Reaction of 1-tert-butyl-3,4-diphenyl-1,2,4-triazol-5-ylidenes with a malonic ester †
Nikolai I.
Korotkikh
*a,
Alan H.
Cowley
b,
Jennifer A.
Moore
b,
Nataliya V.
Glinyanaya
a,
Ilia S.
Panov
a,
Gennady F.
Rayenko
a,
Tatyana M.
Pekhtereva
a and
Oles P.
Shvaika
a
aThe L. M. Litvinenko Institute of Physical Organic and Coal Chemistry, Ukrainian Academy of Sciences, 70, R. Luxemburg, Donetsk, 83114, Ukraine. E-mail: nkorotkikh@ua.fm; Fax: +380 (62)3116830; Tel: +380 (62)3116835
bThe University of Texas at Austin, Department of Chemistry & Biochemistry, 1 University Station A5300, Austin, Texas 78712-0165, USA. E-mail: acowley@cm.utexas.edu; Fax: +1 (512) 471-6822; Tel: +1 (512) 471-7484
First published on 23rd November 2007
Abstract
Four stable carbenes , 1-tert-butyl-3,4-diaryl-1,2,4-triazol-5-ylidenes 1a–d, including new fluorine-containing compounds 1c,d, react with a malonic ester to afford heterocyclic zwitterionic compounds 5a–d. The reactions with more acidic compounds (ethyl acetoacetate, malononitrile and 1,3-dimethylbarbituric acid) proceed with substrate deprotonation to form the respective azolium salts 6a–c. The X-ray crystal structure of 5a was also determined.
Introduction
The reactions of in situ-generated carbenes with C–H acidic compounds are well known. For example, Pazdro and Polaczkova1 showed that dithiol-2-ylidene dimers will insert into the C–H bonds of malononitrile, acetylacetone, ethyl acetoacetate and cyclopentanone to afford the corresponding dihydrodithiol derivatives. However, it is not clear whether such reactions really proceed via the intermediacy of a free carbene . Similar transformations with acetonitrile, dimethyl sulfone and acetylene (pKa = 20–25) have been studied with stable carbenes 2–5 and resulted in the isolation of the respective C–H insertion products, i.e. cyanomethylazolines.To the best of our knowledge,6 the reactions of isolable carbenes with an ester functional group have not been studied thus far. However, it is known that in situ-generated 1,3-diphenylcyclopropene-2-ylidene reacts with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of dimethyl fumarate to form a spirocyclic adduct.7 Likewise, the stable (phosphanyl)(silyl)carbenes react with the double bond of dimethyl fumarate and other electron-poor alkenes to give the corresponding trans-cyclopropanes.8 In the case of the reaction of 1,3,4-triphenyl-1,2,4-triazol-5-ylidene, the initially formed [2 + 1] cycloaddition product undergoes ring opening and a 1,2-hydrogen shift to afford methylenetriazoline derivatives.9
C bond of dimethyl fumarate to form a spirocyclic adduct.7 Likewise, the stable (phosphanyl)(silyl)carbenes react with the double bond of dimethyl fumarate and other electron-poor alkenes to give the corresponding trans-cyclopropanes.8 In the case of the reaction of 1,3,4-triphenyl-1,2,4-triazol-5-ylidene, the initially formed [2 + 1] cycloaddition product undergoes ring opening and a 1,2-hydrogen shift to afford methylenetriazoline derivatives.9
In the present contribution we describe (i) the synthesis of new stable fluorine-containing carbenes of the 1,2,4-triazole series, namely 1-tert-butyl-3-aryl-4-fluoroaryl-1,2,4-triazol-5-ylidenes; (ii) the first Claisen reaction of stable carbenes with an ester functional group of malonic esters to form new heterocyclic zwitterionic compounds; and (iii) the deprotonations of 1,3-dimethylbarbituric acid, malononitrile and ethyl acetoacetate by the carbene .
Results and discussion
The stable carbenes 1a–d, including the new stable fluorine-containing carbenes 1c,d, were synthesized by the ring transformations of 2-phenyl-1,3,4-oxadiazole2a,b with anilines in the presence of trifluoroacetic acid according to the literature method,10 followed by quaternization of the resulting triazoles 3a–d with t-BuI to form the triazolium salts 4a–d (Scheme 1). Deprotonation of the latter salts with potassium tert-butoxide in tetrahydrofuran solution afforded the desired carbenes 1a–d. It is noteworthy that the quaternization of triazoles 3a–d takes place exclusively at the 1-position, hence only one isomer of salt 4a–d is formed. The generation of tert-butyl iodide was carried out in situ by treatment of tert-butyl chloride with sodium iodide in acetic acid solution.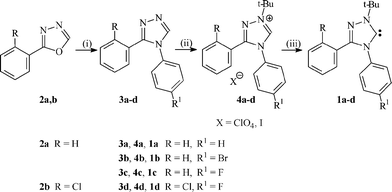 | ||
| Scheme 1 Reagents and conditions: (i) p-H2N-C6H4-R1 (− H2O); (ii) 1. t-BuI; 2. NaClO4; (iii) t-BuOK (− t-BuOH). | ||
A two-stage isolation of carbenes 1a–d by solvent evaporation in the presence of an inorganic salt permitted complete decomposition of the intermediate triazolium alkoxides. In the method described earlier,5 the carbenes were isolated by filtration of the inorganic salt, concentration of the filtrate and recrystallisation of the crystalline residues in order to remove the remaining azolium alkoxide impurities.
The reactions of carbenes 1a–d with diethyl malonate (pKa = 13) were carried out in refluxing toluene for 2–4 h under conditions such that evolved ethanol was removed by a controlled stream of nitrogen. If this procedure is not followed, the reaction times increase to ∼40 h and the transformations are incomplete. Following work-up of the reaction mixtures, the zwitterionic compounds 5a–d were obtained in isolated yields of 51–81%. The outcomes of the reactions can be rationalized on the basis of initial nucleophilic attack of the carbene on the ester functional group to form intermediate 5A (Scheme 2), followed by loss of EtOH to produce the zwitterionic compounds 5a–d. It is noteworthy that C–H insertion products are not observed in these processes. Moreover, this new transformation can be considered to be a carbene analogue of the Claisen reaction of esters .
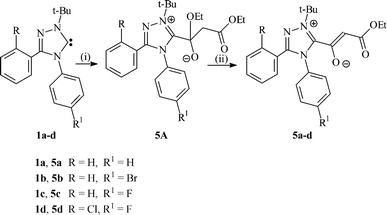 | ||
| Scheme 2 Reagents and conditions: (i) CH2(COOEt)2, Δ; (ii) Δ (− EtOH). | ||
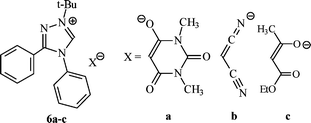
The reaction of 1a with 1,3-dimethylbarbituric acid (pKa = 4.68) results in protonation of the substrate and affords salt 6a. Compound 6a is a stable compound that does not undergo further reaction. Less acidic malononitrile (pKa = 9) and ethyl acetoacetate (pKa = 11) also undergo reaction with carbene 1b to afford salts 6b and 6c, respectively. Compound 6b was isolated as a toluene solvate. Carbon–hydrogen insertion was not observed in any of these reactions.
Compounds 1 and 3–6 are colorless crystalline solids that were characterized by elemental analysis, 1H, 13C NMR, IR and mass spectroscopy. The molecular structure of compound 5a was established by single-crystal X-ray diffraction.
The 1H NMR spectra of salts 4c,d feature the signals for the tert-butyl group (δ 1.72–1.76 ppm), aromatic protons (δ 7.3–7.7 ppm) and the C5-H proton of the triazolium nucleus (δ 10.65–10.73 ppm). The 1H NMR spectra of carbenes 1c,d exhibit similar signals for the tert-butyl (δ 1.78–1.79 ppm) and aromatic protons (δ 6.5–7.3 ppm). However, the chemical shift for the latter is notably upfield relative to that of the salt. The 13C NMR spectra of 1c,d exhibit resonances for the tert-butyl carbon atoms (δ 30.2–30.4 and ipso-C 59.2–59.4 ppm), aromatic nuclei (δ 115.0–164.0 ppm), C3 (δ 148.7–150.7 ppm) and the C5 atoms (δ 206.6–208.6 ppm) of the triazole nucleus. The 1H NMR spectra of zwitterionic compounds 5a–d are characterized by the presence of resonances for the tert-butyl protons (δ 1.80–1.87 ppm), aromatic protons (δ 7.0–7.5 ppm), the ethyl group of an ester fragment (δ 1.12–1.16 and 3.91–3.96 ppm), and the CHC group of an aliphatic fragment (δ 4.78–4.84 ppm). The 13C NMR spectrum (see Fig. 1 for the atom numbering scheme) is distinguished by the presence of resonances for the tert-butyl carbon atoms (δ 28.6–28.8 and 65.8–66.1 ppm), the carbon atoms of the ethyl group (δ 14.5–14.7 and 58.0–58.1 ppm) and benzene nuclei (δ 115.8–134.0 ppm), the triazole atoms C1 and C2 (δ 149.0–150.5; 153.9–154.6 ppm), C21 of the aliphatic fragment (δ 90.3–90.6 ppm), and two signals for the carbonyl group (δ 163.7–164.4 and 170.9–171.2 ppm). The former signal was assigned to the C19O group, and the latter to the C21O moiety. The IR spectrum of 5a shows the absence of typical carbonyl stretching vibrations. The detection of a vibration at 1653 cm−1 is consistent with the proposed delocalized structure for 5a. The mass spectra of compounds 5a,c show molecular ions [MH]+ corresponding to the monomers (m/z 392.5 for 5a; 410.0 for 5c). The molecular ions of compounds 5b–d undergo further decomposition by elimination of isobutene and malonyl fragments (see the Experimental section).
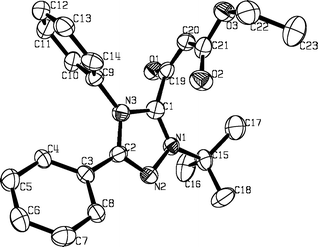
Crystals of compound 5a suitable for X-ray diffraction study were grown from diethyl ether solution. The X-ray data† confirm the ionic character of the triazole ring. Significant structural features are the somewhat elongated bonds of both CO groups (123.6 and 126.6 pm), a shortened C19–C20 bond (137.3 pm) and, to a lesser extent, a shortened C20–C21 aliphatic linkage (141.9 pm) (see Fig. 1 and Fig. 2). The C1–C19 bond distance of 152.6 pm corresponds to that of a typical C–C single bond, and the C19O1 carbonyl group is twisted with respect to the plane of the triazolium nucleus. The benzene rings attached to C2 and N3 subtend dihedral angles of 26.9 and 71.1°, respectively, in relation to the plane of the triazolium ring. All the foregoing structural data are consistent with the proposed zwitterionic structure for 5a.
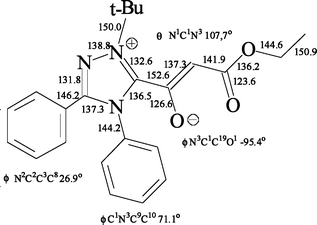 | ||
| Fig. 2 Selected bond lengths (pm) and angles (°) for compound 5a. | ||
The 1H NMR spectra of the triazolium salts 6a–c include signals that can be assigned to the protons of both the cation and the anion. Thus, the singlets at δ 9.70–10.90 ppm can be assigned to the C5-H proton of the cation, and the singlet at δ 3.50–4.05 ppm is attributable to the C2-H proton of the anion. Collectively, these data confirm the ionic structure for 6a–c. The spectra for salts 6b,c exhibit broad C5-H signals due to facile proton exchange.
Conclusion
In summary, we have demonstrated that the stable heteroaromatic carbenes 1-tert-butyl-3,4-diaryl-1,2,4-triazol-5-ylidenes 1a–d react with the ester functional group of diethyl malonate to produce the heterocyclic zwitterionic derivatives 5a–d. These reactions represent the first example of the carbene version of the Claisen reaction of esters . No evidence was found for C–H insertion reactions. It is known that less acidic C–H compounds either undergo insertion reactions with stable carbenes (for example, acetonitrile, ketones , sulfones , and acetylenes ) or do not react (for example, dimethyl sulfoxide, alkylarenes, and saturated hydrocarbons ). The reactions of 1a with the appreciably more acidic C–H compounds, 1,3-dimethylbarbituric acid, malononitrile and ethyl acetoacetate, produce the salts 6a–c via the protonation of the carbene .Experimental
General methods
All experiments with the 1,2,4-triazol-5-ylidenes 1a–d were carried out under an argon atmosphere. All solvents were dried by standard methods prior to use. 1H and 13C NMR chemical shifts are reported relative to tetramethylsilane (TMS, δ = 0.00) as internal standard. Mass spectra were taken on an Agilent 1100 Series chromatomass spectrometer (APCI, 3 kV, chromatography : a column-Zorbax SB-C18, eluent acetonitrile–water 95 : 5 with 0.1% formic acid). IR spectra were measured as Nujol mulls, and thin-layer chromatography was performed on silica gel with chloroform or a 10 : 1 mixture of chloroform and methanol as eluent, followed by development with iodine. Elemental analyses were carried out at the Analytical Laboratory of the Litvinenko Institute of Physical Organic and Coal Chemistry.General procedure for the synthesis of 1-tert-butyl-3,4-diphenyl-1,2,4-triazol-5-ylidenes 1a–d
Potassium tert-butoxide (280 mg, 2.55 mmol) was added to a dispersion of salt 4a–d (2.65 mmol) in anhydrous tetrahydrofuran (10 mL) and stirred at room temperature for 0.5 h. The solvent was evaporated and the product was extracted with a further portion of tetrahydrofuran (10 mL). The latter solution was re-evaporated and the residue was stirred with petroleum ether (5 mL). The resulting solid was filtered off and dried to afford carbenes 1a–d. The new fluorine-containing carbenes 1c,d are characterized as detailed below.Procedure for the synthesis of 3a–d
These were obtained by the ring transformations of the respective oxadiazoles 2a,b with anilines in the presence of trifluoroacetic acid at 180 °C according to the literature method.10,11 Isolation of the new compounds 3c,d was carried out by washing the unpurified products with diethyl ether and then, if necessary, by recrystallization from the indicated solvent.General procedure for the preparation of 1-tert-butyl-3,4-diaryl-1,2,4-triazolium perchlorates (4a–d)
A mixture of sodium iodide (20.2 g, 0.135 mol), tert-butyl chloride (15 mL, 0.135 mol) and the triazole3a–d (0.05 mol) in acetic acid (20 mL) was refluxed until the reaction was complete as monitored by TLC (typically 20 h). The reaction mixture was diluted with 0.5 L of water, heated until boiling, following which a small amount of sodium sulfite along with 1 g of activated carbon was added, and the mixture was filtered. A solution of sodium perchlorate (8.58 g, 0.07 mol) in water (20 mL) was then added and the resulting precipitate was filtered off and dried to give 67–85% of salts 4a–d. The new compounds 4c,d are characterized as indicated below.1-tert-Butyl-3,4-diphenyl-1,2,4-triazolium 5-(2-carbethoxyvinyl-1-oxide) (5a)
Diethyl malonate (0.125 mL, 0.78 mmol) was added to a solution of triazolylidene 1a (100 mg, 0.361 mmol) in anhydrous toluene (2 mL) and the reaction mixture was refluxed for 4 h under conditions such that the evolved ethanol was removed by a stream of dry nitrogen gas. The toluene was evaporated and the solid residue 5a (100 mg, 71%) was washed with petroleum ether, filtered off and recrystallized from n-octane to afford 57.0 mg (40%) of the pure product 5a. Mp 160–162 °C. Found: C 70.7, H 6.6, N 11.0. Calcd for C23H25N3O3: C 70.6, H 6.4, N 10.7%. δH (200 MHz, CDCl3, Me4Si) 1.14 (3H, t, J 7.1 Hz, CH3CH2C), 1.80 (s, 9H, CH3C, t-Bu), 3.93 (2H, quart, J 7.1 Hz, CH3CH2C), 4.81 (1H, s, C5-H–N), 7.42 (10H, m, Ar). δC (50.3 MHz, CDCl3, Me4Si) 14.7 (CH3CH2C), 28.8 (CH3C, t-Bu), 58.0 (CH3CH2C), 65.8 (CH3C, t-Bu), 90.5 (C2–CO), 123.6 (C1, Ar-C), 127.2, 128.8 (enhanced int.), 129.5, 130.8, 131.4 (Ar), 132.1 (C1, ArN), 150.5 (C5), 154.6 (C3), 164.4 (C1O), 171.1 (C3O). m/z (APCI): 392.5 (MH+), C23H26N3O3 requires 392.5. CCDC reference number 607811. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b712885a
1-tert-Butyl-3-phenyl-4-p-bromophenyl-1,2,4-triazolium 5-(2-carbethoxyvinyl-1-oxide) (5b)
Obtained according to the method described above for compound 5a. Yield 50%. Mp 54–56 °C (precipitation from ether by petroleum ether). Found: C 58.8, H 5.0, Br 17.2; N 9.1. Calcd for C23H24BrN3O3: C 58.7, H 5.1, Br 17.0; N 8.9%. δH (200 MHz, CDCl3, Me4Si). 1.15 (3H, t, J 7.1 Hz, CH3CH2), 1.87 (9H, s, CH3C, t-Bu), 3.94 (2H, quart, J 7.1 Hz, CH3CH2C), 4.82 (1H, s, C5-H–N), 7.52 (9H, m, Ar). δC (50.3 MHz, CDCl3, Me4Si) 14.6 (CH3CH2), 28.7 (CH3C, t-Bu), 58.1 (CH3CH2), 65.9 (CH3C, t-Bu), 90.6 (C2–CO), 123.2 (C1, Ar-C), 125.1 (C–Br), 131.5 (C1, ArN), 127.7, 128.7, 130.9, 132.7, 133.0 (Ar), 150.3 (C5), 154.4 (C3), 164.0 (C1O), 171.1 (C3O). m/z (APCI): 356.9 (M+ − Me2CCH2 + H+), 299.9 (3b + H+ or M+ − Me2CCH2 + H+ − COCH2COOEt). C23H24BrN3O3 requires 470.4.
1-tert-Butyl-3-phenyl-4-p-fluorophenyl-1,2,4-triazolium 5-(2-carbethoxyvinyl-1-oxide) (5c)
Obtained according to the method described above for compound 5a. Yield 73%. Mp 143–145 °C (from octane). Found: C 67.8, H 6.0, F 4.7; N 10.3. Calcd for C23H24FN3O3: C 67.5, H 5.9, F 4.6; N 10.3%. δH (200 MHz, CDCl3, Me4Si) 1.12 (3H, t, J 7.1 Hz, CH3CH2), 1.84 (9H, s, CH3C, t-Bu), 3.91 (quart, 2H, J 7.1 Hz, CH3CH2C, Et), 4.78 (1H, s, C5-H–N), 7.11 (2H, dd, 3J 8.0 Hz, 3JF 9.1 Hz, C3-H, ArN), 7.38 (7H, m, Ar). δC (50.3 MHz, CDCl3, Me4Si) 14.6 (CH3CH2, Et), 28.7 (CH3C, t-Bu), 58.0 (CH3CH2), 65.8 (CH3C, t-Bu), 90.3 (C5), 116.4, 116.8 (C3, ArN, J 18.0 Hz), 123.3 (C1, Ar-C), 127.5 (C1, ArN), 129.1 (C2, ArN, J 36.4 Hz), 128.6, 128.7, 131.4 (Ar), 150.4 (C5), 154.6 (C3), 160.8, 165.8 (C–F, J 252.4 Hz), 164.2 (C1O), 171.2 (C3O). m/z (APCI): (MH+) 409.5, C23H26N3O3 requires 410.0; 354.2 (M+ − Me2CCH2), 296.3 (M+ − COCH2COOEt), 240.2 (3c + H+ or M+ − COCH2COOEt − Me2CCH2).
1-tert-Butyl-3-o-chlorophenyl-4-p-fluorophenyl-1,2,4-triazolium 5-(2-carbethoxyvinyl-1-oxide) (5d)
Obtained according to the method described above for compound 5a. Yield 75%. Mp 50–51 °C (precipitation from ether by petroleum ether). Found: C 62.3, H 5.3, Cl 8.1, F 4.3, N 9.4. Calcd for C23H23ClFN3O3: C 62.2, H 5.2, Cl 8.0, F 4.3; N 9.5%. δH (200 MHz, CDCl3, Me4Si) 1.16 (3H, t, J 7.1 Hz, CH3CH2), 1.87 (9H, s, CH3C, t-Bu), 3.96 (2H, quart, J 7.1 Hz, CH3CH2), 4.84 (1H, s, C5-H–N), 7.00 (2H, dd, 3J 8.0 Hz, 3JF 9.1 Hz, C3-H, ArN), 7.39 (m, 6H, Ar). δC (50.3 MHz, CDCl3, Me4Si) 14.5 (CH3CH2), 28.6 (CH3C, t-Bu), 58.0 (CH3CH2), 66.1 (CH3C, t-Bu), 90.6 (C2–CO), 115.8, 116.2 (C3, ArN, J 23.3 Hz), 122.9 (C1, Ar-C), 127.1 (C1, ArN, J 12.6 Hz), 128.8 (C2, ArN, J 36.4 Hz), 126.9, 130.0, 132.3, 132.8 (Ar), 134.0 (C–C1), 149.0 (C5), 153.9 (C3), 160.6, 165.6 (C–F, J 254.5 Hz), 163.7 (C1O), 170.9 (C3O). m/z (APCI): 388.1 (M+ − Me2CCH2), 330.2 (M+ − COCH2COOEt), 274.1 (3d or M+ − COCH2COOEt − Me2CCH2), C23H23ClFN3O3 requires 443.9 (M+).
1-tert-Butyl-3,4-diphenyl-1,2,4-triazolium 1,3-dimethylbarbiturate (6a)
A mixture of 1,3-dimethylbarbituric acid (110 mg, 0.721 mmol) and triazolylidene 1 (200 mg, 0.72 mmol) in anhydrous toluene (3 mL) was stirred for 1.5 h. The volume of the reaction mixture was reduced by 50% and the resulting product was recrystallized from a mixture of toluene and acetonitrile (10 : 1). The solid product was filtered off and washed with petroleum ether to give 250 mg (75%) of salt 6a. Mp 172–174 °C (from 10 : 1 toluene–acetonitrile). Found: C 66.7, H 6.5, N 16.2. Calcd for C24H27N5O3: C 66.5, H 6.3, N 16.2%. δH (200 MHz, CD3CN, Me4Si) 1.77 (9H, s, CH3C), 2.99 (6H, s, CH3N), 4.05 (4H, s, CHC), 7.48 (10H, m, Ar), 10.47 (1H, s, CHN).1-tert-Butyl-3,4-diphenyl-1,2,4-triazolium dicyanomethanide (6b)
Obtained by the same procedure as that described for salt 6a from malononitrile (173 mg, 2.70 mmol) and triazolylidene 1a (500 mg, 1.80 mmol) in toluene (2 mL). Yield 384 mg (63%). Mp 147–149 °C (from 1 : 1 toluene–acetonitrile). Found: C 73.7, H 6.3, N 20.3. Calcd for C21H21N5: C 73.4, H 6.2, N 20.4%. δH (200 MHz, CD3CN, Me4Si) 1.76 (s, 9H, CH3C, t-Bu), 3.50 (s, 1H, CH), 7.4–7.6 (m, 10H, Ar), 9.70 (broad s, 1H, C5-H).1-tert-Butyl-3,4-diphenyl-1,2,4-triazolium ethyl acetoacetate (6c)
Obtained by the same procedure as that described for salt 6a from ethyl acetoacetate (351 mg, 2.70 mmol) and triazolylidene 1a (500 mg, 1.80 mmol) in toluene (2 mL). Yield 420 mg (67%). Mp 131–133 °C (from toluene). Found: C 70.7, H 7.1, N 10.2. Calcd for C24H29N3O3: C 70.7, H 7.2, N 10.3%. δH (200 MHz, DMSO-d6, Me4Si) 1.,02 (3H, s, CH3CH2), 1.76 (9H, s, CH3C, t-Bu), 1.89 (3H, s, CH3CO), 3.74 (2H, m, CH2CH3), 4.00 (1H, broad s, CH), 7.2–7.8 (10H, m, Ar), 10.90 (1H, broad s, C5-H).Acknowledgements
We thank the Ukrainian Academy of Sciences for financial support and the Robert A. Welch Foundation (F-0003) for financial support of the X-ray diffraction studies. We also express gratitude to Dr A. A. Tolmachev (Enamine Ltd, Kiev) for the chromatomass spectrometric data, Dr Chotiy (IPOCC, Donetsk) for recording the IR spectra, and Dr M. Findlater for assistance with the X-ray work.References
- K. M. Pazdro and W. Polaczkowa, Rocz. Chem., 1971, 45, 1487–1494 Search PubMed.
- M. I. Korotkikh, G. F. Rayenko, T. M. Pekhtereva and O. P. Shvaika, Rep. Ukr. Acad. Sci., 1998, 6, 149–152 Search PubMed.
- M. I. Korotkikh, G. F. Rayenko, T. M. Pekhtereva and O. P. Shvaika, Rep. Ukr. Acad. Sci., 2000, 2, 135–140 Search PubMed.
- A. J. Arduengo, III, J. C. Calabrese, F. Davidson, H. V. R. Dias, J. R. Goerlich, R. Krafczyk, W. J. Marshall, M. Tamm and R. Schmutzler, Helv. Chim. Acta, 1999, 82, 2348–2364 CrossRef CAS.
- N. I. Korotkikh, G. F. Rayenko, O. P. Shvaika, T. M. Pekhtereva, A. H. Cowley, J. N. Jones and C. L. B. Macdonald, J. Org. Chem., 2003, 68(14), 5762–5766 CrossRef CAS.
- For reviews, see: (a) W. A. Herrmann and K. Kocher, Angew. Chem., Int. Ed. Engl., 1997, 36, 2162–2187 CrossRef CAS; (b) D. Bourissou, O. Guerret, P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100(1), 39–91 CrossRef; (c) Y. Cheng and O. Meth-Cohn, Chem. Rev., 2004, 104, 2507–2530 CrossRef CAS.
- W. M. Jones, M. E. Stowe, S. E. E. Well and E. W. Lester, J. Am. Chem. Soc., 1968, 90(7), 1849–1859 CrossRef CAS.
- (a) A. Igau, A. Baceiredo, G. Trinquier and G. Bertrand, Angew. Chem., Int. Ed. Engl., 1989, 28, 621–622 CrossRef; (b) S. Goumri-Magnet, T. Kato, H. Gornitzka, A. Baceiredo and G. Bertrand, J. Am. Chem. Soc., 2000, 122, 4464–4470 CrossRef CAS.
- D. Enders, K. Breuer, J. Runsink and J. H. Teles, Liebigs Ann. Chem., 1996, 2019–2028 CAS.
- N. I. Korotkikh, A. V. Kiselyov, A. V. Knishevitsky, G. F. Rayenko, T. M. Pekhtereva and O. P. Shvaika, Chem. Heterocycl. Compd., 2005, 7, 1026–1032.
- N. I. Korotkikh, N. V. Glynyanaya, A. H. Cowley, J. A. Moore, A. V. Knishevitsky, T. M. Pekhtereva, O. P. Shvaika, ARKIVOC, 2007, in press Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal structure data for compound 5a. See DOI: 10.1039/b712885a |
| This journal is © The Royal Society of Chemistry 2008 |