Concise routes to pyrazolo[1,5-a]pyridin-3-yl pyridazin-3-ones†‡
Karen A.
Johnston
a,
Robert W.
Allcock
a,
Zhong
Jiang
a,
Ian D.
Collier§
a,
Haakon
Blakli
a,
Georgina M.
Rosair
a,
Patrick D.
Bailey¶
a,
Keith M.
Morgan
a,
Yasushi
Kohno
b and
David R.
Adams
*a
aChemistry Department, School of Engineering and Physical Sciences, Heriot-Watt University, Riccarton, Edinburgh, UK EH14 4AS. E-mail: D.R.Adams@hw.ac.uk; Fax: +44 (0)131 451 3180; Tel: +44 (0)131 451 8021
bDiscovery Research Laboratories, Kyorin Pharmaceutical Co., Ltd, 2399-1, Nogi, Nogi-machi, Shimotsuga-gun, Tochigi 329-0114, Japan
First published on 22nd November 2007
Abstract
Cycloaddition of pyridine N-imine with 6-alkyl-4-oxohex-5-ynoates followed by condensation with hydrazine provides concise access to pharmacologically active 6-(pyrazolo[1,5-a]pyridin-3-yl)pyridazinones. For the first time alkynyl heterocycles are also shown to be effective dipolarophiles for pyridine N-imine, and analogous compounds can be accessed directly in modest yields through the reaction of 6-(alkyn-1-yl)pyridazin-3-one derivatives.
Introduction
The pyrazolo[1,5-a]pyridine subunit appears in compounds displaying a considerable range of biological activities—in histamine H3 receptor antagonists,1dopamine receptor ligands,2 inhibitors of cyclooxygenase-2,3 serotonin 5-HT3 receptor antagonists,4 P-glycoprotein inhibitors,5 p38 kinase inhibitors,6 phosphodiesterase (PDE) inhibitors,7 and in compounds8 with antiherpetic activity. In many of these compounds, the pyrazolopyridine unit is substituted at the 3-position. Pyrazolopyridines functionalized at this position with a pyridazin-3-one or 4,5-dihydropyridazin-3-one ring in particular have attracted recent interest because of their adenosine receptor antagonist activity, e.g.FK838,9,10 and PDE inhibitory activity, e.g.KCA-1312,11Fig. 1.![Pharmacologically active 6-(pyrazolo[1,5-a]pyridin-3-yl)pyridazin-3-ones, FK838 and KCA-1312.](/image/article/2008/OB/b713638b/b713638b-f1.gif) | ||
| Fig. 1 Pharmacologically active 6-(pyrazolo[1,5-a]pyridin-3-yl)pyridazin-3-ones, FK838 and KCA-1312. | ||
Synthesis of the pyrazolopyridine nucleus is routinely achieved via a 1,3-dipolar cycloaddition strategy that is well suited for the subsequent development of a 3-substituent. This is illustrated for FK838, Scheme 1, where cycloaddition of alkynoate 2 with pyridine N-imine (1), generated in situ by treatment of N-aminopyridinium iodide with base, afforded pyrazolopyridine 3 regioselectively.10 In this case, decarboxylation of the 3-position followed by acetylation yielded acetylpyrazolopyridine 5. A more concise route to 5 was developed on a pilot scale through cycloaddition of 1 with alkynone 6, directly introducing the 3-acetyl substituent. The N-substituted pyridazinone of FK838 was subsequently built up from the methylketone over a number of steps.
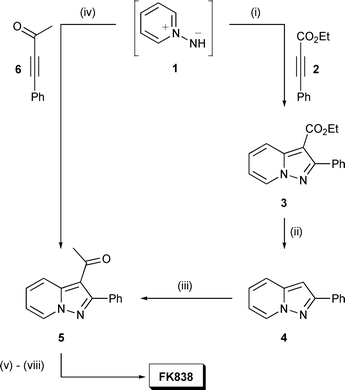 | ||
| Scheme 1 Representative synthesis of FK838.10Reagents and conditions: (i) N-aminopyridinium iodide, KOH, DMF, 32%; (ii) 47% aq. HBr, Δ, 95%; (iii) conc. H2SO4, Ac2O, 41%; (iv) N-aminopyridinium iodide, KOH–CH2Cl2, H2O, 93%; (v) OHCCO2H·2H2O, AcOH, DMF, EtOAc, 65%; (vi) N2H4·H2O, Me2NAc, 105–110 °C, 95%; (vii) Br(CH2)3CO2Et–(PhCH2NEt3)Cl, K2CO3, DMF, MeOH 55 °C; (viii) NaOH–H2O, 89% over 2 steps. | ||
As part of a programme to develop the PDE inhibitory activity of KCA-1312 and analogues, we required a concise route to 6-(pyrazolo[1,5-a]pyridin-3-yl)pyridazin-3-ones that would allow series expansion for structure–activity relationship studies. While the pyridine N-imine cycloaddition is one of the most widely employed routes to pyrazolo[1,5-a]pyridines, it has been largely confined to reactions with simple alkynoates or alkynones such as 2 and 6 as the dipolarophiles.12 We sought to apply the reaction with more advanced alkyne dipolarophiles that would minimize the number of steps required for introduction of the pyridazinone ring following pyrazolopyridine construction. It was envisaged that the cycloaddition of dipolarophiles of type 7 (Scheme 2), containing γ-keto ester functionality, would be straightforward. Subsequent condensation of the immediate cycloaddition product with hydrazine would then complete the formation of the dihydropyridazinone ring in just one additional step to form 8 (R2 = H). We also considered that use of an alkynyl pyridazinone dipolarophile (9) might enable direct access to target compounds of type 10 in a single step. These routes might thus provide complementary access to both pyridazinone and dihydropyridazinone analogues whilst facilitating convenient multiple parallel synthesis of a wide range of pyridine ring-substituted variants through combination of the dipolarophile with a set of substituted pyridine N-imines (11). Reactions of alkynyl heterocycles as dipolarophiles in cycloadditions with pyridine N-imines have not, however, previously been reported in the literature. In this paper we present the first examples of such reactions and the utility of alkynes of types 7 and 9 for the synthesis of pharmacologically active 6-(pyrazolo[1,5-a]pyridin-3-yl)pyridazin-3-ones.
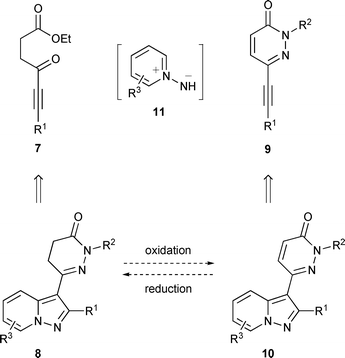 | ||
| Scheme 2 Overall synthetic strategy. | ||
Results and discussion
Beginning with the alkynyl γ-keto ester approach, ethyl 7-methyl-4-oxooct-5-ynoate (13), Scheme 3, was prepared in a single step by reaction of ethyl succinyl chloride (12) with (3-methylbut-1-ynyl)magnesium bromide. Alkyne 13 underwent cycloaddition with pyridine N-imine satisfactorily when treated with a mixture of N-aminopyridinium mesitylenesulfonate13 and K2CO3 in DMF at room temperature, affording pyrazolopyridine 14 in 56% yield. Subsequent condensation with hydrazine generated dihydropyridazinone 15 (92% yield), completing a concise 3-step sequence. Further derivatisation of the top ring was achieved by oxidation to the corresponding pyridazinone (16) and by alkylation to introduce a substituent at the N(2)-position (17 and 18). Attempts to access N(2)-substituted dihydropyridazinones by direct condensation of keto ester 14 with monosubstituted hydrazines (e.g. benzyl hydrazine) were unsuccessful however. The vinylogous amide character of the ketone renders it less susceptible to nucleophilic attack than the ester , and thus the unsubstituted terminus of the hydrazine reacts preferentially with the ester to form a hydrazide .|| The route shown in Scheme 3 was amenable to multiple parallel synthesis of an extensive range of pyridine ring-substituted analogues through combination of the dipolarophile with a set of substituted pyridine N-imines (11) derived in situ from their parent N-aminopyridinium salts.** The requisite salts are readily prepared in a single step by reaction of O-mesitylenesulfonylhydroxylamine with the appropriate pyridine.12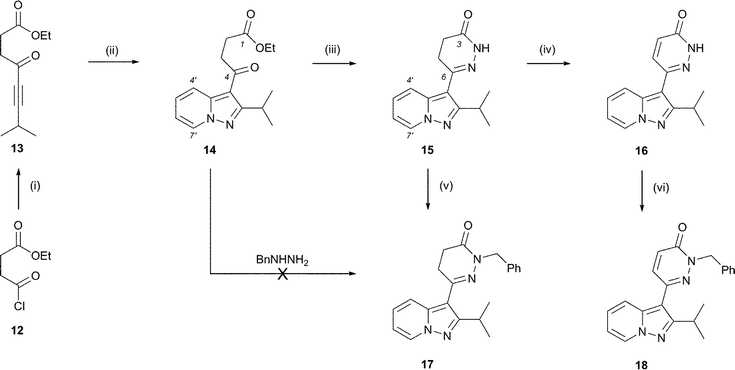 | ||
| Scheme 3 Reagents and conditions: (i) (3-methylbut-1-ynyl)magnesium bromide, THF–Et2O, 0 °C, 48%; (ii) N-aminopyridinium mesitylenesulfonate, K2CO3, DMF, 0 °C–rt, 56%; (iii) N2H4, AcOH (aq.), EtOH, Δ, 92%; (iv) Br2 in AcOH, Δ, 73%; (v) NaH, BnBr, DMF, rt, 70%; (vi) Cs2CO3, BnBr, DMF, rt, 78%. | ||
Attention was next focused upon alkynyl pyridazinone 21 as a dipolarophile, Scheme 4. This compound was prepared by Sonogashira coupling of 3-methyl-1-butyne with excess 3,6-dichloropyridazine (19) followed by treatment of the coupled product (20) with hot acetic acid and aqueous acetate. Attempts to achieve the cycloaddition of 21 with pyridine N-imine under the conditions employed for alkynone 13 (at room temperature in DMF) returned the dipolarophile unreacted. Under forcing conditions (at 120 °C) an intractable mixture of polar components was formed, and it was not possible to achieve the synthesis of pyrazolopyridine 16 by this route. However, the N-benzyl derivative (22) of alkynyl pyridazinone 21 proved more promising. Compound 22 was prepared either by alkylation of 21 with benzyl bromide or by rearranging the synthetic sequence: beginning with hydrolysis of 3,6-dichloropyridazine (19) to 6-chloropyridazin-3(2H)-one (23),14 followed by N-benzylation to 2415 and finally Sonogashira coupling with 3-methyl-1-butyne. Alkyne 22 was also unreactive with pyridine N-imine at room temperature. However, at elevated temperature (120 °C), treatment of 22 with N-aminopyridinium mesitylenesulfonate (2 equivalents) and K2CO3 in DMF over 48 h afforded pyrazolopyridine 18 in 26% yield. Alkynyl pyridazine 20 was also found to react under these conditions and afforded pyrazolopyridine 25 in 20% yield. Treatment of 25 with hot acetic acid and aqueous acetate afforded the pyridazinone (16), which had not been directly accessible from 21. The use of a dipolarophile such as 22 necessarily gives rise to the pyridazinone product. Access to the dihydropyridazinone therefore requires reduction, which can be achieved by treatment with zinc in acetic acid, as illustrated for the conversion of 18 into 17.
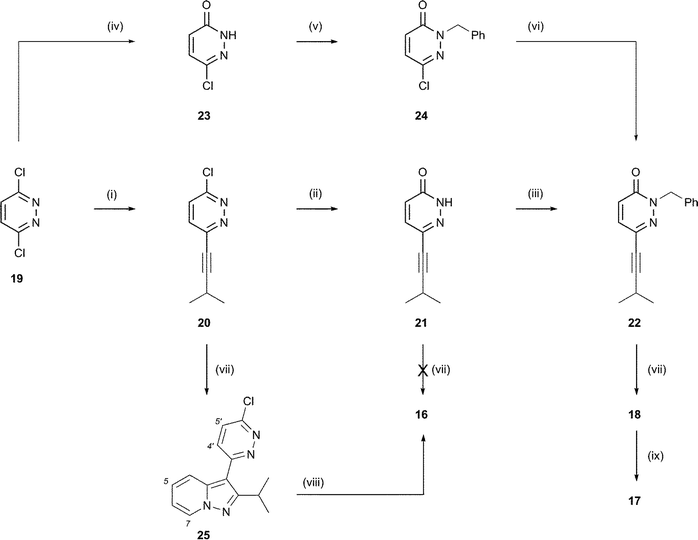 | ||
| Scheme 4 Reagents and conditions: (i) 3-methyl-1-butyne (0.5 equiv.), (i-Pr)2NH, Bu4NI, CuI, Pd(PPh3)4, THF, 90 °C (sealed vessel), 53%; (ii) AcOH, NaOAc (aq.), reflux, 87%; (iii) K2CO3, BnBr, DMF, rt, 82%; (iv) AcOH, Δ, 95%; (v) Cs2CO3, BnBr, DMF, rt, 78%; (vi) 3-methyl-1-butyne, (i-Pr)2NH, Bu4NI, CuI, Pd(PPh3)4, THF, 80 °C (sealed vessel), 83%; (vii) N-aminopyridinium mesitylenesulfonate (2–3 equiv.), K2CO3, DMF, 120 °C; 25, 20%; 16, intractable mixture formed; 18, 26%; (viii) AcOH, NaOAc (aq), Δ, 80%; (ix) Zn, AcOH, Δ, 56%. | ||
Similar chemistry with the N-phenyl analogue (28) of alkynyl pyridazine 22 afforded a concise route to pyrazolopyridines 29 and 30, Scheme 5. In this case the acetylenic dipolarophile was prepared from pyridazinedione 26.16 Thus, chlorination of 26 with POCl3 afforded chloropyridazinone 2717 and subsequent Sonogashira coupling with 3-methyl-1-butyne afforded alkyne 28 in 76% yield for the two steps. The cycloaddition reaction was conducted, as before, with N-aminopyridinium mesitylenesulfonate and K2CO3 in hot DMF, affording pyrazolopyridine 29 in 50% yield. Reduction of the latter with zinc in acetic acid efficiently gave the corresponding dihydropyridazinone (30; 84% yield).
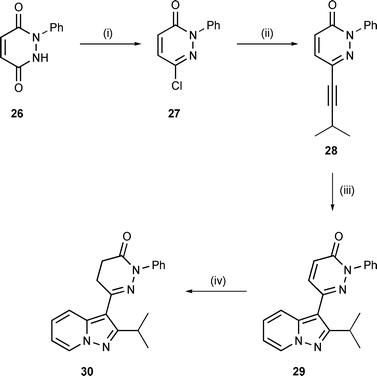 | ||
| Scheme 5 Reagents and conditions: (i) POCl3, Δ, 77%; (ii) 3-methyl-1-butyne, (i-Pr)2NH, Bu4NI, CuI, Pd(PPh3)4, THF, 80 °C (sealed vessel), 99%; (iii) N-aminopyridinium mesitylenesulfonate (2 equiv.), K2CO3, DMF, 120 °C, 50%; (iv) Zn, AcOH, Δ, 84%. | ||
Alkynyl phthalazinone 33 was also found to be effective as a dipolarophile for pyridine N-imine, Scheme 6. This compound was prepared in two steps from phthalazinedione 3118 by chlorination with POCl3 followed by Sonogashira coupling of the intermediate chloride (32) with 3-methyl-1-butyne. The yield (17%) for the first of these steps was compromised by some competing N-debenzylation under the reaction conditions with POCl3. As with the preparation of alkynyl pyridazinones 22 and 28, however, the Sonogashira coupling to 33 was efficient (91%), provided that a sealed reaction vessel was used to prevent loss of the volatile 3-methyl-1-butyne reactant. The cycloaddition step afforded pyrazolopyridine 34 in 40% yield when conducted under similar conditions to the preceding examples in hot DMF.
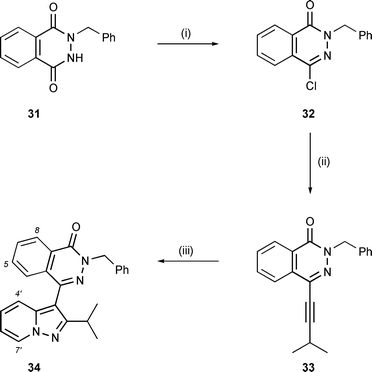 | ||
| Scheme 6 Reagents and conditions: (i) POCl3, Δ, 17%; (ii) 3-methyl-1-butyne, (i-Pr)2NH, Bu4NI, CuI, Pd(PPh3)4, THF, 80 °C (sealed vessel), 91%; (iii) N-aminopyridinium mesitylenesulfonate (2 equiv.), K2CO3, DMF, 100 °C, 40%. | ||
To test further the scope of the cycloaddition reaction of pyridine N-imine with alkynyl heterocycles, attention was turned to alkynyl pyridine 35, prepared in 41% yield by Sonogashira coupling of 2-chloropyridine with 3-methyl-1-butyne (Scheme 7). Reaction of alkyne 35 with N-aminopyridinium mesitylenesulfonate (2 equiv.) and K2CO3 in DMF at 95 °C over 18 h afforded the target pyrazolopyridine (36) in just 3% yield together with 38% recovery of unreacted alkyne . At reduced temperature (80 °C) over 40 h the yield of pyrazolopyridine 36 rose to 10% with 53% recovery of unreacted alkyne , suggesting that the reaction outcome is compromised by competing degradation pathways at higher temperature.††
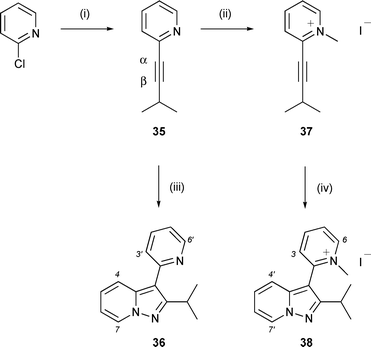 | ||
| Scheme 7 Reagents and conditions: (i) 3-methyl-1-butyne, (i-Pr)2NH, CuI, Pd(PPh3)4, THF, 70 °C (sealed vessel), 41%; (ii) MeI, THF, 60 °C, 72%; (iii) N-aminopyridinium mesitylenesulfonate (2 equiv.), K2CO3 (4 equiv.), DMF, 80 °C, 10%; (iv) N-aminopyridinium mesitylenesulfonate (2 equiv.), K2CO3 (4 equiv.), DMF, 95 °C, 18 h, 39%. | ||
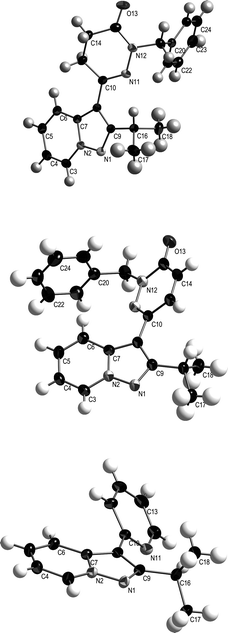
As with the reactions of the alkynyl pyridazine and pyridazinone derivatives, the cycloaddition of alkynyl pyridine 35 proceeded with regiocontrol favouring exclusive formation of a product (36) in which the heterocycle from the dipolarophile is connected to the 3-position of the pyrazolopyridine. The alternative regioisomer was not detected. The regiochemical outcome of this reaction was determined from the NOESY spectrum of 36 and confirmed by the acquisition of an X-ray crystal structure, Fig. 2. The regiochemical outcome for the cycloaddition of alkynyl pyridazinone 22 was similarly confirmed by acquisition of NOESY spectra and X-ray crystal structures for the product (18) and its reduced analogue (17). These structures also served to substantiate the regiochemistry assigned to the reaction of alkynone 13 with pyridine N-imine, Scheme 3.
Cycloaddition reactions of alkynoate and alkynone dipolarophiles with pyridine N-imines are typically conducted within a temperature range of 0–25 °C. In contrast, cycloaddition of the alkynyl heterocycles presented here requires significantly elevated temperatures in order to proceed. This requirement was thought likely to reflect reduced dipolarophilic reactivity due to the comparatively modest electron demand exerted on the alkyne by the heterocycle. Increased electron demand in the heterocycle might therefore be expected to enhance the reactivity of the dipolarophile. In order to verify this expectation at a qualitative level, alkynyl pyridine 35 was quaternised with methyl iodide and the resulting alkynyl pyridinium salt (37) tested in the reaction with pyridine N-imine. When conducted under identical conditions to the reaction of alkynyl pyridine 35, with N-aminopyridinium mesitylenesulfonate (2 equiv.) and K2CO3 in DMF at 95 °C over 18 h, a greatly improved 39% yield of the pyrazolopyridine product (38) was obtained from the cycloaddition step. However, the yields of the cycloaddition reactions of 35 and 37 were not improved with extended reaction times. Indeed, in the case of alkynyl pyridinium salt 37 the reaction time could be reduced substantially without significant reduction in the yield of pyrazolopyridine product. Thus, the yield of 38 was 37% when the reaction was conducted at 95 °C over the reduced duration of 2 h.
Examination of the 13C chemical shifts for the alkyne carbons of compounds 35 and 37 suggests that quaternisation of the pyridine nitrogen exerts a substantial polarizing effect upon the alkyne , which may contribute to the improved reactivity of 37. The alkyne α and β carbons of compound 35 give resonances at δC 80 and 96 respectively, whereas the corresponding carbons in alkynyl pyridinium salt 37 give signals at δC 71 and 117. The enhanced polarisation of alkyne 37 is also reflected in partial charge calculations, which give values of −0.130 and −0.109 for the α and β carbons respectively in alkyne 35 and values of −0.289 and −0.079 for the corresponding carbons in alkyne 37.‡‡
Conclusions
In summary, concise routes to pharmacologically active 6-(pyrazolo[1,5-a]pyridin-3-yl)pyridazin-3-ones have been developed using alkynyl γ-keto ester and alkynyl pyridazinone dipolarophiles for cycloaddition with pyridine N-imines. Use of the alkynyl γ-keto ester in the cycloaddition furnishes the dihydropyridazinone following an additional hydrazine condensation step; the alkynyl pyridazinone affords the pyrazolopyridine-pyridazinone directly. Application of standard reduction and oxidation procedures permits pyridazinone–dihydropyridazinone ring interconversion. Previously reported reactions of pyridine N-imines have been confined to simple alkynone or alkynoate dipolarophiles. We show here for the first time that reactions with alkynyl heterocycles are feasible, albeit requiring elevated temperature. Although yields for the cycloaddition step are modest, the alkynyl heterocycles are readily prepared through robust Sonogashira coupling procedures and allow for very concise routes to pyrazolopyridines substituted at C-3 with a heterocycle.Experimental
General details
Commercially available reagents from Aldrich, Avocado and Lancaster chemical companies were generally used as supplied without further purification. Tetrahydrofuran, diethyl ether and toluene were dried by distillation from sodium-benzophenoneketyl under argon. ‘Light petroleum’ refers to the fraction boiling between 40 °C and 60 °C. Anhydrous N,N-dimethylformamide was purchased from Aldrich and used as supplied from Sure/Seal™ bottles. With the exception of the pyridine N-imine cycloadditions, which were carried out in air to facilitate oxidation of the immediate dihydropyrazolopyridine cycloadduct, reactions were routinely carried out under an inert atmosphere of argon or nitrogen. Analytical thin layer chromatography was carried out using aluminium backed plates coated with Merck Kieselgel 60 GF254 (Art. 05554). Developed plates were visualized under ultra-violet light (254 nm) and/or alkaline potassium permanganate dip. Flash chromatography was performed using DAVISIL® silica (60 Å; 35–70 µm) from Fisher (cat. S/0693/60). Fully characterized compounds were chromatographically homogeneous.Melting points were determined using a Stuart Scientific SMP10 apparatus and are uncorrected. IR spectra were recorded on a Perkin Elmer 1600 FT IR spectrometer. Spectra were recorded as potassium bromide discs, as solutions in CHCl3, or as films between sodium chloride plates. Mass spectra were obtained on Kratos Concept IS EI (electron impact) and Fisons VG Quattro (electrospray) spectrometers. 1H NMR spectra were recorded at 200 and 400 MHz on Bruker AC200 and DPX400 spectrometers; 13C NMR spectra were recorded at 50 and 101 MHz on the same instruments. Chemical shifts are recorded in parts per million (δ in ppm) and are referenced against solvent signals (δC 77.16 for chloroform and δC 39.52 for methyl sulfoxide) for 13C spectra and solvent residual resonances (δH 7.26 for chloroform and δH 2.50 methyl sulfoxide) for 1H spectra.19 Chemical shift values are accurate to ±0.01 ppm and ±0.1 ppm respectively. J values are given in Hz. Multiplicity designations used are: s, d, t, q, sept and m for singlet, doublet, triplet, quartet, septet and multiplet respectively. In 13C NMR spectra, signals corresponding to CH, CH2, or CH3 groups are assigned from DEPT. Elemental analyses were carried out by the analytical service of the Chemistry Department at Heriot-Watt University using an Exeter CE-440 Elemental Analyser.
Ethyl 7-Methyl-4-oxooct-5-ynoate (13)
(Part 1) 3-Methyl-1-butyne (11.2 g, 164 mmol) was dissolved in anhydrous THF (300 mL) and cooled to −15 °C (salt–ice bath) under argon. EtMgBr (3 M solution in Et2O; 55 mL, 165 mmol) was added slowly over 40 min to give a golden brown homogenous solution. The mixture was allowed to attain 15 °C and was stirred at that temperature for 18 h. (Note: the alkynyl magnesium bromide may precipitate!). Further alkyne (ca. 2 mL, 20 mmol) was added; the mixture was heated at 45 °C for 1 h and then cooled to rt.(Part 2) The alkynyl magnesium bromide solution from part 1 was cannulated over a period of 3 h into an ice-cooled solution of ethyl succinyl chloride (71.0 mL, 498 mmol) in anhydrous Et2O (600 mL). The resulting pale yellow heterogenous mixture was quenched by addition of saturated NH4Cl solution (20 mL), concentrated to 200 mL in vacuo and diluted with saturated NaHCO3 solution (200 mL). The mixture was stirred for 1 h. The organic layer was then separated from the basic aqueous phase, washed with brine (40 mL), dried (MgSO4) and concentrated in vacuo to give the product as a red-brown oil. The crude product was subjected to flash column chromatography , eluting with light petroleum–EtOAc (10 : 1). Fractions containing the target material were combined, concentrated in vacuo and then distilled under reduced pressure through a 15 cm fractionating column (84–120 °C, 3 mmHg) to afford a yellow oil (26.3 g). This oil was then dissolved in THF (10 mL) and stirred with saturated NaHCO3 solution (300 mL) for 1 h. The mixture was extracted with CH2Cl2 (100 mL); the separated organic phase was washed with brine, (50 mL), dried (MgSO4) and evaporated to give to give the title compound (15.6 g; 48%) as a pale red oil in >90% purity: δH (200 MHz; CDCl3) 1.20 (6 H, d, J 6.9, CH(CH3)2), 1.22 (3 H, t, J 7.1, OCH2CH3), 2.55–2.63 (2 H, m, CH2-2), 2.69 (1 H, sept, J 6.9, CH(CH3)2), 2.79–2.88 (2 H, m, CH2-3), 4.11 (2 H, q, J 7.1, OCH2CH3); δC (50 MHz; CDCl3) 14.2 (OCH2CH3), 20.8 (CH(CH3)2), 21.9 (CH(CH3)2), 28.2 (CH2-2), 40.1 (CH2-3), 60.9 (OCH2CH3), 79.6 (C-5), 99.7 (C-6), 172.2 (C-1), 185.9 (C-4); m/z (EI) 196 (3%, M+), 167 (6%, M+ − Et), 151 (26%, M+ − OEt), 129 (39%, M+ − C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCH(CH3)2), 43 (100%, C3H7+); (found: M+, 196.1099. C11H16O3 requires 196.1099).
CCH(CH3)2), 43 (100%, C3H7+); (found: M+, 196.1099. C11H16O3 requires 196.1099).
Ethyl 4-(2-isopropylpyrazolo[1,5-a]pyridin-3-yl)-4-oxobutanoate (14)
Powdered K2CO3 (5.64 g, 40.8 mmol) was added to an ice-cooled solution of 1-aminopyridinium mesitylenesulfonate (6.00 g, 20.4 mmol) in DMF (150 mL) to afford a deep purple heterogenous mixture. After 15 min 13 (4.60 g, 23.4 mmol) was added and the mixture stirred for 1 h. The mixture was allowed to attain rt over 18 h and was then partitioned between brine (150 mL) and EtOAc (70 mL). The aqueous phase was further extracted with EtOAc (2 × 70 mL), and the combined organic extracts dried (MgSO4) and evaporated. The residual oil was subjected to flash column chromatography (5 : 2 light petroleum–EtOAc) to afford the title compound (3.29 g; 56%) as a colourless powder: mp 72–73 °C (from EtOAc–light petroleum); νmax(KBr)/cm−1 2975, 2935, 1738, 1640, 1618, 1503, 1462, 1441, 1418, 1382, 1368, 1286, 1217, 1178, 1155, 1089, 1018, 998, 804, 763; δH (400 MHz; CDCl3) 1.25 (3 H, t, J 7.1, OCH2CH3), 1.38 (6 H, d, J 6.9, CH(CH3)2), 2.77 (2 H, t, J 6.6, CH2-2), 3.23 (2 H, t, J 6.6, CH2-3), 3.78 (1 H, sept, J 6.9, CH(CH3)2), 4.15 (2 H, q, J 7.1, OCH2CH3), 6.88 (1 H, dt, J6′,4′ 1.3, J6′,5′ 6.9, J6′,7′ 6.9, H-6′), 7.37 (1 H, ddd, J5′,7′ 1.2, J5′,6′ 6.9, J5′,4′ 9.0, H-5′), 8.09 (1 H, dt, J4′,5′ 9.0, J4′,6′ 1.1, J4′,7′ 1.1, H-4′), 8.45 (1 H, dt, J7′,6′ 6.9, J7′,5′ 1.1, J7′,4′ 1.1, H-7′); δC (101 MHz; CDCl3) 14.3 (OCH2CH3), 22.3 (CH(CH3)2), 27.9 (CH(CH3)2), 28.3 (CH2-2), 37.0 (CH2-3), 60.7 (OCH2CH3), 109.6 (C-3′), 113.4 (CH-6′), 119.2 (CH-4′), 127.9 (CH-5′), 129.3 (CH-7′), 141.8 (C-3a′), 164.1 (C-2′), 173.3 (C-1), 192.0 (C-4); m/z (EI) 288 (61%, M+), 243 (52%, M+ − OEt), 215 (55%, M+ − CO2Et), 187 (93%, M+ − CH2CH2CO2Et); (found: M+, 288.1474. C16H20N2O3 requires 288.1474); (found C, 66.71; H, 7.03; N, 9.74. C16H20N2O3 requires: C, 66.65; H, 6.99; N, 9.72%).§§6-(2-Isopropylpyrazolo[1,5-a]pyridin-3-yl)-4,5-dihydropyridazin-3(2H)-one (15)
Keto ester 14 (2.80 g, 9.71 mmol) was boiled for 18 h in a mixture of aqueous hydrazine buffered to pH 5 with AcOH (1.84 M in H2NNH2; 53 mL, 97.5 mmol) and EtOH (30 mL). The mixture was then concentrated in vacuo to afford an aqueous slurry that was extracted with CH2Cl2 (2 × 50 mL). The combined organic extracts were washed with brine (2 × 20 mL), dried (MgSO4) and evaporated to give the crude product (2.45 g) as a buff powder. Trituration of the latter with hot Et2O (50 mL) afforded the title compound (2.30 g; 92%) as a colourless powder: mp 211–212 °C (from EtOAc–light petroleum); νmax(KBr)/cm−1 3212, 3076, 2958, 1661, 1632, 1537, 1519, 1373, 1325, 1258, 1223, 951, 757, 736; δH (200 MHz; CDCl3) 1.40 (6 H, d, J 6.9, CH(CH3)2), 2.58–2.67 (2 H, m, CH2-4), 3.00–3.09 (2 H, m, CH2-5), 3.45 (1 H, sept, J 6.9, CH(CH3)2), 6.79 (1 H, dt, J6′,4′ 1.4, J6′,5′ 6.9, J6′,7′ 6.9, H-6′), 7.21 (1 H, ddd, J5′,7′ 1.2, J5′,6′ 6.9, J5′,4′ 8.9, H-5′), 7.78 (1 H, dt, J4′,5′ 8.9, J4′,6′ 1.1, J4′,7′ 1.1, H-4′), 8.43 (1 H, dt, J7′,6′ 6.9, J7′,5′ 1.1, J7′,4′ 1.1, H-7′), 8.58 (1 H, br s, NH); δC (50 MHz; 20% CD3OD/CDCl3) 22.2 (CH(CH3)2), 25.2 (CH2), 26.0 (CH2), 26.9 (CH(CH3)2), 105.0 (C-3′), 112.3 (CH-6′), 117.8 (CH-4′), 125.3 (CH-5′), 128.1 (CH-7′), 138.8 (C-3a′), 148.2 (C-6), 159.6 (C-2′), 167.9 (C-3); m/z (EI) 256 (34%, M+), 198 (20%), 170 (13%), 184 (70%); (found: M+, 256.1322. C14H16N4O requires 256.1324); (found C, 65.51; H, 6.25; N, 21.72. C14H16N4O requires: C, 65.61; H, 6.29; N, 21.86%).6-(2-Isopropylpyrazolo[1,5-a]pyridin-3-yl)pyridazin-3(2H)-one (16)
2-Benzyl-6-(2-isopropylpyrazolo[1,5-a]pyridin-3-yl)-4,5-dihydropyridazin-3(2H)-one (17)
2-Benzyl-6-(2-isopropylpyrazolo[1,5-a]pyridin-3-yl)pyridazin-3(2H)-one (18)
3-Chloro-6-(3-methylbut-1-ynyl)pyridazine (20)
A sealable, heavy-walled flask (∼750 mL capacity) was charged with 3,6-dichloropyridazine (41.6 g, 279 mmol), Bu4NI (37.0 g, 100 mmol), CuI (1.30 g, 6.83 mmol), Pd(PPh3)4 (1.66 g, 1.44 mmol), anhydrous THF (400 mL), 3-methyl-1-butyne (9.50 g, 140 mmol) and iPr2NH (20.0 mL, 143 mmol). The flask was sealed and the mixture stirred magnetically in an oil bath thermostatted at 90 °C for 24 h [SAFETY SCREEN]. The flask was then cooled and opened; the mixture was diluted with light petroleum (600 mL) and passed through a column of Merck kieselgel 60H (120 mm diameter × 90 mm length), eluting with 1 : 1 EtOAc–light petroleum (2 L). The eluate was evaporated and the resulting dark brown residue subjected to flash column chromatography (30% EtOAc–light petroleum). Fractions containing the target material were evaporated and the resulting light brown powder crystallised from Et2O–light petroleum to afford the title compound (13.4 g; 53%) as a colourless solid: mp 49–51 °C (Et2O–light petroleum); νmax (CHCl3 film)/cm−1 3099, 3067, 2981, 2238, 1560, 1395, 1145, 1067, 857; δH (200 MHz; CDCl3) 1.28 (6 H, d, J 6.9, CH(CH3)2), 2.83 (1 H, sept, J 6.9, CH(CH3)2), 7.44 (2 H, app. s, H-4 and H-5); δH (200 MHz; C6D6) 1.03 (6 H, d, J 6.9, CH(CH3)2), 2.48 (1 H, sept, J 6.9, CH(CH3)2), 6.32 (1 H, d, J 8.8, H-4), 6.51 (1 H, d, J 8.8, H-5); δC (50 MHz; CDCl3) 21.2 (CH(CH3)2), 22.2 (CH(CH3)2), 75.9 (CCCH), 102.7 (CCCH), 127.6 (CH-4), 131.7 (CH-5), 147.5 (C-6), 154.6 (C-3); δC (50 MHz; C6D6) 21.5 (CH(CH3)2), 22.4 (CH(CH3)2), 77.2 (CCCH), 101.7 (CCCH), 126.8 (CH-4), 131.0 (CH-5), 147.6 (C-6), 154.7 (C-3); m/z (EI) 180 (35Cl; 100%, M+), 179 (35Cl; 64%, M+ − H), 145 (37%, M+ − Cl); (found: C, 59.87; H, 4.94; N, 15.55. C9H9ClN2 requires C, 59.84; H, 5.02; N, 15.51%).§§
6-(3-Methylbut-1-ynyl)pyridazin-3(2H)-one (21)
Chloropyridazine 20 (6.07 g, 33.6 mmol) was heated in a mixture of AcOH (140 mL), water (18 mL) and NaOAc·3H2O (5.30 g) at 100 °C for 15 h. The mixture was then cooled and decanted into an ice-cooled solution of NaOH (100 g) in water (700 mL). The resulting alkaline solution was washed with CH2Cl2 (2 × 200 mL) and the organic phase discarded. The pH of the aqueous mixture was adjusted to ∼6 by addition of conc. hydrochloric acid and it was extracted with CH2Cl2 (3 × 200 mL). The combined organic extract was dried (MgSO4) and evaporated. The title compound (4.73 g; 87%) was isolated from the resulting residue as a colourless solid by flash column chromatography (50% EtOAc–light petroleum) followed by crystallisation from CHCl3–light petroleum: mp 96–97 °C (CHCl3–light petroleum); νmax(KBr)/cm−1 3468, 2976, 2925, 2233, 1682, 1648, 1585, 1547, 1442, 1005, 849, 657, 638; δH (200 MHz; CDCl3) 1.27 (6 H, d, J 6.9, CH(CH3)2), 2.79 (1 H, sept, J 6.9, CH(CH3)2), 6.95 (1 H, d, J 9.7, H-4), 7.28 (1 H, d, J 9.7, H-5), 12.65 (1 H, br s, NH); δC (50 MHz; CDCl3) 21.0 (CH(CH3)2), 22.4 (CH(CH3)2), 75.0 (CCCH), 98.9 (CCCH), 129.6 (CH-4), 133.3 (C-6), 136.2 (CH-5), 161.5 (C-3); EIMS: m/z (EI) 162 (83%, M+), 161 (100%, M+ − H), 147 (71%, M+ − CH3), 133 (38%, M+ − C2H5), 119 (33%, M+ − C3H7); (found: C, 66.44; H, 6.19; N, 17.31. C9H10N2O requires C, 66.65; H, 6.21; N, 17.27%).§§
2-Benzyl-6-(3-methylbut-1-ynyl)pyridazin-3(2H)-one (22)
CCH), 98.7 (CCCH), 128.0 (Ph CH), 128.7 (2× Ph CH), 128.8 (2× Ph CH), 129.7 (CH-4), 132.3 (Ph C), 135.1 (CH-5), 136.0 (C-6), 159.2 (C-3); m/z (EI) 252 (100%, M+), 237 (36%, M+ − CH3), 91 (79%, C7H7+); (found: M+, 252.1265. C16H16N2O requires 252.1263); (found: C, 75.86; H, 6.35; N, 11.10. C16H16N2O requires C, 76.16; H, 6.39; N, 11.10%).§§
3-(6-Chloropyridazin-3-yl)-2-isopropylpyrazolo[1,5-a]pyridine (25)
A stirred mixture of 1-aminopyridinium mesitylenesulfonate12 (1.32 g, 4.47 mmol), 20 (254 mg, 1.41 mmol) and powdered K2CO3 (1.12 g, 8.08 mmol) in DMF (15 mL) was heated at 120 °C for 30 h. The mixture was then evaporated to dryness and the residue partitioned between EtOAc (50 mL) and water (50 mL). The organic phase was separated, washed with brine (50 mL), dried (MgSO4) and evaporated. The resulting residue was subjected to flash column chromatography (2 : 3 EtOAc–light petroleum) to afford the title compound (78.0 mg; 20%) as a pale yellow powder: mp 147–148 °C (from hexane–EtOAc); νmax(KBr)/cm−1 3080, 2969, 1632, 1576, 1543, 1531, 1506, 1465, 1400, 1359, 1214, 1146, 1092, 771, 754; δH (200 MHz; CDCl3) 1.38 (6 H, d, J 6.9, CH(CH3)2), 3.48 (1 H, sept, J 6.9, CH(CH3)2), 6.79 (1 H, dt, J6,4 1.4, J6,5 6.9, J6,7 6.9, H-6), 7.20 (1 H, ddd, J5,7 1.1, J5,6 6.8, J5,4 9.0, H-5), 7.49 (1 H, d, J5′,4′ 9.0, H-5′), 7.62 (1 H, d, J4′,5′ 9.0, H-4′), 7.99 (1 H, dt, J4,5 9.0, J4,6 1.2, J4,7 1.2, H-4), 8.43 (1 H, dt, J7,6 6.9, J7,5 1.1, J7,4 1.1, H-7); δC (50 MHz; CDCl3) 22.6 (CH(CH3)2), 27.0 (CH(CH3)2), 104.4 (C-3), 112.8 (CH-6), 118.0 (CH-4), 125.9 (CH-5), 127.5 (CH-4′), 128.0 (CH-5′), 128.8 (CH-7), 139.6 (C-3a), 153.2 (C-6′), 155.6 (C-3′), 160.1 (C-2); m/z (EI) 272 (35Cl; 19%, M+), 271 (35Cl; 18%, M+ − H), 237 (44%, M+ − Cl), 69 (100%); (found: C, 61.69; H, 4.69; N, 20.78. C14H13ClN4 requires C, 61.65; H, 4.80; N, 20.54%).6-(3-Methylbut-1-ynyl)-2-phenylpyridazin-3(2H)-one (28)
A sealable, heavy-walled flask (∼50 mL capacity) was charged with 2717 (1.06 g, 5.13 mmol), Bu4NI (2.08 g, 5.64 mmol), CuI (58.6 mg, 0.31 mmol), Pd(PPh3)4 (232 mg, 201 µmol), anhydrous THF (16 mL), 3-methyl-1-butyne (927 mg, 13.6 mmol) and iPr2NH (1.00 mL, 7.13 mmol) under argon. The flask was sealed and the mixture was stirred magnetically in an oil bath thermostatted at 80 °C for 19 h [SAFETY SCREEN]. The flask was then cooled and opened; the mixture was filtered and the filtrate evaporated to afford a residue that was dissolved in EtOAc (15 mL). The resulting solution was washed with water (2 × 10 mL) followed by brine (2 × 10 mL), dried (Na2SO4) and evaporated. The residue was subjected to flash column chromatography (1 : 1 Et2O–light petroleum gradient elution). Fractions containing the target material were evaporated to afford the title compound (1.22 g; 99%) as a buff powder: νmax(KBr)/cm−1 3300, 3045, 2970, 2237, 1677, 1589, 1312, 1198, 1022, 851, 767, 692; δH (200 MHz; CDCl3) 1.26 (6 H, d, J 6.9, CH(CH3)2), 2.78 (1 H, sept, J 6.9, CH(CH3)2), 6.97 (1 H, d, J 9.6, H-4), 7.26 (1 H, d, J 9.6, H-5), 7.33–7.59 (5 H, complex overlapping m, Ph); δC (50 MHz; CDCl3) 21.2 (CH(CH3)2), 22.5 (CH(CH3)2), 75.2 (CCCH), 99.2 (CCCH), 125.7 (2× Ph ortho-CH), 128.6 (Ph para-CH), 129.0 (2× Ph meta-CH), 130.7 (CH-4), 132.8 (C-6), 135.0 (CH-5), 141.4 (Ph CH), 159.0 (C-3); m/z (EI) 238 (65%, M+), 223 (29%, M+ − CH3), 209 (11%, M+ − C2H5), 195 (7%, M+ − C3H7), 77 (100%, C6H5+); (found: M+, 238.1106. C15H14N2O requires 238.1106).
6-(2-Isopropylpyrazolo[1,5-a]pyridin-3-yl)-2-phenylpyridazin-3(2H)-one (29)
A stirred mixture of 1-aminopyridinium mesitylenesulfonate12 (753 mg, 2.56 mmol), 28 (500 mg, 2.10 mmol) and powdered K2CO3 (597 mg, 4.32 mmol) in DMF (10 mL) was heated at 120 °C for 9 h. Additional pyridinium salt (506 mg, 1.72 mmol) and K2CO3 (535 mg, 3.87 mmol) were then added and heating continued for a further 12 h. The mixture was then evaporated to dryness and the resulting residue partitioned between EtOAc (75 mL) and water (40 mL). The organic phase was separated, washed with water (2 × 40 mL) followed by brine (4 × 40 mL), dried (Na2SO4) and evaporated. The residue was subjected to flash column chromatography (1 : 1 EtOAc–light petroleum) to afford the title compound (346 mg; 50%) as a pale yellow powder: mp 174–176 °C (from petroleum 60–80 °C); νmax(KBr)/cm−1 3053, 2969, 1670, 1590, 1527, 1476, 1310, 1043, 852, 745, 695; δH (400 MHz; CDCl3) 1.43 (6 H, d, J 6.9, CH(CH3)2), 3.46 (1 H, sept, J 6.9, CH(CH3)2), 6.79 (1 H, dt, J6′,4′ 1.4, J6′,5′ 6.9, J6′,7′ 6.9, H-6′), 7.15 (1 H, d, J4,5 9.7, H-4), 7.20 (1 H, ddd, J5′,7′ 1.1, J5′,6′ 6.8, J5′,4′ 9.0, H-5′), 7.38–7.42 (1 H, complex m, Ph para-H), 7.48–7.53 (2 H, complex m, 2× Ph meta-H), 7.56 (1 H, d, J5,4 9.7, H-5), 7.70–7.72 (3 H, complex overlapping m, H-4′ and 2× Ph ortho-H), 8.45 (1 H, dt, J7′,6′ 7.0, J7′,5′ 1.0, J7′,4′ 1.0, H-7′); δC (50 MHz; CDCl3) 22.8 (CH(CH3)2), 27.2 (CH(CH3)2), 103.9 (C-3′), 112.4 (CH-6′), 117.2 (CH-4′), 125.4 (2× ortho-CH and CH-5′), 128.2 (Ph para-CH), 128.9 (2× Ph meta-CH and CH-7′), 131.5 (CH-4), 132.8 (CH-5), 139.2 (C-3a′), 141.7 (C-6), 141.9 (Ph C), 159.2 (C-3), 159.7 (C-2′); m/z (EI) 330 (36%, M+), 315 (9%, M+ − CH3), 197 (100%); (found: C, 72.48; H, 5.52; N, 16.69. C20H18N4O requires C, 72.71; H, 5.49; N, 16.96%).§§6-(2-Isopropylpyrazolo[1,5-a]pyridin-3-yl)-2-phenyl-4,5-dihydropyridazin-3(2H)-one (30)
A mixture of Zn dust (51.6 mg, 789 µmol) and 29 (250 mg, 757 µmol) in AcOH (12 mL) was boiled for 37 h, adding further zinc dust (145 mg, 2.22 mmol) in increments until TLC indicated consumption of 29. The mixture was then cooled, filtered through celite and evaporated to afford a residue that was partitioned between EtOAc (30 mL) and saturated NaHCO3 solution (10 mL). The organic phase was separated, washed with additional volumes of NaHCO3 solution (2 × 10 mL) followed by brine (3 × 10 mL), dried (Na2SO4) and evaporated. The resulting residue was subjected to flash column chromatography (3 : 2 Et2O–light petroleum) to afford the title compound (212 mg; 84%) as a pale yellow powder: mp 129–130 °C (from petroleum 60–80 °C–EtOAc); νmax(CHCl3 film)/cm−1 3019, 2975, 1670, 1634, 1523, 1496, 1357, 1216, 753, 669; δH (200 MHz; CDCl3) 1.42 (6 H, d, J 6.9, CH(CH3)2), 2.75–2.83 (2 H, complex m, CH2), 3.10–3.18 (2 H, complex m, CH2), 3.52 (1 H, sept, J 6.9, CH(CH3)2), 6.78 (1 H, dt, J6′,4′ 1.4, J6′,5′ 6.9, J6′,7′ 6.9, H-6′), 7.20 (1 H, ddd, J5′,7′ 1.1, J5′,6′ 6.8, J5′,4′ 9.0, H-5′), 7.21–7.30 (1 H, complex m, Ph para-H), 7.37–7.46 (2 H, complex m, 2× Ph meta-H), 7.58–7.65 (2 H, complex m, 2× Ph ortho-H), 7.80 (1 H, dt, J4′,5′ 9.0, J4′,6′ 1.2, J4′,7′ 1.2, H-4′), 8.42 (1 H, dt, J7′,6′ 6.9, J7′,5′ 1.1, J7′,4′ 1.1, H-7′); δC (101 MHz; CDCl3) 22.8 (CH(CH3)2), 25.9 (CH2), 27.7 (CH(CH3)2), 28.5 (CH2), 105.2 (C-3′), 112.4 (CH-6′), 118.3 (CH-4′), 124.9 (2× Ph ortho-CH), 125.5 (CH), 126.5 (CH), 128.6 (2× Ph meta-CH), 129.0 (CH), 139.3 (C-3a′), 141.4 (Ph C), 149.7 (C-6), 160.4 (C-2′), 165.4 (C-3); m/z (EI) 332 (100%, M+), 303 (23%, M+ − C2H5); (found: C, 72.09; H, 5.99; N, 16.58; C20H20N4O requires C, 72.27; H, 6.06; N, 16.86%).2-Benzyl-4-chlorophthalazin-1(2H)-one (32)
Phthalazinedione 3117 (4.93 g, 19.5 mmol) was boiled in POCl3 (70 mL). After 5 h the POCl3 was removed by distillation. The residue was dissolved in EtOAc (30 mL) and washed with saturated NaHCO3 (3 × 30 mL). The organic layer was then washed with brine (3 × 30 mL), dried (Na2SO4) and evaporated to give a brown solid that was subjected to flash column chromatography (1 : 9 EtOAc–light petroleum) to afford the title compound (905 mg; 17%) as a colourless powder: mp 99–101 °C (from EtOH); νmax(KBr)/cm−1 3033, 2958, 1656, 1578, 1495, 1344, 1295, 1128, 1074, 998, 767, 752, 682; δH (200 MHz; CDCl3) 5.37 (2 H, s, CH2Ph), 7.26–7.38 (3 H, complex overlapping m), 7.46–7.52 (2 H, complex m), 7.77–7.90 (2 H, complex overlapping m), 7.95–8.00 (1 H, complex m), 8.42–8.47 (1 H, complex m); δC (50 MHz; CDCl3) 55.0 (CH2Ph), 125.8 (CH), 127.7 (CH + C), 128.1 (CH), 128.7 (2× CH), 128.9 (2× CH), 132.7 (CH), 133.8 (CH), 136.4 (2× C), 137.8 (C), 158.9 (C); m/z (EI) 270 (35Cl; 19%, M+), 235 (44%, M+ − Cl), 91 (38%, C7H7+); (found: C, 66.50; H, 4.09; N, 10.14. C15H11ClN2O requires C, 66.55; H, 4.03; N, 10.35%).2-Benzyl-4-(3-methylbut-1-ynyl)phthalazin-1(2H)-one (33)
A sealable, heavy-walled flask (∼100 mL capacity) was charged with 32 (3.07 g, 11.3 mmol), Bu4NI (4.39 g, 11.9 mmol), CuI (141 mg, 740 µmol), Pd(PPh3)4 (524 mg, 453 µmol), anhydrous THF (50 mL), 3-methyl-1-butyne (2.74 g, 40.2 mmol) and iPr2NH (3.00 mL, 21.4 mmol). The flask was sealed and the mixture stirred magnetically in an oil bath thermostatted at 80 °C for 21 h [SAFETY SCREEN]. The flask was then cooled and opened; the mixture was evaporated to afford a residue that was dissolved in CH2Cl2 (30 mL). The resulting solution was washed with water (2 × 20 mL) followed by brine (2 × 20 mL), dried (Na2SO4) and evaporated. The residue was subjected to flash column chromatography (gradient elution from light petroleum to 1 : 9 EtOAc–light petroleum) to afford the title compound (3.12 g; 91%) as a colourless powder: mp 82–83 °C (from EtOAc–light petroleum); νmax(KBr)/cm−1 3090, 2972, 2873, 2230, 1661, 1607, 1582, 1454, 1348, 1301, 1112, 778, 701; δH (200 MHz; CDCl3) 1.36 (6 H, d, J 6.9, CH(CH3)2), 2.92 (1 H, sept, J 6.9, CH(CH3)2), 5.41 (2 H, s, CH2Ph), 7.25–7.36 (3 H, complex overlapping m), 7.43–7.50 (2 H, complex m), 7.70–7.86 (2 H, overlapping m), 7.99–8.04 (1 H, complex m), 8.37–8.42 (1 H, complex m, CH); δC (50 MHz; CDCl3) 21.4 (CH(CH3)2), 22.7 (CH(CH3)2), 55.5 (CH2Ph), 73.6 (CCCH), 101.5 (CCCH), 126.4 (CH), 127.0 (CH), 127.6 (C), 127.8 (CH), 128.6 (2× Ph CH), 128.7 (2× Ph CH), 130.4 (C), 131.8 (CH), 132.7 (C), 133.4 (CH), 136.8 (C), 159.0 (C); m/z (EI) 302 (100%, M+), 287 (4%, M+ − CH3), 91 (40%, C7H7+), 77 (9%, C6H5+); (found: M+, 302.1451. C20H18N2O requires 302.1419); (found: C, 79.05; H, 5.89; N, 9.19. C20H18N2O requires C, 79.44; H, 6.00; N, 9.26%).
2-Benzyl-4-(2-isopropylpyrazolo[1,5-a]pyridin-3-yl)phthalizin-1(2H)-one (34)
A stirred mixture of 1-aminopyridinium mesitylenesulfonate12 (498 mg, 1.69 mmol), 33 (503 mg, 1.66 mmol) and powdered K2CO3 (457 mg, 3.31 mmol) in DMF (16 mL) was heated at 100 °C for 20 h. Additional pyridinium salt (515 mg, 1.75 mmol) and K2CO3 (470 mg, 3.40 mmol) were then added and heating continued for a further 20 h. The mixture was then evaporated to dryness and the resulting residue partitioned between EtOAc (30 mL) and water (20 mL). The organic phase was separated, washed with water (20 mL) followed by brine (2 × 20 mL), dried (Na2SO4) and evaporated. The residue was subjected to flash column chromatography (gradient elution from light petroleum to 1 : 9 Et2O–light petroleum), affording the starting alkyne (194 mg; 39%) followed by the title compound (262 mg; 40%) as a colourless powder: mp 198–199 °C (from MeOH); νmax(KBr)/cm−1 3040, 2958, 1651, 1581, 1532, 1350, 1302, 1256, 1158, 992, 766, 702; δH (200 MHz; CDCl3) 1.24 (3 H, d, J 6.9, CH(CH3)), 1.28 (3 H, d, J 6.9, CH(CH3)), 3.11 (1 H, sept, CH(CH3)2), 5.36 (1 H, AB d, Jgem 13.8, CHHPh), 5.62 (1 H, AB d, Jgem 13.9, CHHPh), 6.77 (1 H, ddd, J6′,7′ 7.0, J6′,5′ 5.9, J6′,4′ 2.2, H-6′), 7.03–7.17 (2 H, complex overlapping m), 7.23–7.38 (3 H, complex overlapping m), 7.45–7.53 (3 H, complex overlapping m), 7.70 (1 H, td, J 7.2 and 1.6, H-6 or H-7), 7.78 (1 H, td, J 7.2 and 1.4, H-6 or H-7), 8.49 (1 H, dt, J7′,6′ 7.0, J7′,5′ 1.0, J7′,4′ 1.0, H-7′), 8.53–8.58 (1 H, complex m, H-8); δC (50 MHz; CDCl3) 22.5 (CH(CH3)), 23.0 (CH(CH3)), 27.1 (CH(CH3)2), 54.9 (CH2Ph), 102.8 (C-3′), 111.9 (CH-6′), 116.8 (CH-4′), 124.5 (CH), 126.7 (CH), 127.5 (CH), 127.8 (CH), 128.5 (C), 128.6 (2× Ph CH), 128.8 (CH), 128.9 (2× Ph CH), 130.4 (C), 131.7 (CH), 133.1 (CH), 137.2 (C), 140.1 (C), 140.8 (C), 159.2 (C), 160.8 (C); m/z (EI) 394 (100%, M+), 91 (56%, C7H7+); (found: M+, 394.1793. C25H22N4O requires 394.1778); (found: C, 75.78; H, 5.52; N, 14.18. C25H22N4O requires C, 76.12; H, 5.62; N, 14.20%).2-(3-Methylbut-1-ynyl)pyridine (35)
A sealable, heavy-walled flask (∼100 mL capacity) was charged with CuI (1.08 g, 5.67 mmol), Pd(PPh3)4 (227 mg, 196 µmol), anhydrous THF (50 mL), 2-bromopyridine (1.20 mL, 12.6 mmol), 3-methyl-1-butyne (1.08 g, 15.9 mmol) and iPr2NH (3.00 mL, 21.4 mmol). The flask was sealed and the mixture stirred magnetically in an oil bath thermostatted at 70 °C for 40 h [SAFETY SCREEN]. The flask was then cooled and opened; the mixture was evaporated to afford a residue that was partitioned between CH2Cl2 (75 mL) and water (75 mL). The organic phase was separated and washed with water (2 × 75 mL) followed by brine (2 × 20 mL), dried (MgSO4) and evaporated. The residue was subjected to flash column chromatography (15 : 40 : 45 CH2Cl2–Et2O–pentane) to afford the title compound (750 mg; 41%) as a pale yellow oil: νmax(neat)/cm−1 3053, 2972, 2933, 2235, 1584, 1464, 1428, 1322, 1149, 991, 780; δH (200 MHz; CDCl3) 1.25 (6 H, d, J 6.9, CH(CH3)2), 2.77 (1 H, sept, J 6.9, CH(CH3)2), 7.13 (1 H, ddd, J5,4 7.6, J5,6 4.9, J5,3 1.2, H-5), 7.33 (1 H, dt, J3,4 7.8, J3,5 1.1, J3,6 1.1, H-3), 7.56 (1 H, td, J4,3 7.7, J4,5 7.7, J4,6 1.8, H-4), 8.50 (1 H, ddd, J6,5 4.9, J6,4 1.7, J6.3 0.9, H-6); δC (50 MHz; CDCl3) 21.0 (CH(CH3)2), 22.7 (CH(CH3)2), 79.6 (CCCH), 96.1 (CCCH), 122.3 (CH-5), 126.8 (CH-3), 136.1 (CH-4), 143.9 (C-2), 149.8 (CH-6); m/z (ESI) 146 (96%, M+ + H), 130 (21%, M+ − CH3), 118 (93%, M+ − CHN), 78 (70%, C5H4N+); (found: M+, 145.0903. C10H11N requires 145.0892).
2-Isopropyl-3-(pyridin-2-yl)pyrazolo[1,5-a]pyridine (36)
A stirred mixture of 1-aminopyridinium mesitylenesulfonate12 (596 mg, 2.02 mmol), 35 (158 mg, 1.09 mmol) and powdered K2CO3 (570 mg, 4.12 mmol) in DMF (12 mL) was heated at 80 °C for 40 h. The mixture was then evaporated to dryness and the residue partitioned between EtOAc (20 mL) and water (20 mL). The organic phase was separated, washed with brine (20 mL), dried (MgSO4) and evaporated. The resulting residue was subjected to flash column chromatography (gradient elution from light petroleum to 7 : 3 EtOAc–light petroleum) to recover starting alkyne (83.0 mg; 53%) and afford the title compound (24.6 mg; 10%) as a pale yellow powder: mp 82–83 °C (from CHCl3–hexane), νmax(thin film)/cm−1 3019, 2972, 1635, 1590, 1533, 1216, 756, 669; δH (400 MHz; CDCl3) 1.42 (6 H, d, J 6.9, CH(CH3)2), 3.63 (1 H, sept, J 6.9, CH(CH3)2), 6.75 (1 H, td, J6,5 6.9, J6,7 6.9, J6,4 1.4, H-6), 7.13–7.17 (2 H, overlapping m, H-5 and H-5′), 7.48 (1 H, br d, J3′,4′ 8.0, H-3′), 7.74 (1 H, td, J4′,3′ 7.8, J4′,5′ 7.8, J4′,6′ 1.9, H-4′), 7.91 (1 H, dt, J4,5 9.0, J4,6 1.2, J4,6 1.7, H-4), 8.44 (1 H, dt, J7,6 7.0, J7,4 1.1, J7,5 1.1, H-7), 8.71 (1 H, br d, J6′,5′ 4.8, H-6′); δC (101 MHz; CDCl3) 22.9 (CH(CH3)2), 26.7 (CH(CH3)2), 109.2 (C-3), 112.0 (CH-6), 118.1 (CH-4), 120.5 (CH-5′), 123.0 (CH-3′), 124.6 (CH-5), 128.6 (CH-7), 136.4 (CH-4′), 139.6 (C-3a), 149.9 (CH-6′), 153.8 (C-2′), 159.5 (C-2); m/z (EI) 237 (91%, M+), 236 (100%, M+ − H), 222 (74%, M+ − CH3), 194 (26%, M+ − C3H7), 78 (16%, C5H4N+); (found: M+, 237.1252. C15H15N3 requires 237.1266); (found: C, 75.97; H, 6.34; N, 17.65. C15H15N3 requires C, 75.92; H, 6.37; N, 17.71%).‡,§§1-Methyl-2-(3-methylbut-1-ynyl)pyridinium iodide (37)
A solution of methyl iodide (90.0 µL, 1.45 mmol) and 35 (200 mg, 1.38 mmol) in dry THF (2 mL) was heated at 60 °C for 1.5 h. Upon cooling and standing the mixture separated into two phases. The upper phase was decanted off and the lower phase pumped (0.1 mm Hg) to afford the title salt (283 mg; 72%) as a brown oil: νmax(neat)/cm−1 3041, 2974, 2223, 1618, 1509, 1457, 1270, 1180, 754; δH (200 MHz; CDCl3) 1.21 (6 H, d, J 6.9, CH(CH3)2), 2.87 (1 H, sept, J 6.9, CH(CH3)2), 4.42 (3 H, s, NCH3), 7.89–8.00 (2 H, overlapping m, H-3 and H-5), 8.52 (1 H, br td, J4,3 8.0, J4,5 8.0, J4,6 0.8, H-4), 9.42 (1 H, br d, J6,5 5.7, H-6); δC (50 MHz; CDCl3) 21.6 (CH(CH3)2), 21.7 (CH(CH3)2), 47.9 (NCH3), 71.4 (CCCH), 117.0 (CCCH), 126.6 (CH), 131.5 (CH), 138.0 (C-2), 145.3 (CH-4), 147.2 (CH-6); m/z (+ve ion ESI) 160 (100%, C11H14N+), 117 (22%, C11H14N+ − C3H7); m/z (−ve ion ESI) 127 (100%, I−); (found: organic cation, 160.1120. C11H14N+ requires 160.1121).§§
2-(2-Isopropylpyrazolo[1,5-a]pyridin-3-yl)-1-methylpyridinium iodide (38)
A stirred mixture of 1-aminopyridinium mesitylenesulfonate12 (581 mg, 1.97 mmol), 37 (283 mg, 986 µmol) and powdered K2CO3 (555 mg, 4.01 mmol) in DMF (5 mL) was heated at 95 °C for 18 h. The mixture was then evaporated to dryness and the residue directly subjected to flash column chromatography (gradient elution from CH2Cl2 to 1 : 9 MeOH–CH2Cl2). Relevant fractions were combined, evaporated and the resulting residue triturated with 2 : 1 CH2Cl2–Et2O to afford the title salt (146 mg; 39%) as a buff coloured powder: νmax(thin film)/cm−1 3019, 2972, 1632, 1530, 1216, 756, 754; δH (400 MHz; CDCl3) 1.23 (3 H, d, J 6.9, CHCH3), 1.30 (3 H, d, J 6.9, CHCH3), 2.97 (1 H, sept, J 6.9, CH(CH3)2), 4.30 (3 H, s, NMe), 6.94 (1 H, td, J6′,5′ 6.9, J6′,7′ 6.9, J6′,4′ 1.3, H-6′), 7.36 (1 H, ddd, J5′,4′ 8.9, J5′,6′ 6.9, J5′,7′ 1.1, H-5′), 7.71 (1 H, dt, J4′,5′ 8.9, J4′,6′ 1.1, H-4′), 7.87 (1 H, dd, J3,4 7.9, J3,5 1.4, H-3), 8.14 (1 H, ddd, J5,4 7.7, J5,6 6.3, J5,3 1.4, H-5), 8.48 (1 H, d6, J7′6,′ 7.0, J7′,4′ 1.0, J7′,5′ 1.0, H-7′), 8.55 (1 H, td, J4,3 7.9, J4,5 7.9, J4,6 1.2, H-4), 9.62 (1 H, br d, J6,5 6.2, H-6); δC (101 MHz; CDCl3) 22.6 (CHCH3), 22.9 (CHCH3), 27.0 (CH(CH3)2), 47.2 (NMe), 98.9 (C-3′), 113.7 (CH-6′), 117.0 (CH-4′), 126.9 (CH-5), 127.6 (CH-5′), 129.2 (CH-7′), 131.5 (CH-3), 139.3 (C-3a′), 145.2 (CH-4), 148.5 (CH-6), 149.3 (C-2), 159.9 (C-2′); m/z (+ve ion ESI) 252 (100%, C16H18N+); m/z (−ve ion ESI) 127 (100%, I−); (found: organic cation, 252.1494. C16H18N+ requires 252.1495).§§Acknowledgements
We thank the EPSRC Mass Spectrometry Centre at Swansea for mass spectra.Notes and references
- W. Y. Chai, J. G. Breitenbucher, A. Kwok, X. B. Li, V. Wong, N. I. Carruthers, T. W. Lovenberg, C. Mazur, S. J. Wilson, F. U. Axe and T. K. Jones, Bioorg. Med. Chem. Lett., 2003, 13, 1767–1770 CrossRef CAS.
- P. Gmeiner, J. Sommer, G. Höfner and J. Mierau, Arch. Pharm. (Weinheim, Ger.), 1992, 325, 649–655 CrossRef CAS; P. Gmeiner, J. Sommer, J. Mierau and G. Höfner, Bioorg. Med. Chem. Lett., 1993, 3, 1477–1483 CrossRef CAS; S. Löber, H. Hübner, W. Utz and P. Gmeiner, J. Med. Chem., 2001, 44, 2691–2694 CrossRef CAS; L. Bettinetti, K. Schlotter, H. Hübner and P. Gmeiner, J. Med. Chem., 2002, 45, 4594–4597 CrossRef CAS; S. Löber, T. Aboul-Fadl, H. Hübner and P. Gmeiner, Bioorg. Med. Chem. Lett., 2002, 12, 633–636 CrossRef CAS; S. Löber, B. Ortner, L. Bettinetti, H. Hübner and P. Gmeiner, Tetrahedron: Asymmetry, 2002, 13, 2303–2310 CrossRef CAS; F. Boeckler, H. Russig, W. N. Zhang, S. Löber, J. Schetz, H. Hübner, B. Ferger, P. Gmeiner and J. Feldon, Psychopharmacology (Berlin, Ger.), 2004, 175, 7–17 Search PubMed; L. Bettinetti, H. Hübner and P. Gmeiner, Arch. Pharm. (Weinheim, Ger.), 2005, 338, 276–280 CrossRef CAS; O. Prante, C. Hocke, S. Löber, H. Hübner, P. Gmeiner and T. Kuwert, Nuklearmedizin, 2006, 45, 41–48 Search PubMed.
- P. Beswick, S. Bingham, C. Bountra, T. Brown, K. Browning, I. Campbell, I. Chessell, N. Clayton, S. Collins, J. Corfield, S. Guntrip, C. Haslam, P. Lambeth, F. Lucas, N. Mathews, G. Murkit, A. Naylor, N. Pegg, E. Pickup, H. Player, H. Price, A. Stevens, S. Stratton and J. Wiseman, Bioorg. Med. Chem. Lett., 2004, 14, 5445–5448 CrossRef CAS; S. Bingham, P. J. Beswick, C. Bountra, T. Brown, I. B. Campbell, I. P. Chessell, N. Clayton, S. D. Collins, P. T. Davey, H. Goodland, N. Gray, C. Haslam, J. P. Hatcher, A. J. Hunter, F. Lucas, G. Murkitt, A. Naylor, E. Pickup, B. Sargent, S. G. Summerfield, A. Stevens, S. C. Stratton and J. Wiseman, J. Pharmacol. Exp. Ther., 2005, 312, 1161–1169 CAS.
- J. B. Hansen, J. Weis, P. D. Suzdak and K. Eskesen, Bioorg. Med. Chem. Lett., 1994, 4, 695–698 CrossRef.
- H. Shinoda, M. Inaba and T. Tsuruo, Cancer Res., 1989, 49, 1722–1726 CAS; H. Shinoda, H. Ebisu, J. Mitsuhashi, M. Inaba and T. Tsuruo, Cancer Chemother. Pharmacol., 1992, 30, 335–340 CrossRef CAS; Y. Kobayashi, T. Yamashiro, H. Nagatake, T. Yamamoto, N. Watanabe, H. Tanaka, K. Shigenobu and T. Tsuruo, Biochem. Pharmacol., 1994, 48, 1641–1646 CrossRef CAS; H. Tanaka, T. Sekine, M. Terada, Y. Kobayashi and K. Shigenobu, Naunyn–Schmiedeberg’s Arch. Pharmacol., 1997, 356, 853–855 CrossRef CAS; T. Yamashiro, N. Watanabe and Y. Kobayashi, Biochem. Pharmacol., 1997, 54, 143–148 CrossRef CAS; H. Tanaka and K. Shigenobu, Cardiovasc. Drug Rev., 2000, 18, 93–102 Search PubMed.
- K. L. Stevens, D. K. Jung, M. J. Alberti, J. G. Badiang, G. E. Peckham, J. M. Veal, M. Cheung, P. A. Harris, S. D. Chamberlain and M. R. Peel, Org. Lett., 2005, 7, 4753–4756 CrossRef CAS.
- K. Awano and S. Suzue, Chem. Pharm. Bull., 1986, 34, 2833–2839 CAS; K. Awano, S. Suzue and M. Segawa, Chem. Pharm. Bull., 1986, 34, 2828–2832 CAS; L. C. D. Gibson, S. F. Hastings, I. McPhee, R. A. Clayton, C. E. Darroch, A. Mackenzie, F. L. MacKenzie, M. Nagasawa, P. A. Stevens and S. J. MacKenzie, Eur. J. Pharmacol., 2006, 538, 39–42 CrossRef CAS; K. Nishino, H. Ohkubo, M. Ohashi, S. Hara, J. Kito and T. Irikura, Jpn. J. Pharmacol., 1983, 33, 267–278 Search PubMed; S. Ohmuro, M. Komuro, K. Momo, H. Ohkubo and K. Nishino, Cardiovasc. Drug Rev., 1993, 11, 466–478 Search PubMed; S. Ohmuro, T. Sasahara, F. Taga, H. Ohkubo and H. Uchida, Arzneim. Forsch., 1992, 42–1, 43–47; J. E. Souness, M. E. Villamil, L. C. Scott, A. Tomkinson, M. A. Giembycz and D. Raeburn, Br. J. Pharmacol., 1994, 111, 1081–1088 CAS.
- B. A. Johns, K. S. Gudmundsson, E. M. Turner, S. H. Allen, D. K. Jung, C. J. Sexton, F. L. Boyd and M. R. Peel, Tetrahedron, 2003, 59, 9001–9011 CrossRef CAS; K. S. Gudmundsson, B. A. Johns, Z. C. Wang, E. M. Turner, S. H. Allen, G. A. Freeman, F. L. Boyd, C. J. Sexton, D. W. Selleseth, K. R. Moniri and K. L. Creech, Bioorg. Med. Chem., 2005, 13, 5346–5361 CrossRef CAS; B. A. Johns, K. S. Gudmundsson, E. M. Turner, S. H. Allen, V. A. Samano, J. A. Ray, G. A. Freeman, F. L. Boyd, C. J. Sexton, D. W. Selleseth, K. L. Creech and K. R. Moniri, Bioorg. Med. Chem., 2005, 13, 2397–2411 CrossRef CAS; S. H. Allen, B. A. Johns, K. S. Gudmundsson, G. A. Freeman, F. L. Boyd, C. H. Sexton, D. W. Selleseth, K. L. Creech and K. R. Moniri, Bioorg. Med. Chem., 2006, 14, 944–954 CrossRef CAS.
- A. Akahane, H. Katayama, T. Mitsunaga, Y. Kita, T. Kusunoki, T. Terai, K. Yoshida and Y. Shiokawa, Bioorg. Med. Chem. Lett., 1996, 6, 2059–2062 CrossRef CAS; T. Maemoto, K. Finlayson, H. J. Olverman, A. Akahane, R. W. Horton and S. P. Butcher, Br. J. Pharmacol., 1997, 122, 1202–1208 CrossRef CAS; D. Shi, W. L. Padgett, K. D. Hutchinson, S. P. Moore and J. W. Daly, Drug Dev. Res., 1997, 42, 41–56 CrossRef CAS; A. Akahane, H. Katayama, T. Mitsunaga, T. Kato, T. Kinoshita, Y. Kita, T. Kusunoki, T. Terai, K. Yoshida and Y. Shiokawa, J. Med. Chem., 1999, 42, 779–783 CrossRef CAS; H. Ito, T. Maemoto, A. Akahane, S. P. Butcher, H. J. Olverman and K. Finlayson, Eur. J. Pharmacol., 1999, 365, 309–315 CrossRef CAS; S. Kuroda, A. Akahane, H. Itani, S. Nishimura, K. Durkin, T. Kinoshita, I. Nakanishi and K. Sakane, Tetrahedron, 1999, 55, 10351–10364 CrossRef CAS; S. Kuroda, A. Akahane, H. Itani, S. Nishimura, K. Durkin, T. Kinoshita, Y. Tenda and K. Sakane, Bioorg. Med. Chem. Lett., 1999, 9, 1979–1984 CrossRef CAS; S. Kuroda, A. Akahane, H. Itani, S. Nishimura, K. Durkin, Y. Tenda and K. Sakane, Bioorg. Med. Chem., 2000, 8, 55–64 CrossRef CAS; S. Kuroda, F. Takamura, Y. Tenda, H. Itani, Y. Tomishima, A. Akahane and K. Sakane, Chem. Pharm. Bull., 2001, 49, 988–998 CrossRef CAS; T. Maemoto, M. Tada, T. Mihara, N. Ueyama, H. Matsuoka, K. Harada, T. Yamaji, K. Shirakawa, S. Kuroda, A. Akahane, A. Iwashita, N. Matsuoka and S. Mutoh, J. Pharmacol. Sci., 2004, 96, 42–52 CrossRef CAS.
- A. Zanka, N. Hashimoto, R. Uematsu and T. Okamoto, Org. Process Res. Dev., 1998, 2, 320–324 Search PubMed.
- Y. Kohno, T. Ogata, K. Awano, K. Matsuzawa and T. Tooru, PCT Int. Appl., WO9814448, 1998 Search PubMed; N. Yoshida, M. Aono, T. Tsubuki, K. Awano and T. Kobayashi, Tetrahedron: Asymmetry, 2003, 14, 529–535 Search PubMed; N. Yoshida, K. Awano, T. Kobayashi and K. Fujimori, Synthesis, 2004, 1554–1556 CrossRef CAS.
- V. Boekelheide and N. A. Fedoruk, J. Org. Chem., 1968, 33, 2062–2064 CrossRef CAS; R. Grashey and M. Baumann, Tetrahedron Lett., 1969, 10, 2173–2175 CrossRef; S. Suzue, M. Hirobe and T. Okamoto, Chem. Pharm. Bull., 1973, 21, 2146–2160 CAS; K. T. Potts, H. P. Youzwak and S. J. Zurawel, J. Org. Chem., 1980, 45, 90–97 CrossRef CAS; P. L. Anderson, J. P. Hasak, A. D. Kahle, N. A. Paolella and M. J. Shapiro, J. Heterocycl. Chem., 1981, 18, 1149–1155 CrossRef CAS; Y. Miki, J. Tasaka, K. Uemura, K. Miyazeki and J. Yamada, Heterocycles, 1996, 43, 2249–2256 CrossRef CAS; Z. Maqbool, M. Hasan, K. T. Potts, A. Malik, T. A. Nizami and W. Voelter, Z. Naturforsch., B: Chem. Sci., 1997, 52, 1393–1400 CAS; M. Bencková and A. Krutošíková, Collect. Czech. Chem. Commun., 1999, 64, 539–547 CrossRef CAS.
- Y. Tamura, M. Ikeda, J. Minamika, Y. Miki and S. Matsugas, Tetrahedron Lett., 1972, 13, 4133–4135 CrossRef; Y. Tamura, J. Minamikawa, K. Sumoto, S. Fujii and M. Ikeda, J. Org. Chem., 1973, 38, 1239–1241 CrossRef CAS; Y. Tamura, J. Minamikawa and M. Ikeda, Synthesis, 1977, 1–17 CrossRef CAS.
- H. Feuer and H. Rubinstein, J. Org. Chem., 1959, 24, 811–813 CrossRef CAS; F. Yoneda and Y. Nitta, Chem. Pharm. Bull., 1963, 11, 269–271 CAS; W. J. Coates and A. McKillop, Heterocycles, 1993, 35, 1313–1329 CrossRef CAS; A. Katrusiak and A. Katrusiak, J. Mol. Struct., 2003, 647, 203–210 CrossRef CAS.
- F. Yoneda and Y. Nitta, Chem. Pharm. Bull., 1963, 11, 737–740.
- W. H. Pirkle and P. L. Gravel, J. Org. Chem., 1977, 42, 1367–1370 CrossRef CAS.
- J. Druey, A. Hüni, K. Meier, B. H. Ringier and A. Staehelin, Helv. Chim. Acta, 1954, 37, 510–523 CrossRef CAS.
- R. Wegler, J. Prakt. Chem., 1937, 148, 135–160 CrossRef CAS; N. P. Buu-Hoï, H. Le Bihan and F. Binon, Recl. Trav. Chim. Pays-Bas, 1951, 70, 1099–1103 CAS.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Procedures for preparation of N-aminopyridinium mesitylenesulfonate and compounds 23, 24, 26, 27 and 31; crystallographic data for compounds 17, 18 and 36. See DOI: 10.1039/b713638b |
| ‡ CCDC reference numbers 659630–659632. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b713638b |
| § Deceased. |
| ¶ Current address: School of Chemistry, University of Manchester, Oxford Road, Manchester, UK, M13 9PL. |
| || D. R. Adams, R. W. Allcock, P. D. Bailey, I. D. Collier, Z. Jiang and K. M. Morgan, unpublished results. |
| ** Detailed PDE inhibitory activities for our 6-(pyrazolo[1,5-a]pyridin-3-yl)pyridazin-3-one series will be presented elsewhere. |
| †† Some unreacted dipolarophile was also recovered from the reactions of the two N-benzyl diazinones (22 and 33), though not from the reactions of the N-phenyl pyridazinone (28) or the chloropyridazine (20). Alkyne recovery was respectively 43% and 39% from the reactions of 22 and 33, which were conducted with two equivalents of the N-aminopyridinium mesitylenesulfonate. Attempts to improve the yield of isolated product in these reactions with prolonged reaction times were ineffective. Use of larger excesses of N-aminopyridinium salt in the reaction complicated product isolation by generating quantities of tarry residue. |
| ‡‡ Partial charges calculated using standard semi-empirical AM1 methods within CAChe software. |
| §§ NMR assignments for compounds 14, 16, 18, 25, 29 and 36 were supported by the acquisition of NOESY, HSQC and HMBC spectra; assignments for compounds 20 and 21 were supported by the acquisition of HSQC and HMBC spectra; assignments for salt 38 were supported by the acquisition of COSY, NOESY, HSQC and HMBC spectra; 13C NMR assignments for compound 22 were supported by the acquisition of an HSQC spectrum. 1H NMR spectra of salts 37 and 38 are shown in the accompanying ESI file.† |
| This journal is © The Royal Society of Chemistry 2008 |