 showed low activity as an inhibitor of MraY.
Experimental
Solubilisation of MraY
100 µl of Micrococcus flavus membranes23 (19 mg protein ml−1) was added to 150 µl of solubilisation buffer (50 mM Tris-HCl pH 7.5, 1 mM MgCl2, 2 mM 2-mercaptoethanol). The mixture was shaken at 4 °C for 30 minutes and then centrifuged at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 minutes. The supernatant had a protein concentration of 1.5 mg ml−1 and was used directly in the radiochemical assay .
000 rpm for 30 minutes. The supernatant had a protein concentration of 1.5 mg ml−1 and was used directly in the radiochemical assay .
Coupled MraY–MurG radiochemical assay
12.5 µl of freshly solubilised MraY was added directly to undecaprenyl phosphate (0.25 µg). 12.5 µl of buffer (400 mM Tris-HCl pH 7.5, 100 mM MgCl2) was added followed by 9 µl of water, 5 µl of UDP-MurNAc-pentapeptide solution (1 mM), 1 µl of MurG solution (100 µg protein ml−1) and 5 µl of DMSO or inhibitor solution (127 mM in DMSO). The mixture was incubated at 35 °C for 15 minutes and then 5 µl of UDP-[3H]GlcNAc (10 µM, 500 mCi mmol−1) was added and the mixture was incubated for a further 15 minutes. The reaction was stopped by the addition of 50 µl of pyridinium acetate pH 4.6. 100 µl of n-butanol and 100 µl of water were then added and the layers were mixed and then separated by centrifugation. 100 µl of the top n-butanol phase was removed, 50 µl of fresh n-butanol was added to it, and it was then extracted with 100 µl of water. 100 µl of the n-butanol phase was then removed and analyzed for radioactivity. Typically, this procedure yielded 1000–2000 cpm per assay ; duplicate assays were routinely carried out, and yielded consistent data (±10%). Control incubations were carried out containing no inhibitor, no enzyme, and 50 µM ramoplanin (MurG inhibitor). Micrococcus flavus membranes, UDP-MurNAc-pentapeptide and Escherichia coli MurG were all prepared as described previously.23–25Acknowledgements
The National Natural Science Foundation of China, the Ministry of Education of China and Shanghai Municipal Scientific Committee are greatly acknowledged for funding this work.References
- W. G. Struve, R. K. Sinha and F. C. Neuhaus, Biochemistry, 1966, 5, 82 CrossRef CAS.
- D. S. Boyle and W. D. Donachie, J. Bacteriol., 1998, 180, 6429 CAS.
- L. A. Mitscher, S. P. Pillai, E. J. Gentry and D. M. Shankel, Med. Res. Rev., 1999, 19, 477 CrossRef CAS.
- K. Isono, M. Uramoto, H. Kusakabe, K. Kimura, K. Izaki, C. C. Nelson and J. A. McCloskey, J. Antibiot., 1985, 38, 1617 CAS.
- K. Kimura, Y. Ikeda, S. Kagami, M. Yoshihama, K. Suzuki, H. Osada and K. Isono, J. Antibiot., 1998, 51, 1099 CAS.
- C. Dini, P. Collette, N. Drochon, J. C. Guillot, G. Lemoine, P. Mauvais and J. Aszodi, Bioorg. Med. Chem. Lett., 2000, 10, 1839 CrossRef CAS.
- A. Takatsuki, K. Kawamura, M. Okina, Y. Kodama, T. Ito and G. Tamura, Agric. Biol. Chem., 1977, 41, 2307 CAS.
- (a) C. Dini, N. Drochon, S. Feteanu, J. C. Guillot, C. Peixoto and J. Aszodi, Bioorg. Med. Chem. Lett., 2001, 11, 529 CrossRef CAS; (b) C. Dini, N. Drochon, J. C. Guillot, P. Mauvais, P. Walter and J. Aszodi, Bioorg. Med. Chem. Lett., 2001, 11, 533 CrossRef CAS; (c) C. Dini, S. Didier-Laurent, N. Drochon, S. Feteanu, J. C. Guillot, F. Monti, E. Uridat, J. Zhang and J. Aszodi, Bioorg. Med. Chem. Lett., 2002, 12, 1209 CrossRef CAS.
- (a) Organofluorine Chemistry: Principles and Commerical Applications, ed. R. E. Banks, B. E. Smart and J. C. Tatlow, Plenum, New York, 1994 Search PubMed; (b) J. T. Welch, S. Eswaraksrishnan, Fluorine in Bioorganic Chemistry, Wiley, New York, 1991 Search PubMed; (c) Biomedical Frontiers of Fluorine Chemistry (ACS Symposium Series 639), ed. I. Ojima, J. R. McCarthy and J. T. Welch, American Chemical Society, Washington, DC, 1996 Search PubMed; (d) Organofluorine Chemicals and Their Industrial Applications, ed. R. E. Banks, Ellis Harwood, New York, 1979 Search PubMed; (e) R. Peters, Carbon-Fluorine Compounds: Chemistry, Biochemistry and Biological Activities. A Ciba Foundation Symposium, Elsevier, Amsterdam, 1972 Search PubMed.
- L. Pauling, The Nature of the Chemical Bond, 3rd edn, Cornell University Press, Ithaca, NY, 1960, p. 93 Search PubMed.
- (a) L. W. Hertel, J. S. Kroin, J. W. Missner and J. M. Tustin, J. Org. Chem., 1988, 52, 2406 CrossRef; (b) W. Plunkett, V. Gandhi, C. Chubb, B. Nowak, V. Heinemann, S. Mineishi, A. Sen, L. W. Hertel and G. B. Grindey, Nucleosides Nucleotides, 1989, 8, 775; (c) V. W. T. Ruiz, V. Haperen, G. Veerman, J. B. Vermorken and G. J. Peters, Biochem. Pharmacol., 1993, 46, 762 CrossRef.
- X.-H. Xu, X.-L. Qiu, X.-G. Zhang and F.-L. Qing, J. Org. Chem., 2006, 71, 2820 CrossRef CAS.
- M. Cornia, M. Menozzi, E. Ragg, S. Mazzini, A. Scarafoni, F. Zanardic and G. Casiraghi, Tetrahedron, 2000, 56, 3977 CrossRef CAS.
- (a) W. J. Middleton, J. Org. Chem., 1975, 40, 574 CrossRef CAS; (b) M. Hudlicky, Org. React., 1988, 35, 513.
- (a) K. Biggadike, A. D. Borthwick, D. Evans, A. M. Exall, B. E. Kirk, S. M. Roberts, L. Stephenson, P. Youds, A. M. Z. Slawin and D. J. Williams, J. Chem. Soc., Chem. Commun., 1987, 251 RSC; (b) A. D. Borthwick, D. N. Evans, B. E. Kirk, K. Biggadike, A. M. Exall, P. Youds, S. M. Roberts, D. J. Knight and J. A. V. Coates, J. Med. Chem., 1990, 33, 179 CrossRef CAS.
- (a) L. P. Kotra, Y. Xiang, M. G. Newton, R. F. Schinazi, Y.-C. Cheng and C. K. Chu, J. Med. Chem., 1997, 40, 3635 CrossRef CAS; (b) L. P. Kotra, M. G. Newton and C. K. Chu, Carbohydr. Res., 1998, 306, 69 CrossRef CAS; (c) W. Zhou, G. Gumina, Y. Chong, J.-N. Wang, R. F. Schinazi and C. K. Chu, J. Med. Chem., 2004, 47, 3399 CrossRef CAS; (d) X.-G. Zhang, H.-R. Xia, X.-C. Dong, J. Jin, W.-D. Meng and F.-L. Qing, J. Org. Chem., 2003, 68, 9026 CrossRef CAS.
- M. Ebner and A. E. Stütz, Carbohydr. Res., 1998, 305, 331.
- (a) S. Mehta, M. Meldal, V. Ferro, J. Ø. Duus and K. Bock, J. Chem. Soc., Perkin Trans. 1, 1997, 1365 RSC; (b) J. Wang, J. Li, D. Tuttle, J. Y. Takemoto and T. C.-W. Chang, Org. Lett., 2002, 4, 3997 CrossRef CAS; (c) B. Elchert, J. Li, J. Wang, Y. Hui, R. Rai, R. Ptak, P. Ward, J. Y. Takemoto, M. Bensaci and C.-W. T. Chang, J. Org. Chem., 2004, 69, 1513 CrossRef CAS.
- S.-J. Deng, B. Yu, Y.-Z. Hui, H. Yu and X.-W. Han, Carbohydr. Res., 1999, 317, 53 CrossRef CAS.
- C. G. Boojamra, R. C. Lemoine, J. C. Lee, R. Léger, K. A. Stein, N. G. Vernier, A. Magon, O. Lomovskaya, P. K. Martin, S. Chamberland, M. D. Lee, S. J. Hecker and V. J. Lee, J. Am. Chem. Soc., 2001, 123, 870 CrossRef CAS.
- R. R. Schmidt, M. Behrendt and A. Toepfer, Synlett, 1990, 694 CrossRef CAS.
- S. P. Vincent, M. D. Burkart, C.-Y. Tsai, Z. Zhang and C.-H. Wong, J. Org. Chem., 1999, 64, 5264 CrossRef CAS.
- E. Breukink, H. E. van Heusden, P. J. Vollmerhaus, E. Swiezewska, L. Brunner, S. Walker, A. J. R. Heck and B. de Kruijff, J. Biol. Chem., 2003, 278, 19898 CrossRef CAS.
- P. E. Brandish, M. Burnham, J. T. Lonsdale, R. Southgate, M. Inukai and T. D. H. Bugg, J. Biol. Chem., 1996, 271, 7609 CrossRef CAS.
- S. Ha, D. Walker, Y. Shi and S. Walker, Protein Sci., 2000, 9, 1045 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional synthesis and characterization data; COSY and NOESY spectra of compounds 4 and 11. See DOI: 10.1039/b713068f |
| This journal is © The Royal Society of Chemistry 2008 |