A compact water-soluble porphyrin bearing an iodoacetamido bioconjugatable site†
K. Eszter Borbasa, Hooi Ling Keeb, Dewey Holten*b and Jonathan S. Lindsey*a
aDepartment of Chemistry, North Carolina State University, Raleigh, NC, 27695-8204, USA. E-mail: jlindsey@ncsu.edu; Fax: +1-919-513-2830; Tel: +1-919-515-6406
bDepartment of Chemistry, Washington University, St. Louis, MO 63130-4889, USA. E-mail: holten@wustl.edu; Fax: +1-314-935-4481; Tel: +1-314-935-6502
First published on 23rd November 2007
Abstract
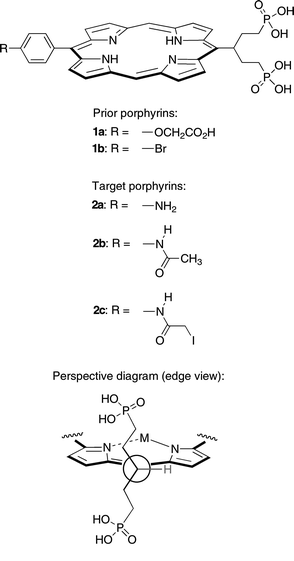
A broad range of applications requires access to porphyrins that are compact, water-soluble, and bioconjugatable. A symmetrically branched hydrocarbon chain (‘swallowtail’) bearing polar end groups imparts high (>10 mM) aqueous solubility upon incorporation at one of the meso positions of a trans-AB-porphyrin. Two such swallowtail-porphyrins (1a, 1b) equipped with a conjugatable group (carboxylic acid, bromophenyl) have been prepared previously. The synthesis of three new water-soluble trans-AB-porphyrins is reported, where each porphyrin bears a diphosphonate-terminated swallowtail group and an amino (2a), acetamido (2b), or iodoacetamido (2c) group. The amine affords considerable versatility for functionalization. The iodoacetamide provides a sulfhydryl-reactive site for bioconjugation. Porphyrins 2a–2c were fully characterized in aqueous solution by 1H NMR spectroscopy (in D2O), ESI-MS, static absorption spectroscopy, and static and time-resolved fluorescence spectroscopy. Porphyrins 2a–2c exhibit characteristic porphyrin absorption and emission bands in aqueous solution, with a strong, sharp absorption band in the blue region (∼401 nm) and emission in the red region (∼624, 686 nm). Porphyrin 2b in aqueous phosphate buffer or phosphate-buffered saline solution exhibits a fluorescence quantum yield of ∼0.04 and an excited singlet-state lifetime of ∼11 ns. Collectively, the facile synthesis, amenability to bioconjugation, large spacing between the main absorption and fluorescence features, and long singlet excited-state lifetime make this molecular design quite attractive for a range of biomedical applications.
Introduction
Diverse biological and medicinal problems require the labeling of biomolecules with fluorophores.1–6 The most important characteristics of such labels are the photophysical features (absorption strength, absorption/emission maxima, excited-state lifetime, fluorescence quantum yield) of the fluorophore and the functional-group specificity of the bioconjugatable moiety. It is also desirable that the fluorophores be soluble in aqueous solution, including various buffers to facilitate the labeling process.7The availability of bioconjugatable (hydro)porphyrins would provide fluorophores that (1) absorb strongly in the visible-to-red region, which is ideal for biomedical applications, and (2) can be tuned to the desired wavelengths by introduction of suitable peripheral substituents.8–10 A serious drawback of using porphyrins as bioconjugatable fluorophores until recently was their insolubility in aqueous media. We recently reported the synthesis of highly water-soluble porphyrins 1a and 1b (∼20 mM at pH 7 in pure water).11 The key to the molecular design was the presence of a symmetrically branched polar-terminated alkyl chain (‘swallowtail motif’), which projects above and below the porphyrin macrocycle and thereby suppresses intermolecular π–π stacking (Chart 1). Concentrated aqueous solutions of such swallowtail porphyrins were stable for extended periods of time (several weeks) without any sign of aggregation or precipitation. The carboxylate moiety of porphyrin 1a was exploited for bioconjugation to an antibody for a series of biological studies. A particularly appealing aspect of the bioconjugation was the facile handling of the porphyrins in aqueous solution without the need for organic cosolvents.
 | ||
| Chart 1 | ||
We were interested in extending the scope of functional group specificity for the swallowtail porphyrins. We envisaged that an amino analogue of porphyrins 1a and 1b would be exceptionally versatile given the broad range of derivatization chemistry that can be carried out at the amine site, including traditional acylations or more recent “click” chemistry. One example of the former is provided by the iodoacetamide moiety, which enables sulfhydryl-specific bioconjugation.12 A protein or a peptide with a single cysteine unit can be labeled in a site-specific manner.13–15 At physiological pH, iodoacetamides react selectively with cysteine residues, and only above pH 9 does labeling through lysine amino residues occur. Unlike maleimides, iodoacetamides are not prone to nucleophilic attack by amino residues in the proteins, which can result in protein cross-linking, or to hydrolysis.16 Under acidic conditions, selenocysteine residues in selenoproteins can be selectively labeled without interference from cysteine.17 Iodoacetamide moieties have been incorporated into many common fluorophores and related bioprobes, including cyanine,7,18,19 fluorescein,20 rhodamine,21 coumarin,22 and dansyl derivatives;23 luminescent complexes of iridium,24 osmium,25 and platinum;26 redox-active ferrocene derivatives;27 and spin-traps.28 Because of the site-specificity afforded by iodoacetamide labeling, sulfhydryl-labeled compounds have been extensively used in resonance energy-transfer experiments. Thus, time-resolved fluorescence energy transfer studies on a site-specific labeled two-cysteine-containing mutant of adenylate kinase with 5-iodoacetamidofluorescein (acceptor) and 5-iodoacetamidosalicylic acid (donor) enabled the elucidation of the substrate-binding mechanism.29 Stopped-flow distance-change measurements were carried out on phosphoglycerate kinase labeled with 5-[2-(iodoacetamido)ethylamino]naphthalenesulfonic acid (donor) and 5-iodoacetamidofluorescein (acceptor) to monitor in real-time the unfolding of the protein,30 and to determine interdomain distance distributions.31 A crucial aspect of the labeling experiments is the availability of suitable donor–acceptor pairs, and, if necessary, the preparation of the iodoacetamides.31
In this paper, we report the synthesis of a trans-AB-porphyrin that bears a diphosphonate-terminated swallowtail group and a p-aminophenyl unit (2a). This versatile molecular design has been extended to include an acetamido-porphyrin (2b) and an iodoacetamido-porphyrin analogue (2c). A relatively concise synthetic approach was devised that circumvents the use of an unprotected aminophenyl unit, which is not compatible with current porphyrin synthesis methods.32,33 The photophysical features of the acetamido-porphyrin analogue (2b) have been characterized. This work should facilitate the use of porphyrins as fluorophores in a range of biomedical applications.
Results and discussion
1. Synthesis
The key intermediate building block was porphyrin 3, which contains the protected diphosphonate-derivatized swallowtail group and the p-aminophenyl unit in a trans-AB arrangement (Chart 2). We considered three possible porphyrin precursors to 3, including A-porphyrin 4 and trans-AB-porphyrins 5 and 6. Porphyrin 4 was subjected to bromination with the expectation of subsequent Suzuki coupling to introduce the p-aminophenyl unit, but the bromination was not regioselective and afforded an inseparable mixture. Porphyrin 5 was reacted with PPh3/CBr4 in CH2Cl2 to obtain the porphyrin-dibromide, but the Boc protecting group was lost and no porphyrin-dibromide could be isolated. Hence we turned to the synthesis of porphyrin 6, which is described below. The syntheses of porphyrins 4 and 5 are described in the Supplementary Material.†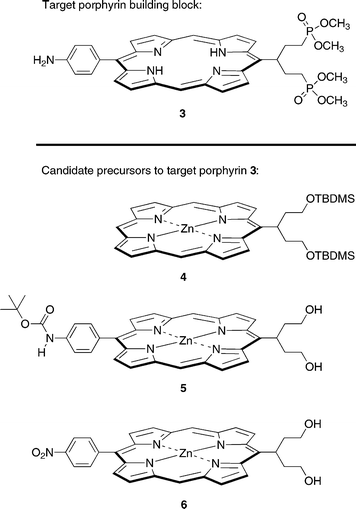 | ||
| Chart 2 | ||
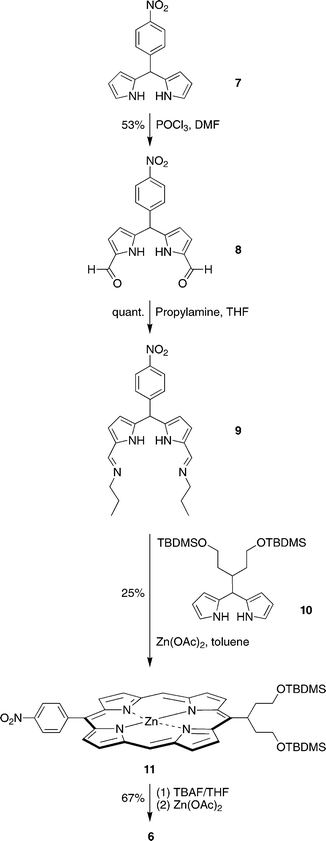
Dipyrromethane 7,34 obtained by reaction of 4-nitrobenzaldehyde with excess pyrrole in the presence of a mild Lewis acid catalyst (InCl3),35 was subjected to Vilsmeier formylation to obtain 1,9-diformyldipyrromethane 8 (Scheme 1). Vilsmeier formylation of 7 under standard conditions (room temperature, work-up with 10 M aqueous NaOH)36 gave a tar and only trace amounts of the desired diformyldipyrromethane 8. Both chromatographic purification and complexation with dibutyltin dichloride (to yield the dibutyltin–diformyldipyrromethane complex37) were attempted to no avail. Conducting the reaction at 0 °C and quenching with portions of ice-cold aqueous NaOH yielded 8 in a reproducible 53% yield after silica column chromatography. Treatment of 8 with excess propylamine in THF quantitatively afforded the 1,9-bis(propyliminomethyl)dipyrromethane 9, which upon isolation was used immediately in the porphyrin-forming step.
 | ||
| Scheme 1 | ||
The condensation of 9 and swallowtail-dipyrromethane 10 was carried out in the presence of zinc acetate in toluene11 (rather than ethanol36), affording the corresponding 4-nitrophenyl-substituted swallowtail-porphyrin 11. Treatment of a THF solution of 11 with excess TBAF caused straightforward cleavage of the TBDMS groups but also partial demetalation of the zinc porphyrin [as indicated by laser desorption mass spectrometric (LD-MS) analysis]. Treatment of the sample with zinc acetate prior to chromatography on neutral alumina afforded zinc porphyrin 6.
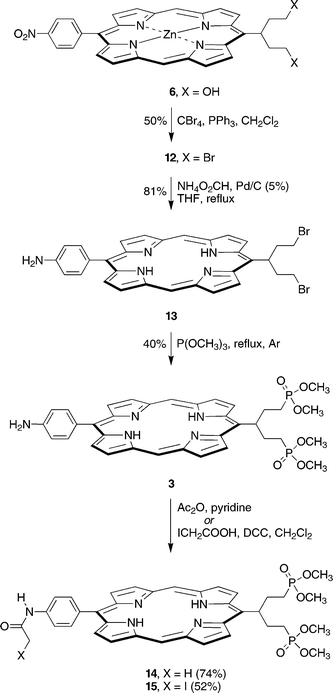
The reaction of 6 with PPh3/CBr4 in CH2Cl2 following reported procedures11,38 gave the porphyrin-dibromide 12 (Scheme 2). Initially we attempted phosphonation of 12 via the Arbuzov reaction (Br![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) →P(O)(OCH3)2), but found that the major product was not the porphyrin-alkyldiphosphonate, but presumably the ipso-substituted (de-nitro) derivative, as indicated by 1H NMR and LD-MS analysis (see Supplementary Material). To avoid the putative ipso-substitution pathway during phosphonation, 12 was reduced to the amine derivative prior to phosphonation. Catalytic transfer hydrogenation39–41 of 12 was carried out in THF using ammonium formate and palladium supported on carbon (5 wt%). The starting porphyrin-dibromide 12 and the product 13 are somewhat labile in solution, and partial debromination was observed [1H NMR, LD-MS, m/z 547.0 (13− Br), m/z 467.8 (13− 2Br), m/z 576.4 (12− Br)]. Prolonged reduction resulted in further losses of the bromine substituents. Attempts at reducing the amount of debrominated product by replacing the ammonium formate with cyclohexene as the hydrogen source were unsuccessful (Supplementary Material). Therefore, the hydrogenation was closely monitored and stopped immediately after the disappearance of the starting material 12 [TLC, silica, CH2Cl2, Rf(12) = 0.70, Rf(13) = 0.17] to minimize debromination. Aqueous–organic extraction yielded the product in 81% yield and ∼85% purity with debrominated products as the major contaminants as determined by 1H NMR spectroscopy. The sample thus obtained was pure enough for use in the Arbuzov reaction. Further purification of 13 was possible by flash silica column chromatography, but was usually avoided, given the lability of the compound. Trace amounts of a dimeric porphyrin were detected in the purified sample by LD-MS analysis (m/z 1177.3), formed presumably via N-alkylation with one of the swallowtail units. Thus, refluxing the crude sample of 13 in a mixture of THF and P(OCH3)3 (1 : 5 v/v) for 36 h under an argon atmosphere yielded the valuable target porphyrin 3 in 40% yield.
→P(O)(OCH3)2), but found that the major product was not the porphyrin-alkyldiphosphonate, but presumably the ipso-substituted (de-nitro) derivative, as indicated by 1H NMR and LD-MS analysis (see Supplementary Material). To avoid the putative ipso-substitution pathway during phosphonation, 12 was reduced to the amine derivative prior to phosphonation. Catalytic transfer hydrogenation39–41 of 12 was carried out in THF using ammonium formate and palladium supported on carbon (5 wt%). The starting porphyrin-dibromide 12 and the product 13 are somewhat labile in solution, and partial debromination was observed [1H NMR, LD-MS, m/z 547.0 (13− Br), m/z 467.8 (13− 2Br), m/z 576.4 (12− Br)]. Prolonged reduction resulted in further losses of the bromine substituents. Attempts at reducing the amount of debrominated product by replacing the ammonium formate with cyclohexene as the hydrogen source were unsuccessful (Supplementary Material). Therefore, the hydrogenation was closely monitored and stopped immediately after the disappearance of the starting material 12 [TLC, silica, CH2Cl2, Rf(12) = 0.70, Rf(13) = 0.17] to minimize debromination. Aqueous–organic extraction yielded the product in 81% yield and ∼85% purity with debrominated products as the major contaminants as determined by 1H NMR spectroscopy. The sample thus obtained was pure enough for use in the Arbuzov reaction. Further purification of 13 was possible by flash silica column chromatography, but was usually avoided, given the lability of the compound. Trace amounts of a dimeric porphyrin were detected in the purified sample by LD-MS analysis (m/z 1177.3), formed presumably via N-alkylation with one of the swallowtail units. Thus, refluxing the crude sample of 13 in a mixture of THF and P(OCH3)3 (1 : 5 v/v) for 36 h under an argon atmosphere yielded the valuable target porphyrin 3 in 40% yield.
 | ||
| Scheme 2 | ||
Acetylation of 3 with acetic anhydride in pyridine42 afforded acetamido-porphyrin 14. In addition, reaction of 3 with excess iodoacetic acid in the presence of DCC gave iodoacetamido-porphyrin 15 in 52% yield. It is important to use DCC as the carbodiimide rather than the water-soluble EDCI, even though the latter simplifies purification of the product. Replacing DCC with EDCI resulted in partial halogen exchange between the hydrochloride salt of the coupling reagent and the iodoacetamide, giving small amounts of the chloroacetamido-porphyrin (as shown by LD-MS, m/z 764.5). Removal of the excess iodoacetic acid, DCC, and the urea side-product was achieved with aqueous washing and flash column chromatography. Small quantities (10–20 mg) of 15 were easily purified on a plug of alumina, but larger amounts (50–60 mg) required a silica stationary phase. Surprisingly, 15 proved quite stable under these conditions, and, if heating during solvent removal was avoided, was isolated without apparent loss of the iodine.
The final step for porphyrins 3, 14, and 15 was methyl→silyl exchange followed by hydrolysis of the phosphonate silyl ethers. Previously we used bromotrimethylsilane in refluxing CHCl3.11 Here we employed the more reactive iodotrimethylsilane to enable reaction at room temperature in CH2Cl2 and avoid both thermal deiodination and undesired halogen exchange. The silylation reaction was complete in 30 min. Removal of the volatile components under vacuum, hydrolysis of the silyl ethers under mild conditions (5 wt% NaHCO3, room temperature, 20 min), and preparative reverse-phase silica column chromatography in each case yielded the desired water-soluble porphyrin-swallowtail-diphosphonate 2a, 2b or 2c in good yield (Scheme 3).
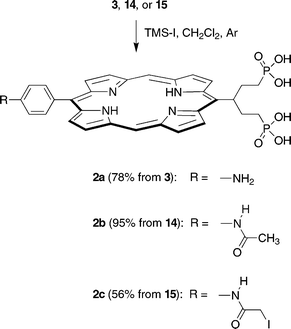 | ||
| Scheme 3 | ||
2. Characterization and water-solubility
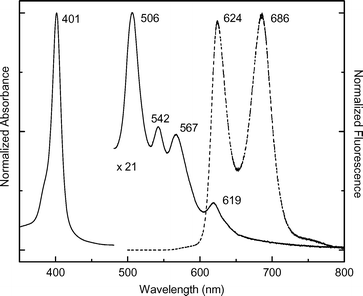 | ||
| Fig. 1 Normalized absorption (solid) and fluorescence (dashed) spectra of 2b in PBS at room temperature. The Q-band region of the absorption spectrum has been multiplied by the factor indicated. | ||
The fluorescence quantum yield (Φf) for deoxygenated 2b is 0.042 ± 0.006 in PBS and 0.023 ± 0.005 in PB. The smaller value for 2b in PB is qualitatively consistent with the reduced oscllator strength of the Qx absorption features (and thus the radiative rate) compared to PBS. The corresponding lifetime of the lowest-energy excited singlet state (Qx), measured by fluorescence methods, is τ = 10.9 ± 0.4 ns in PBS and 11.3 ± 0.2 ns in PB. In comparison, the values for H2TPP are Φf = 0.09 and τ = 13 ns in toluene.43,44 Thus, the average fluorescence yield of 2b is roughly one-third that of H2TPP while the excited-state lifetime is reduced by ∼20%. These modest differences are due in part to a reduced radiative rate constant for 2b versus H2TPP, which can be calculated from the formula kf = Φf/τ. The values are kf∼(260 ns)−1 and (490 ns)−1 for 2b in PBS and PB, respectively, compared to ∼(145 ns)−1 for H2TPP. The yield of the lowest triplet excited state of 2b in PB is ΦT = 0.7 ± 0.1. This value is comparable to the reported values of 0.67 to 0.84 for H2TPP in organic solvents.43
Collectively, the photophysical properties of acetamido-porphyrin 2b are generally similar to those of typical free base porphyrins in organic solvents. The characteristics include strong absorption in the near UV, weak absorption in the blue–green to orange–red regions, weak-modest fluorescence, and a long lifetime of the lowest-energy singlet excited state.
Outlook
A versatile trans-AB-porphyrin bearing a diphosphonate-terminated swallowtail unit and a p-aminophenyl unit has been prepared by reaction of a dihydroxy-terminated swallowtail-dipyrromethane and a 1,9-bis(propyliminomethyl)-5-nitrophenyldipyrromethane followed by functional group transformations. Derivatization of the amine to give the α-iodoacetamido moiety affords a sulfhydryl-specific functionality for bioconjugation. The synthesis is readily implemented to obtain 20–50 mg of target porphyrin. The protected dimethylphosphonate groups in each of three free base porphyrins were converted under mild conditions to the corresponding phosphonic acid moieties. The diphosphonate-terminated swallowtail moiety imparts high aqueous solubility (>10 mM).The design and synthesis of p-iodoacetamido trans-AB-porphyrin 2c can be compared with that of an earlier A3B-porphyrin bearing a p-chloroacetamido group and three mesityl groups; the latter was not soluble in water and the synthesis of the macrocycle relied on a statistical reaction.45 Extension of the design and synthesis methodology reported herein to hydroporphyrins (e.g., chlorins and bacteriochlorins) is expected to open access to a wide range of porphyrinic fluorophores that absorb across the near-UV, visible or near-IR range; emit in the red or near-IR region; exhibit high water solubility; and can be derivatized at the amine functionality as desired for bioconjugation purposes.
We note that the fluorescence quantum yield of porphyrin 2b is substantially less than that of many commonly employed fluorophores. However, for a given illumination, brightness is the product of absorption intensity and fluorescence quantum yield.46 In this regard, the effect of the low fluorescence quantum yield of porphyrins such as 2b can be mitigated by exploiting the extraordinary intensity of the Soret absorption band. Illumination in this manner results in a profoundly large effective “Stokes” shift. Moreover, such porphyrins exhibit a long fluorescence lifetime. Porphyrin fluorophores of this type are expected to be well suited for applications such as flow cytometry where minimization of scattered excitation light into the detection channel is important and where the incorporation of large numbers of highly water-soluble fluorophores is desired.
Experimental
General procedures
1H NMR (300 MHz) and 13C NMR (75 MHz) spectra were recorded in CDCl3 unless noted otherwise. Absorption spectra and fluorescence spectra were collected at room temperature in CH2Cl2 unless noted otherwise. Infrared absorption spectra were recorded as thin films. Hydrophobic porphyrins were analyzed in neat form by laser desorption mass spectrometry (LD-MS) without a matrix.47 The water-soluble porphyrins were analyzed by direct infusion of water–acetonitrile (40 : 60) solutions by atmospheric pressure electrospray mass spectrometry (ESI-MS). Both in LD-MS and ESI-MS analyses, positive ions were detected unless noted otherwise. Melting points are uncorrected. Solvents were dried according to standard procedures. Compounds 734 and 1011 were synthesized as described in the literature.Chromatography
Preparative chromatography was performed using silica or alumina (80–200 mesh). Thin layer chromatography was performed on silica or alumina. Samples were visualized by UV-light (254 nm and 365 nm), Br2-vapor or KMnO4/K2CO3. Dipyrromethanes were analyzed by GC as described previously.35 Reverse-phase preparative column chromatography was carried out using C-18-coated silica and eluants based on water admixed with methanol. Analytical HPLC in all cases was carried out using a reverse phase C-18 column (5 μm, 125 mm × 4 mm) with the following elution program: flow rate = 1.0 mL min−1; 0–2 min, 0% B; 2–20 min, 0 → 90% B; 20–23 min, 90% B; A = water (0.1% TFA), B = methanol (0.1% TFA); detection at 254, 410 and 417 nm; void volume typically 1.1 min.Photophysical measurements
The photophysical properties of acetamido-porphyrin 2b were investigated at room temperature using as the aqueous solvent either 1x PBS (phosphate-buffered saline; 138 mM NaCl, 2.7 mM KCl, 10 mM Na3PO4, pH 7.4) or PB (phosphate buffer; 10 mM Na3PO4, pH 7.4). Static absorption (Varian Cary 100) and fluorescence (Spex Fluorolog Tau 2) measurements were performed using dilute (μM) solutions, as described previously.48,49 Fluorescence lifetimes were obtained using a phase modulation technique.48 Argon-purged solutions with an absorbance of ≤0.10 at the Soret-band excitation wavelength were used for the fluorescence spectral and lifetime measurements. For fluorescence spectra, the excitation and detection monochromators had a band pass of 1.5 and 3.3 nm, respectively, and spectra were obtained using 0.2 nm data intervals. The fluorescence spectra were corrected for detection-system spectral response. Fluorescence quantum yields were determined using argon-purged solutions and several excitation wavelengths (395, 401, 408 nm) for 2b relative to both chlorophyll-a in benzene (Φf = 0.325)50 and H2TPP in toluene (Φf = 0.09),43 and the results averaged. An estimate for the triplet excited-state yield of 2b in PB was obtained using a transient-absorption technique similar to that described previously.51,52 The extent of bleaching of the ground-state Qy(1,0) band (relative to the featureless transient absorption) due to the lowest singlet excited state was measured immediately following a 130 fs flash at 570 nm. The amplitude of this signal was compared to that due to the lowest triplet excited state measured at a long (∼20 ns) time delay. The latter value was derived from the long-time asymptote of the singlet-excited-state bleaching decay (4 ns time course) derived from an exponential fit with the time constant fixed at the fluorescence lifetime.5-(4-Aminophenyl)-15-(1,5-bis(dihydroxyphosphoryl)pent-3-yl)porphyrin (2a)
A sample of 3 (49 mg, 0.071 mmol) in anhydrous CH2Cl2 (2 mL) under argon was treated with trimethylsilyl iodide (100 μL). The reaction was allowed to proceed for 30 min. The reaction was quenched with MeOH (0.5 mL). The sample was concentrated. The solid residue was treated with dilute aqueous sodium bicarbonate (2 mL of a 5 wt% solution). Chromatography (C-18 silica, water–MeOH, 0→50%) followed by evaporation of the solvent and freeze-drying from water afforded a deep red voluminous solid (35 mg, 78%):1H NMR (D2O) δ 1.34–1.39 (m, 2H), 2.13–2.17 (m, 2H), 3.41–3.52 (m, 4H), 5.69 (m, 1H), 7.30–7.32 (m, 2H), 7.66–7.76 (m, 2H), 8.85–8.93 (m, 2H), 9.34–9.43 (m, 2H), 9.87–9.94 (m, 2H), 10.34–10.54 (m, 4H); ESI-MS obsd (+) 316.6 (M + 2H)2+, 632.0 (M + H)+, 654.0 (M + Na)+; calcd 631.18 (C31H31N5O6P2); λabs (H2O) 402, 508, 546, 567 nm; λem (H2O, λexc 402 nm) 625, 688 nm; HPLC tR= 13.98 min.5-(4-Acetamidophenyl)-15-(1,5-bis(dihydroxyphosphoryl)pent-3-yl)porphyrin (2b)
A sample of 14 (20.4 mg, 0.0279 mmol) in anhydrous CH2Cl2 (2 mL) under argon was treated with trimethylsilyl iodide (150 μL). The reaction was allowed to proceed for 30 min. The reaction was quenched with MeOH (0.5 mL). The sample was concentrated. The solid residue was treated with dilute aqueous sodium bicarbonate (2 mL of a 5 wt% solution). Chromatography (C-18 silica, water–MeOH, 0→50%) followed by evaporation of the solvent and freeze-drying from water afforded a deep red solid (17.9 mg, 95%): 1H NMR (D2O) δ 1.66 (br, 2H), 2.15–2.28 (m, 5H), 3.55–3.60 (m, 4H), 5.69 (br, 1H), 6.66 (br, 2H), 7.15 (br, 2H), 7.66 (br, 2H), 7.92–8.12 (m, 2H), 9.29–9.53 (m, 4H), 10.12–10.22 (m, 2H); ESI-MS obsd (+) 688.0 (M + Na)+, 674.2 (M + H)+, 344.5 (M + Na + H)2+, 337.5 (M + 2H)2+; calcd 673.19 (C33H33N5O7P2); λabs (H2O) 401, 505 nm; λem (H2O, λexc 401 nm) 625, 686 nm; HPLC tR = 10.09 min.5-(1,5-Bis(dihydroxyphosphoryl)pent-3-yl)-15-(4-α-iodoacetamidophenyl)porphyrin (2c)
A sample of 15 (58 mg, 0.068 mmol) in anhydrous CH2Cl2 (2 mL) under argon was treated with trimethylsilyl iodide (100 μL). The reaction was allowed to proceed for 30 min. The reaction was quenched with MeOH (0.5 mL). The sample was concentrated. The solid residue was treated with dilute aqueous sodium bicarbonate (2 mL of a 5 wt% solution). Chromatography (C-18 silica, water–MeOH, 0→50%) followed by evaporation of the solvent and freeze-drying from water afforded a deep red voluminous solid (30.4 mg, 56%): 1H NMR (D2O) δ 1.41–1.67 (m, 2H), 2.08–2.30 (m, 2H), 3.39–3.60 (m, 4H), 5.60–5.62 (m, 1H), 6.52 (br, 2H), 7.05 (br, 2H), 7.44–7.58 (m, 2H), 7.75 (br, 1H), 8.00 (br, 1H), 9.16 (br, 1H), 9.26 (br, 1H), 9.48 (br, 2H), 10.05 (br, 1H), 1014 (br, 1H); ESI-MS obsd (+) 674.1 (M − I + H)+, 800.0 (M + H)+; (−) 672.1 (M − I − H)−, 798.0 (M − H)−; calcd 799.08 (C33H32IN5O7P2); λabs (H2O) 402, 508, 542, 568 nm; λem (H2O, λexc 402 nm) 625, 688 nm; HPLC tR = 17.41 min.5-(4-Aminophenyl)-15-(1,5-bis(dimethoxyphosphoryl)pent-3-yl)porphyrin (3)
A solution of 13 (168 mg, 0.268 mmol) in THF (7 mL) was treated with P(OCH3)3 (35 mL). The reaction mixture was refluxed under argon for 36 h. The sample was concentrated at reduced pressure. The residue was dissolved in a mixture of CH2Cl2 and water. The phases were separated and the aqueous layer was extracted with CH2Cl2. The organic extract was dried (Na2SO4). Chromatography (silica, CH2Cl2–MeOH, 0→7%) afforded a deep red solid (72.8 mg, 40%): 1H NMR δ−2.84 (s, 1H), −2.77 (s, 1H), 1.25–1.46 (m, 2H), 1.90–2.01 (m, 2H), 3.12–3.15 (m, 2H), 3.30–3.49 (m, 14H), 5.42 (m, 1H), 7.11–7.13 (m, 2H), 8.02–8.04 (m, 2H), 9.14–9.16 (m, 2H), 9.36–9.37 (m, 2H), 9.47–9.49 (m, 2H), 9.70–9.72 (m, 2H), 10.27–10.28 (m, 2H); LD-MS obsd 687.2; FAB-MS obsd 688.2479, calcd 688.2454 [(M + H)+, M = C35H39N5O6P2]; λabs 409, 505 nm; λem (λexc 409 nm) 640, 702 nm.Zn(II)-5-(1,5-dihydroxypent-3-yl)-15-(4-nitrophenyl)porphyrin (6)
A solution of 11 (130 mg, 0.158 mmol) was dissolved in THF containing TBAF (3.0 mL of a 1.0 M solution, water content ∼5%). The reaction was allowed to proceed for 12 h. Then the mixture was poured into ethyl acetate, and the solution was washed with water. The aqueous layer was extracted with ethyl acetate. The organic extract was washed (water), dried (Na2SO4), and concentrated. The solid residue was dissolved in a small volume of THF. Anhydrous zinc acetate (200 mg, 1.09 mmol) was added, and the mixture was stirred at room temperature for 15 min. Column chromatography [neutral alumina, CH2Cl2–MeOH–THF (49 : 1 : 0 → 7 : 1 : 2)] afforded a bright purple solid (62.8 mg, 67%): 1H NMR δ 3.07–3.11 (m, 2H), 3.26–3.28 (m, 2H), 3.55–3.59 (m, 4H), 5.97 (m, 1H), 8.46–8.49 (m, 2H), 8.66–8.69 (m, 2H), 8.96 (s, 2H), 9.40–9.47 (m, 4H), 9.94–9.95 (m, 2H), 10.06–10.07 (m, 2H), 10.23 (s, 2H); LD-MS obsd 595.1; FAB-MS obsd 595.1183, calcd 595.1198 (C31H25N5O4Zn); λabs [CH2Cl2–MeOH (95 : 5)] 412, 545 nm; λem (λexc 412 nm) 598, 641 nm. Note: it is important to load the sample onto the column as a concentrated solution in THF, as the porphyrin-diol has very poor solubility in many other organic solvents, including CH2Cl2. As the initial eluant mixture (CH2Cl2–MeOH) does not contain THF, the desired porphyrin-diol precipitates. After elution of the less polar contaminants (unidentified), the porphyrin should be redissolved by the addition of 20–30 mL of neat THF onto the column. Afterwards the desired compound can be isolated by elution with a mixture of CH2Cl2–MeOH–THF (7 : 1 : 2).1,9-Diformyl-5-(4-nitrophenyl)dipyrromethane (8)
A solution of 7 (2.79 g, 10.4 mmol) in DMF (10.4 mL) at 0 °C under argon was treated with phosphorous oxychloride (2.04 mL, 22.3 mmol). Stirring was continued for 1 h at 0 °C. The solution was poured into ice-cold aqueous sodium hydroxide (80 mL of 10 wt% solution). The mixture was extracted with ethyl acetate (5 × 80 mL). The organic extract was washed (water and brine), dried (Na2SO4), and concentrated. Chromatography on silica (CH2Cl2–ethyl acetate, 1 : 3 → 1 : 2) gave a pale brown solid (1.80 g, 53%): mp 73–75 °C (dec.); IR (film, νmax, cm−1) 1486, 1519, 1645, 2845, 3129, 3240; 1H NMR δ 5.68 (s, 1H), 6.02–6.04 (m, 2H), 6.87–6.89 (m, 2H), 7.54–7.56 (m, 2H), 8.22–8.24 (m, 2H), 9.15 (s, 2H), 10.89 (s, 2H); 13C NMR δ 44.50, 112.38, 122.66, 124.22, 124.39, 133.09, 140.66, 146.80, 147.57, 179.52; FAB-MS obsd 324.0996, calcd 324.0984 [(M + H)+, M = C17H13N3O4]; λabs 443 nm.5-(4-Nitrophenyl)-1,9-bis[(N-propylimino)methyl]dipyrromethane (9)
A solution of 8 (1.50 g, 4.64 mmol) in THF (16 mL) was treated with propylamine (7.9 mL). The solution was stirred at room temperature for 1 h. The volatile components were evaporated, and the sample was dried at reduced pressure, affording a pale brown solid (quantitative): mp 54–56 °C (dec.); 1H NMR δ 0.87–0.92 (m, 6H), 1.57–1.64 (m, 4H), 3.38–3.43 (m, 4H), 5.50 (s, 1H), 5.90–5.92 (m, 2H), 6.36 (m, 2H), 7.32–7.35 (m, 2H), 7.92 (s, 2H), 8.12–8.15 (m, 2H); 13C NMR δ 11.99, 24.45, 62.63, 109.81, 114.96, 123.90, 129.49, 130.91, 135.21, 147.11, 148.90, 151.86; FAB-MS obsd 406.2239, calcd. 406.2243 [(M + H)+, M = C23H27N5O2]. Note: it is possible to store rigorously solvent-free samples of 9 for a few days at −4 °C without significant loss of reactivity. However, 9 proved quite unstable at room temperature, and generally was used immediately upon isolation.Zn(II)-5-[1,5-bis(tert-butyldimethylsilyloxy)pent-3-yl]-15-(4-nitrophenyl)porphyrin (11)
A solution of 9 (405 mg, 1.00 mmol) and 10 (476 mg, 1.00 mmol) in toluene (92 mL) was treated with Zn(OAc)2 (1.68 g, 10.0 mmol). The mixture was refluxed for 18 h open to the air. The toluene was evaporated, and the residue was chromatographed (silica, CH2Cl2) to give a purple solid (208 mg, 25%): 1H NMR δ−0.12 (s, 12H), 0.82 (s, 18H), 3.08–3.16 (m, 2H), 3.29–3.40 (m, 2H), 3.61–3.77 (m, 4H), 5.80–6.03 (m, 1H), 8.18–8.21 (m, 2H), 8.60–8.63 (m, 2H), 8.76–8.78 (m, 2H), 9.18–9.21 (m, 2H), 9.43–9.48 (m, 2H), 9.92–9.93 (m, 1H), 10.07–10.12 (m, 3H); LD-MS obsd 824.0; FAB-MS obsd 823.2927, calcd 823.2928 (C43H53N5O4Si2Zn); λabs 408, 537 nm; λem (λexc 408 nm) 583, 633 nm.5-(1,5-Dibromopent-3-yl)-15-(4-nitrophenyl)porphyrin (12)
Following a reported method,11,38 a suspension of 6 (62.3 mg, 0.104 mmol) in dry CH2Cl2 (23 mL) was treated with CBr4 (100 mg, 0.301 mmol). The mixture was cooled in an ice-water bath for 10 min. Triphenylphosphine (155 mg, 0.591 mmol) was added. The solution was allowed to warm to room temperature. The reaction was allowed to proceed at room temperature for 12 h. Water was added, and the phases were separated. The aqueous phase was extracted with CH2Cl2. The organic extract was washed with water and dried (Na2SO4). Chromatography (silica, CH2Cl2) afforded a dark green solid (34.4 mg, 50%): 1H NMR δ−2.87 (s, 1H), −2.83 (s, 1H), 3.20–3.42 (m, 6H), 3.67–3.77 (m, 2H), 5.82–5.86 (m, 1H), 8.41–8.44 (m, 2H), 8.67–8.69 (m, 2H), 8.96–8.99 (m, 2H), 9.41–9.64 (m, 5H), 10.01–10.02 (m, 1H), 10.34 (s, 2H); LD-MS obsd 657.2, also obsd 578.8 (M − Br)−; FAB-MS obsd 658.0472, calcd 658.0453 [(M + H)+, M = C31H25Br2N5O2]; λabs 407, 504 nm; λem (λexc 407 nm) 634, 699 nm. Note: prolonged reaction times resulted in loss of the bromine substituents. In a typical reaction sequence, isolation of 12 was followed immediately by reduction to 13, which in turn was used immediately upon isolation for the synthesis of 3.5-(4-Aminophenyl)-15-(1,5-dibromopent-3-yl)porphyrin (13)
A sample of 12 (93.0 mg, 0.141 mmol) in THF (14 mL) was treated with Pd/C (5 wt%, 90.1 mg) and ammonium formate (89.0 mg, 1.41 mmol). The reaction mixture was refluxed for 2 h. The reaction mixture was filtered. The filtered solid was washed with CH2Cl2 and water. The aqueous layer was extracted with CH2Cl2. The organic extract was washed with water and dried (Na2SO4). Evaporation of the solvent afforded a deep red solid that was used without further purification (71.7 mg, 81%): 1H NMR δ 3.16–3.21 (m, 4H), 3.59–3.71 (m, 4H), 5.75–5.80 (m, 1H), 7.10–7.13 (m, 2H), 7.97–8.00 (m, 2H), 9.09–9.12 (m, 2H), 9.32–9.36 (m, 2H), 9.43–9.48 (m, 2H), 9.57–9.58 (m, 1H), 9.92–9.94 (m, 1H), 10.24–10.26 (m, 2H); LD-MS obsd 627.0, calcd 627.1 (C31H27Br2N5); λabs 407, 504 nm; λem (λexc 407 nm) 634, 699 nm.5-(4-Acetamidophenyl)-15-(1,5-bis(dimethoxyphosphoryl)pent-3-yl)porphyrin (14)
A sample of 3 (28.2 mg, 0.0410 mmol) in pyridine (1.0 mL) was treated with acetic anhydride (38.5 μL, 0.410 mmol). Stirring was continued for 12 h at room temperature. The reaction mixture was diluted with CH2Cl2 and washed with water. The aqueous layer was extracted with CH2Cl2. The organic extract was washed with water, dried (Na2SO4), and concentrated. Chromatography (neutral alumina, CH2Cl2–MeOH, 0→3%) afforded a deep red solid (22 mg, 74%): 1H NMR δ−3.94 (s, 1H), −2.79 (s, 1H), 1.34–1.44 (m, 2H), 1.95–2.00 (m, 2H), 2.34 (s, 3H), 3.15–3.52 (m, 16H), 5.34–5.46 (m, 1H), 7.89–7.91 (m, 2H), 8.00–8.03 (m, 2H), 8.24 (s, 1H), 8.84–8.85 (m, 1H), 8.95–8.96 (m, 1H), 9.08 (app s, 1H), 9.19 (app s, 1H), 9.47 (s, 2H), 9.74–9.75 (m, 2H), 10.17 (s, 2H); LD-MS obsd 730.1; FAB-MS obsd 730.2540, calcd 730.2560 [(M + H)+, M = C37H41N5O7P2]; λabs 407, 504, 538, 576, 631 nm; λem (λexc 407 nm) 635, 702 nm.5-(1,5-Bis(dimethoxyphosphoryl)pent-3-yl)-15-(4-α-iodoacetamidophenyl)porphyrin (15)
A sample of 3 (52 mg, 0.076 mmol) in CH2Cl2 (5 mL) was treated with DCC (158 mg, 0.767 mmol) and iodoacetic acid (539 mg, 2.89 mmol). Stirring was continued for 3 h at room temperature. The reaction mixture was diluted with CH2Cl2 and washed with water. The aqueous layer was extracted with CH2Cl2. The organic extract was washed with water, dried (Na2SO4), and concentrated. Chromatography (neutral alumina, CH2Cl2–MeOH, 0→3%) afforded a deep red solid (33.6 mg, 52%): 1H NMR δ−3.05 (s, 1H), −2.83 (s, 1H), 1.33–1.53 (m, 2H), 1.93–2.08 (m, 2H), 3.18–3.49 (m, 16H), 4.06 (s, 2H), 5.41–5.43 (m, 1H), 7.82–7.96 (m, 4H), 8.65–8.66 (m, 1H), 8.79 (s, 1H), 8.89–8.91 (m, 1H), 8.94–8.96 (m, 1H), 9.20–9.21 (m, 1H), 9.47–9.50 (m, 2H), 9.72–9.74 (m, 2H), 10.14 (s, 1H), 10.22 (s, 1H); LD-MS obsd 855.0; FAB-MS obsd 856.1556, calcd 856.1526 [(M + H)+, M = C37H40IN5O7P2]; λabs 405, 504, 539, 576 nm; λem (λexc 405 nm) 635, 700 nm.
Acknowledgements
This work was supported by the NIH (GM36238). Mass spectra were obtained at the Mass Spectrometry Laboratory for Biotechnology at North Carolina State University. Partial funding for the facility was obtained from the North Carolina Biotechnology Center and the NSF. We thank Dr J. Bruce Pitner (Becton, Dickinson & Co., Research Triangle Park, North Carolina) for stimulating discussions.References
- T. Morii, K. Sugimoto, K. Makino, M. Otsuka, K. Imoto and Y. Mori, J. Am. Chem. Soc., 2002, 124, 1138–1139 CrossRef CAS.
- P. Panizzi, R. Friedrich, P. Fuentes-Prior, H. K. Kroh, J. Briggs, G. Tans, W. Bode and P. E. Bock, J. Biol. Chem., 2006, 281, 1169–1178 CAS.
- Y. Park, K. Y. Kwok, C. Boukarim and K. G. Rice, Bioconjugate Chem., 2002, 13, 232–239 CrossRef CAS.
- M. M. Mhlanga, D. Y. Vargas, C. W. Fung, F. R. Kramer and S. Tyagi, Nucleic Acids Res., 2005, 33, 1902–1912 CrossRef CAS.
- T. Nagase, E. Nakata, S. Shinkai and I. Hamachi, Chem. Eur. J., 2003, 9, 3660–3669 CrossRef CAS.
- Y. Adachi, W. Chen, W. H. Shang and T. Kamata, Anal. Biochem., 2005, 342, 348–351 CrossRef CAS.
- A. Toutchkine, P. Nalbant and K. M. Hahn, Bioconjugate Chem., 2002, 13, 387–391 CrossRef CAS.
- M. Taniguchi, M. Ptaszek, B. E. McDowell, P. D. Boyle and J. S. Lindsey, Tetrahedron, 2007, 63, 3850–3863 CrossRef CAS.
- J. K. Laha, C. Muthiah, M. Taniguchi and J. S. Lindsey, J. Org. Chem., 2006, 71, 7049–7052 CrossRef CAS.
- J. K. Laha, C. Muthiah, M. Taniguchi, B. E. McDowell, M. Ptaszek and J. S. Lindsey, J. Org. Chem., 2006, 71, 4092–4102 CrossRef CAS.
- K. E. Borbas, P. Mroz, M. R. Hamblin and J. S. Lindsey, Bioconjugate Chem., 2006, 17, 638–653 CrossRef CAS.
- G. T. Hermanson, Bioconjugate Techniques, Academic Press, San Diego, CA, 1996 Search PubMed.
- L. Tolosa, I. Gryczynski, L. R. Eichhorn, J. D. Dattelbaum, F. N. Castellano, G. Rao and J. R. Lakowicz, Anal. Biochem., 1999, 267, 114–120 CrossRef CAS.
- K. K.-W. Lo, L.-L. Wong and H. A. O. Hill, FEBS Lett., 1999, 451, 342–346 CrossRef CAS.
- B. D. Hamman, A. V. Oleinikov, G. G. Jokhadze, R. R. Traut and D. M. Jameson, Biochemistry, 1996, 35, 16672–16679 CrossRef CAS.
- R. P. Haugland, Handbook of Fluorescent Probes and Research Chemicals, Molecular Probes Inc., Eugene, OR, 1996 Search PubMed.
- L. Johansson, C. Chen, J.-O. Thorell, A. Fredriksson, S. Stone-Elander, G. Gafvelin and E. S. J. Arnér, Nat. Methods, 2004, 1, 1–6 CrossRef.
- L. A. Ernst, R. K. Gupta, R. B. Mujumdar and A. S. Waggoner, Cytometry, 1989, 10, 3–10 CrossRef CAS.
- A. Toutchkine, D.-V. Nguyen and K. M. Hahn, Bioconjugate Chem., 2007, 18, 1344–1348 CrossRef CAS.
- (a) Y. Wu, K.-S. Kwon and S. G. Rhee, FEBS Lett., 1998, 440, 111–115 CrossRef CAS; (b) C. Bayle, C. Issac, R. Salvayre, F. Couderc and E. Caussé, J. Chromatogr., A, 2002, 979, 255–260 CrossRef CAS.
- (a) J. E. T. Corrie and J. S. Craik, J. Chem. Soc., Perkin Trans. 1, 1994, 2967–2973 RSC; (b) M. J. Blackman, J. E. T. Corrie, J. C. Croney, G. Kelly, J. F. Eccleston and D. M. Jameson, Biochemistry, 2002, 41, 12244–12252 CrossRef CAS.
- M. Sinev, P. Landsmann, E. Sineva, V. Ittah and E. Haas, Bioconjugate Chem., 2000, 11, 352–362 CrossRef CAS.
- P. Hammarström, R. Owenius, L.-G. Mårtensson, U. Carlsson and M. Lindgren, Biophys. J., 2001, 80, 2867–2885 CrossRef CAS.
- (a) K. K.-W. Lo, D. C.-M. Ng and C.-K. Chung, Organometallics, 2001, 20, 4999–5001 CrossRef CAS; (b) K. K.-W. Lo, C.-K. Chung, T. K.-M. Lee, L.-H. Lui, K. H.-K. Tsang and N. Zhu, Inorg. Chem., 2003, 42, 6886–6897 CrossRef CAS.
- C. Garino, S. Ghiani, A. Gobetto, C. Nervi, L. Salassa, V. Ancarani, P. Neyroz, L. Franklin, J. B. A. Ross and E. Seibert, Inorg. Chem., 2005, 44, 3857–3879.
- K. M.-C. Wong, W.-S. Tang, B. W.-K. Chu, N. Zhu and V. W.-W. Yam, Organometallics, 2004, 23, 3459–3465 CrossRef CAS.
- K. K.-W. Lo, J. S.-Y. Lau, D. C.-M. Ng and N. Zhu, J. Chem. Soc., Dalton Trans., 2002, 1753–1756 RSC.
- (a) P. G. Fajer, Proc. Natl. Acad. Sci. U. S. A., 2007, 91, 937–941; (b) Y. P. Liu, Y. Q. Ji, Y. G. Song, K. J. Liu, B. Liu, Q. Tian and Y. Liu, Chem. Commun., 2005, 4943–4945 RSC.
- M. A. Sinev, E. V. Sineva, V. Ittah and E. Haas, FEBS Lett., 1996, 397, 273–276 CrossRef CAS.
- M. P. Lillo, B. K. Szpikowska, M. T. Mas, J. D. Sutin and J. M. Beechem, Biochemistry, 1997, 36, 11273–11281 CrossRef CAS.
- G. Haran, E. Haas, B. K. Szpikowska and M. T. Mas, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 11764–11768 CAS.
- S. H. H. Zaidi, R. S. Loewe, B. A. Clark, M. J. Jacob and J. S. Lindsey, Org. Process Res. Dev., 2006, 10, 304–314 Search PubMed.
- S. H. H. Zaidi, R. M. Fico, Jr and J. S. Lindsey, Org. Process Res. Dev., 2006, 10, 118–134 Search PubMed.
- B. J. Littler, M. A. Miller, C.-H. Hung, R. W. Wagner, D. F. O'Shea, P. D. Boyle and J. S. Lindsey, J. Org. Chem., 1999, 64, 1391–1396 CrossRef CAS.
- J. K. Laha, S. Dhanalekshmi, M. Taniguchi, A. Ambroise and J. S. Lindsey, Org. Process Res. Dev., 2003, 7, 799–812 Search PubMed.
- M. Taniguchi, A. Balakumar, D. Fan, B. E. McDowell and J. S. Lindsey, J. Porphyrins Phthalocyanines, 2005, 9, 554–574 CrossRef CAS.
- S.-i. Tamaru, L. Yu, W. J. Youngblood, K. Muthukumaran, M. Taniguchi and J. S. Lindsey, J. Org. Chem., 2004, 69, 765–777 CrossRef CAS.
- M. Wedel, A. Walter and F.-P. Montforts, Eur. J. Org. Chem., 2001, 1681–1687 CrossRef CAS.
- D. C. Tabor, F. H. White, L. W. Collier, IV and S. A. Evans, Jr., J. Org. Chem., 1983, 48, 1638–1643 CrossRef CAS.
- T. Gartiser, C. Selve and J.-J. Delpuech, Tetrahedron Lett., 1983, 24, 1609–1610 CrossRef CAS.
- P. M. G. Bavin, Org. Synth., 1973, Coll. Vol. 5, 30–31.
- D. Horton, W. Priebe and O. Varela, Carbohydr. Res., 1985, 144, 317–324 CrossRef CAS.
- V. S. Chirvony, A. van Hoek, V. A. Galievsky, I. V. Sazanovich, T. J. Schaafsma and D. Holten, J. Phys. Chem. B, 2000, 104, 9909–9917 CrossRef CAS.
- S. I. Yang, J. Seth, J.-P. Strachan, S. Gentemann, D. Kim, D. Holten, J. S. Lindsey and D. F. Bocian, J. Porphyrins Phthalocyanines, 1999, 3, 117–147 CrossRef CAS.
- J. S. Lindsey, S. Prathapan, T. E. Johnson and R. W. Wagner, Tetrahedron, 1994, 50, 8941–8968 CrossRef CAS.
- B. Valeur, Molecular Fluorescence (1st edn, 3rd corrected reprint), Wiley-VCH, Weinheim, 2006, pp. 50–52 Search PubMed.
- N. Srinivasan, C. A. Haney, J. S. Lindsey, W. Zhang and B. T. Chait, J. Porphyrins Phthalocyanines, 1999, 3, 283–291 CrossRef CAS.
- H. L. Kee, C. Kirmaier, L. Yu, P. Thamyongkit, W. J. Youngblood, M. E. Calder, L. Ramos, B. C. Noll, D. F. Bocian, W. R. Scheidt, R. R. Birge, J. S. Lindsey and D. Holten, J. Phys. Chem. B, 2005, 43, 20433–20443 CrossRef.
- F. Li, S. Gentemann, W. A. Kalsbeck, J. Seth, J. S. Lindsey, D. Holten and D. F. Bocian, J. Mater. Chem., 1997, 7, 1245–1262 RSC.
- G. Weber and F. W. J. Teale, Trans. Faraday Soc., 1957, 53, 646–655 RSC.
- C. D. Tait and D. Holten, Photobiochem. Photobiophys., 1983, 6, 201–209 Search PubMed.
- D. Magde, M. W. Windsor, D. Holten and M. Gouterman, Chem. Phys. Lett., 1974, 29, 183–188 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Discussion of synthetic routes I and II; experimental procedures and characterization data. See DOI: 10.1039/b715072e |
| This journal is © The Royal Society of Chemistry 2008 |