RNA-selective cross-pairing of backbone-extended pyrrolidine-amide oligonucleotide mimics (bePOMs)†
Received
20th September 2007
, Accepted 8th October 2007
First published on 1st November 2007
Abstract
Pyrrolidine-amide oligonucleotide mimics (POMs) can cross-pair strongly with complementary parallel and antiparallel DNA and RNA targets in a sequence-specific fashion. As a result POMs have significant potential for applications including in vivogene silencing, diagnostics and bioanalysis. To further modulate the DNA- and RNA-recognition properties and fine-tune the physiochemical properties of POMs for nucleic acid targeting, backbone-extended pyrrolidine-amide oligonucleotide mimics (bePOM I and II) were introduced. The bePOMs differ from the original POMs through the insertion of an additional methylene group into the backbone units, which increases the flexibility of the oligomers. bePOM I and II oligomers were synthesised using solid-phase peptide chemistry. Interestingly, UV thermal denaturation and circular dichroism studies reveals bePOM I and II can hybridise with complementary RNA , but not DNA.
Introduction
Nucleic acid mimics that can selectively hybridise with complementary DNA and RNA can be used to down regulate gene expression in vivo, which is particularly valuable for functional genomics.1 In addition, nucleic acid mimics have been employed as bioanalytical tools or diagnostic agents,2 as well as building blocks in the programmed assembly of nanostructures.3 Moreover, a study of the properties of nucleic acid mimics can also provide a valuable alternative insight into the structure, recognition properties, function and origins of the natural genetic material.4
Previously we introduced the pyrrolidine-amide oligonucleotide mimics (POMs) 3 (Fig. 1). Notably it was shown that fully modified short POM homopolymers5 and longer mixed sequences6 are capable of cross-pairing with both complementary DNA and RNA , exhibiting UV transition melting temperatures (Tm) that on the whole are higher than isosequential peptide nucleic acids (PNAs).7 Interestingly, mixed sequence POMs (e.g.Lys–TCACAACTT–NH2)6 cross-pair strongly with parallel and antiparallel DNA as well as RNA , but with rates of association/dissociation that are noticeably slower than those typically observed with short oligonucleotides or PNA. One possible reason for this could be the rigidity of the POM backbone compared to other more flexible mimics such as PNA. Indeed, nucleic acid mimics with more rigid backbone structure could favour formation of stable secondary structures in the single-stranded state, which are not optimal for hybridisation.8 As a consequence, the conformational reorganisation of the backbone into a structure that enables base pairing to take place may be slow and rate-limiting. In light of this it was decided to investigate the effects of increasing the flexibility of the POM backbone, by introducing an additional methylene group into the backbone. This leads to the backbone extended POMs (bePOM I (1) and II (2), Fig. 1), which both possess repeating 7-atom linkages and differ only in the relative position of the amide linkage. Previous studies have established that it is not necessary to match the six-atom linkage of the backbone of native nucleic acids in order to retain base pairing. In fact, modified nucleic acids with units containing five-9 and seven-atom10 linkages are also capable of cross-pairing with complementary DNA and RNA . Accordingly, oligomers of bePOM I (1) and II (2) were synthesised, using solid-phase peptide chemistry and their hybridisation properties explored using UV thermal denaturation experiments and CD spectroscopy.
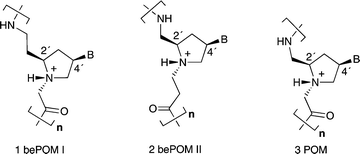 |
| | Fig. 1 Pyrrolidine-amide oligonucleotide mimics (POM) and backbone-extended POMs (bePOM I and II). The protonated pyrrolidine N1′-substituent prefers the less sterically demanding trans-configuration and POMs are thus stereochemically equivalent to natural nucleic acids .5,6 | |
Results and discussion
Synthesis of backbone-extended POM monomers
It was envisaged that the synthesis of bePOM oligomers would be accomplished using Boc-Z solid-phase peptide synthesis protocols, similar to those developed previously for the original fully modified mixed sequence POM.6 In order to test this the bePOM I thymine monomer was first prepared from the ethyl ester hydrochloride salt 4 (Scheme 1).11 Protection of the pyrrolidine nitrogen of 4 with a benzyloxycarbonyl group gave 5 in 91% yield and subsequent tert-butyldimethylsilyl (TBDMS)-protection of the secondary alcohol provided pyrrolidine 6 in 85% yield. This allowed reduction of the ethyl ester of 6 with LiBH4 resulting in the primary alcohol 7 in 70% yield, which was then transformed to the mesylate 8. The mesylate was not isolated but treated with sodium cyanide to give nitrile 9 in a yield of 68% over the two steps. Reduction of nitrile 9 was then achieved with NaBH4 in the presence of CoCl2·6H20.12 The resulting primary amine 10 was then Boc-protected using di-tert-butyldicarbonate to give Boc-amine 11 in 62% overall yield. Deprotection of the pyrrolidine nitrogen by hydrogenation over 10% Pd–C afforded amine 12, which was alkylated with methyl bromoacetate to afford methyl ester 13 in 65% yield. Following TBDMS-deprotection with tetrabutylammoniumfluoride (TBAF), it was necessary to invert the stereochemistry of the C4 alcohol of 14 in order to obtain the desired (2S,4R) configuration of the bePOM I monomer. Accordingly (4R)-alcohol 14 was transformed to the (4S)-formyl ester 15 under Mitsunobu conditions13 in 70% yield. Cleavage of the formyl ester with sodium methoxide in anhydrous methanol gave the (4S)-alcohol 16 in 90% yield. Thymine was then introduced onto the pyrrolidine ring as N3-benzoylthimine14 to ensure the desired N1-alkylation is obtained. This was achieved using another Mitsunobu reaction to form the N1-thyminyl derivative 17 in a yield of 67%. Treatment with aqueous sodium hydroxide in THF, followed by neutralisation provided the bePOM I thyminyl acid 18 in 71% yield.
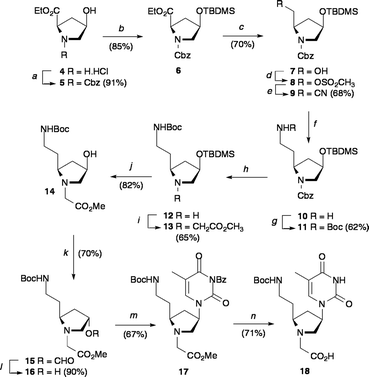 |
| | Scheme 1 Synthesis of bePOM I thyminyl monomer: (a) benzylchloroformate, Et3N, 1 : 1 water–1,4-dioxane, 50 °C for 2 h then rt for 18 h; (b) TBDMS–Cl, imidazole, DIEA, DMF, rt, 18 h; (c) LiBH4, THF, 0 °C → rt, 18 h; (d) MsCl, DIEA, CH2Cl2, 0 °C → rt, 3 h; (e) NaCN, DMF, 75 °C, 20 h; (f) NaBH4, CoCl2·6H2O, CH3OH, rt, 4 h; (g) Boc anhydride, Et3N, 1 : 1 water–1,4-dioxane, rt, 18 h; (h) 10% Pd–C, CH3OH, H2, rt, 18 h; (i) BrCH2CO2CH3, DIEA, CH2Cl2, 0 °C → rt, 18 h; (j) TBAF, THF, rt, 4 h; (k) HCO2H, PPh3, DIAD, THF,–20 °C → rt, 18 h; (l) NaOCH3, CH3OH, rt, 5 h; (m) N3-benzoylthymine, PPh3, DIAD, THF,–20 °C → rt, 18 h; (n) 1 M NaOH (aq), THF, rt for 18 h, then 0.1 M HCl (aq). | |
The synthesis of the bePOM II thymine monomer is achieved via a conjugate addition between the reported5d amine HCl salt 19 and methyl acrylate, which gave methyl ester 20 in 85% yield (Scheme 2). Azide-reduction with trimethylphosphine under Staudinger conditions, and in situ Boc-protection of the resulting amine with 2-(tert-butoxycarbonyloxyimino)-2-phenylacetonitrile (Boc-ON)15 afforded the Boc-amine 21 in 89% yield. The C4-OH group of the pyrrolidine ring was again inverted via the (4S)-formyl ester 22, which was formed in 65% yield then cleaved with sodium methoxide to give (4S)-alcohol 23 in 82% yield. Introduction of N3-benzoylthimine under Mitsunobu conditions, similarly gave the thyminyl derivative 24 in 64% yield and saponification and neutralisation as before provided the bePOM II thyminyl acid 25 in 69% yield.
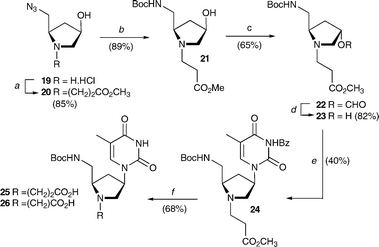 |
| | Scheme 2 Synthesis of bePOM II thyminyl monomer: (a) methyl acrylateDIEA, CH2Cl2, 0 °C for 30 min then rt for 18 h; (b) PMe3, THF, rt, 1.5 h, then 2-(tert-butoxycarbonyloxyimino)-2-phenylacetonitrile (Boc-ON), −20 °C, 15 min, then rt for 1 h; (c) HCO2H, PPh3, DIAD, THF, −20 °C → rt, 18 h; (d) NaOCH3, CH3OH, rt, 2 h; (e) N3-benzoylthymine, PPh3, DIAD, THF, 0 °C → rt, 18 h; (f) 1M NaOH (aq), THF, rt for 3 h, then 0.1 M HCl (aq). | |
Boc-Z solid-phase synthesis of POM, bePOM I and bePOM II Lys–(T)8–NH2 oligomers
POM Lys–(T)8–NH227, bePOM I Lys–(T)8–NH228 and bePOM II Lys–(T)8–NH229 were synthesised following the Boc-Z POM synthetic protocol.6,16 The octamers were prepared on methylbenzhydrylamine (MBHA LL)-functionalised resin, adjusted at the first coupling to give a loading of 0.12 mmol.g−1. Unreacted amino groups were capped with acetic anhydride. In the case of POM Lys–(T)8–NH227 subsequent couplings employed four equivalents of Boc-protected POM thyminyl acid 26,6 preactivated with 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethylguanidinium hexafluorophosphate (HBTU) (3.8 equiv.) and diisopropylethylamine (DIEA) (4.4 equiv.). Coupling reactions proceeded for 2 h and were monitored by the Kaiser test.17 Capping of any unreacted oligomer was carried out by reaction with acetic anhydride again in the presence of DMF and collidine. Boc-deprotection with trifluoroacetic acid (TFA) and m-cresol as a scavenger was followed by repeated coupling, capping and deprotection steps. In the case of bePOM I Lys–(T)8–NH228 and bePOM II Lys–(T)8–NH229 five equivalents of thyminyl acid 18 or 25 were preactivated with HBTU (4.75 equiv.) and DIEA (5.5 equiv.) prior to the second and subsequent coupling reactions. Cleavage of the oligomers from the resin was carried out by the “low–high” TFMSA method16 and the crude oligomers were analysed by analytical C18 HPLC and MALDI-TOF mass spectrometry (Fig. 2). POM Lys–(T)8–NH227 was synthesised in a yield of 52%, as determined by analytical C18 HPLC, this corresponds to an average coupling efficiency of 93.0%. The yield for the synthesis of bePOM I Lys–(T)8–NH228 was 86%, equating to an average coupling efficiency of 98.3%. The yield for the synthesis of bePOM II Lys–(T)8–NH229 was 60%, equating to an average coupling efficiency of 94.5%. Oligomers were purified by semi-preparative C18 HPLC and the purity of oligomers was estimated to be higher than 95% based on analytical C18 HPLC.
![(A) MALDI-MS of crude POM Lys–(T8)–NH227 showing m/z 2260.1 ([M + H]+ 100%, C111H154N51O19 requires m/z, 2260.2); (B) MALDI-MS of crude POM bePOM I Lys–(T8)–NH228 showing m/z 2372.1 ([M + H]+ 100%, C119H160N51O19 requires m/z, 2372.2); (C) MALDI-MS of crude POM bePOM I Lys–(T8)–NH229 showing m/z 2372.2 ([M + H]+ 100%, C119H160N51O19 requires m/z, 2372.2).](/image/article/2008/OB/b714580m/b714580m-f2.gif) |
| | Fig. 2 (A) MALDI-MS of crude POM Lys–(T8)–NH227 showing m/z 2260.1 ([M + H]+ 100%, C111H154N51O19 requires m/z, 2260.2); (B) MALDI-MS of crude POM bePOM I Lys–(T8)–NH228 showing m/z 2372.1 ([M + H]+ 100%, C119H160N51O19 requires m/z, 2372.2); (C) MALDI-MS of crude POM bePOM I Lys–(T8)–NH229 showing m/z 2372.2 ([M + H]+ 100%, C119H160N51O19 requires m/z, 2372.2). | |
Nucleic acid -binding properties of bePOM I and II: UV thermal denaturation and renaturation experiments
The POM and bePOM oligomers were next subjected to UV thermal denaturation/renaturation experiments. In line with earlier observations,5 the prototype POM Lys–(T)8–NH227 hybridises strongly with both RNA and DNA, exhibiting transition melting temperatures (Tm heating) of 41.5 and 36.4 °C with r(CGCA8CGC) and d(CGCA8CGC), respectively, under close to physiological conditions (Table 1). With longer homopolymers poly(rA) and poly(dA), higher Tm were observed, which are accompanied by more pronounced hysteresis, indicative of slow rates of association/dissociation. Interestingly the extent of this hysteresis, is similar for both poly(rA) and poly(dA), suggesting that the kinetic selectivity for RNA over DNA observed previously with thyminyl POM pentamers,5 is not evident with the octameric POM 27.
Table 1 UV thermal denaturation/renaturation transition temperatures (Tm) for POM 27, bePOM I 28 and bePOM II 29vs. complementary nucleic acids
|
T
m/°Ca (hyperchromicb and hypochromicc shifts (%)) |
|
|
| POM Lys–(T)8–NH227 |
bePOM I Lys–(T)8–NH228 |
bePOM II Lys–(T)8–NH229 |
| Heating |
Cooling |
Heating |
Cooling |
Heating |
Cooling |
|
Experiments were carried out with 84 µM total conc. in bases, in a 1 : 1 ratio of strands for d(CGCA8CGC) and r(CGCA8CGC) and 1 : 1 ratio of bases for poly(rA) and poly(dA), and 10 mM K2HPO4, 0.12 M K+, pH 7.0 (total volume 1.0 cm3). UV absorbance (A260) was recorded with heating at 5 °C min−1 from 23 to 93 °C, cooling at 0.2 °C min−1 to 15 °C and heating at 0.2 °C min−1 to 93 °C. The Tm was determined from the 1st derivative of the slow heating and cooling curve.
Hyperchromic and are indicated in parentheses and were calculated as follows: [Abs 93 °C − Abs 15 °C] × 100/Abs 93 °C.
Hypochromic shifts are indicated in parentheses and were calculated as follows: [Abs 93 °C − Abs 15 °C] × 100/Abs 93 °C.
Samples were incubated with nucleic acid for 24 h before being subjected to slow thermal denaturation (0.2 °C min−1).
n.t. = no transition evident. Note that no transitions are evident in control experiments where POMs are subjected to UV thermal denaturation in the absence of complementary nucleic acid targets.
|
| r(CGCA8CGC) |
41.5a (12.6)b |
37.9a (12.2)c |
36.7a (9.1)b |
32.5a (7.7)c |
31.2 and 48.8a (13.5)b |
38.6a (7.7)b |
| |
43.8d (13.4) |
|
34.8d (13.4) |
|
n.t.de (16.2) |
|
| d(CGCA8CGC) |
36.4a (12.3) |
35.6a (10.6) |
n.t.e (4.0) |
n.t.e (1.0) |
n.t.e (5.4) |
n.t.e (3.0) |
| |
41.0d (20.5) |
|
n.t.de (12.5) |
|
n.t.de (12.7) |
|
| Poly(rA) |
52.4a (22.0) |
38.2a (21.5) |
n.t.e (17.1) |
n.t. (14.5) |
44.4a (9.2)b |
33.3a (8.8)b |
| |
54.0d (24.7) |
|
n.t.de (17.6) |
|
48.2d (13.6) |
|
| Poly(dA) |
53.4a (10.0) |
41.4a (12.1) |
n.t.e (12.7) |
n.t. (11.0) |
n.t.e (9.4) |
n.t.e (8.3) |
| |
53.4d (15.8) |
|
n.t.de (12.9) |
|
n.t.de (17.7) |
|
The oligomer with the type-I extended-backbone bePOM I Lys–(T)8–NH228 hybridises with r(CGCA8CGC) with a Tm (heating) of 36.7 °C (Fig. 3) and exhibits slight hysteresis with Tm (cooling) of 32.5 °C. Incubation at room temperature for 24 h prior to denaturation has little effect on Tm or hyperchromic shift (Table 1). Under identical conditions bePOM I Lys–(T)8–NH228 shows no evidence of cooperative melting transitions with d(CGCA8CGC), poly(dA) or poly(rA) and there is no significant hyperchromic shifts. For the type-II backbone-extended POM (29) apparent hybridisation with r(CGCA8CGC) is observed (Fig. 3), with two distinct transitions in the denaturation curve, at 31.2 °C and 48.8 °C, possibly indicative of transitions from triplex to duplex to single strands. However attempts to define stoichiometry of binding through Job plots were inconclusive. It was previously noted that the prototype POM can hybridise with DNA and RNA in a parallel or antiparallel fashion.6 Therefore the presence of a mixture of parallel and antiparallel complexes of differing thermodynamic stability, may also account for the observed double transition. Clearly this issue is best resolved using mixed-sequence oligomers, with defined orientations and modes of hybridisation. Cross-pairing between bePOM II 29 and poly(rA) was also apparent. In this case, notable hysteresis was observed with Tm of 44.4 and 33.3 °C (ΔTm = 11.1 °C) for the denaturation and renaturation curves, respectively. On the other hand, bePOM II 29 shows no evidence of thermal denaturation/renaturation d(CGCA8CGC) or poly(dA) even after the complementary strands are incubated at room temperature for a prolonged period of time. These experiments indicate that both the type-I and type-II backbone-extended POMs (28 and 29) can hybridise with complementary RNA , but not DNA. This apparent cross-pairing selectivity of bePOM I and II for RNA over DNA was further investigated using circular dichroism (CD) spectroscopy.
 |
| | Fig. 3 UV thermal denaturation curves and first derivatives for POM Lys–(T8)–NH227, bePOM I Lys–(T8)–NH228 and bePOM II Lys–(T8)–NH229vs. r(CGCA8CGC) at 7.6 µM (total conc. in strands, 1 : 1 ratio of strands) and 10 mM K2HPO4, 0.12 M K+, pH 7.0 (total volume 1.0 cm3): (A) slow heating (denaturation) curves for POM 27 (i), bePOM I 28 (ii) and bePOM II 39 (iii) vs. r(CGCA8CGC); (B) the corresponding first derivatives for POM 27 (i), bePOM I 28 (ii) and bePOM II 39 (iii) vs. r(CGCA8CGC). | |
Circular dichroism experiments
Initially the CD spectra of POM Lys–(T)8–NH227, bePOM I Lys–(T)8–NH228 and bePOM II Lys–(T)8–NH229 single strands were recorded (Fig. 4). In the case of prototype, POM Lys–(T)8–NH227 shows a strong negative bands at 215 and 285 nm and a strong positive band at 260 nm. Interestingly, the sign of the bands for the single stranded POM 27 are opposite to those typically observed in the CDs of short RNA and DNA strands (see ESI† ).18 This might suggest that POM 27 is preorganised into a base-stacked conformation with a left-handed helical sense that is opposite to that typically observed with right-handed helical DNA and RNA . The bePOM I and II oligomers (28 and 29) have CD spectra that exhibit bands of lower intensity than the original POM 27. This is indicative of bePOM I and II possessing less structurally ordered single strands than the more rigid POM 27, which is presumably due to the extra methylene group increasing the intrinsic flexibility of the backbone. Also notable is the fact that bePOM II 29 possesses bands, which are opposite in sign to those observed with POM 27, suggesting that the single strands possess opposite helical sense.
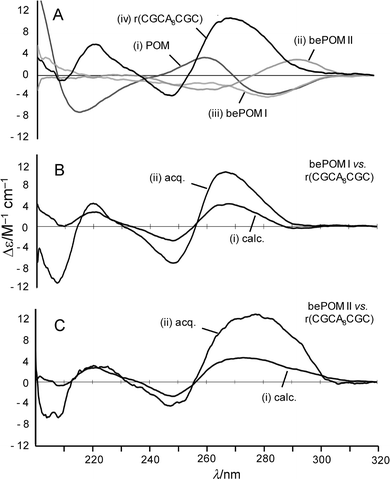 |
| | Fig. 4 CD spectra of POM 27, bePOM I 28 and bePOM II 29vs. r(CGCA8CGC) at 7.6 µM (total conc. in strands, 1 : 1 ratio of strands) and 10 mM K2HPO4, 0.12 M K+, pH 7.0 (total volume 1.0 cm3): (A) single strands (i) POM 27, (ii) bePOM I 28, (iii) bePOM II 29, (iv) r(CGCA8CGC); (B) bePOM I 28vs. r(CGCA8CGC), (i) calculated, (ii) acquired; (C) bePOM II 29vs. r(CGCA8CGC), (i) calculated, (ii) acquired. | |
The CD spectra for the equimolar complexes of oligomers (27, 28 and 29) with r(CGCA8CGC) and d(CGCA8CGC) were next compared against the calculated CD spectra resulting from the average of the CD spectra obtained for the corresponding separate single strands. For POM 27 with RNA and DNA (CGCA8CGC) significant difference in both the wavelength and intensity of the CD bands is observed between the calculated and observed spectra (Fig. 5). Notably, the overall CD spectra of the complex between POM 27 and r(CGCA8CGC) closely resembles that of typical A-type RNA helices.18 This suggests that the RNA strand has greater influence over the final conformation of the POM–RNA complex. In the case of bePOM I and II (28 and 29) with r(CGCA8CGC) a noticeable increase in band intensity is evident for the observed CD compared with the calculated CD, and again the CD spectra closely resemble those observed for A-type helical RNA (Fig. 4). In contrast, there are essentially no differences between the observed and calculated CDs of equimolar mixtures of bePOM I and II (28 and 29) with d(CGCA8CGC) (see ESI† ). This fully supports the earlier observations, showing that whilst bePOM I and II can cross-pair with RNA , no hybridisation is evident with isosequential DNA. Of course the interpretation of the CD spectra is only qualitative and NMR or X-ray crystallography are required for more detailed structural and conformational analysis .
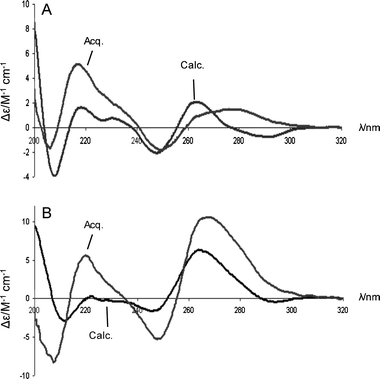 |
| | Fig. 5 CD spectra for POM Lys–(T)8–NH227vs. d(CGCA8CGC) and r(CGCA8CGC) 7.6 µM (total conc. in strands, 1 : 1 ratio of strands) and 10 mM K2HPO4, 0.12 M K+, pH 7.0 (total volume 1.0 cm3). (A) CD spectra of acquired and calculated for POM 27vs. d(CGCA8CGC); (B) CD spectra of acquired and calculated for POM 27vs. r(CGCA8CGC). | |
Conclusion
Boc-protected thyminyl monomers were prepared and used for the solid-phase synthesis of backbone-extended pyrrolidine-amide oligonucleotide mimics. The synthetic bePOMs POM I and II thyminyl octamers (28 and 29) were purified by RP-HPLC and characterised by MALDI mass spectrometry and analytical RP-HPLC. The DNA- and RNA-hybridisation properties of bePOMs POM I and II thyminyl octamers (28 and 29) were then compared with the prototype POM oligomer, using UV thermal denaturation and renaturation experiments and CD spectroscopy. This showed that bePOM I thyminyl octamer 28 binds to r(CGCA8CGC) with a slightly lower Tm (heating) than that of the prototype POM, but shows no evidence of hybridisation with d(CGCA8CGC). The bePOM II thyminyl octamer 29 similarly exhibits hybridisation with RNA , but not DNA. Hybridisation of bePOM II with r(CGCA8CGC) is accompanied by two transitions in the denaturation curve. This could be due to triplex formation or the formation of a mixture of parallel and antiparallel complexes of differing thermodynamic stability. Circular dichroism experiments also show additional evidence of complex formation between the bePOM I and II oligomers with RNA but not DNA. These findings are consistent with the earlier observation that a backbone-extended nucleic acid mimic containing (2′S,4′S)-pyrrolidine units, which is the enantiomer of the bePOM II presented here, is also capable of selective cross-pairing with RNA .10d Currently, the synthesis of longer mixed-sequence bePOMs is under way in order to fully investigate the recognition of more biologically relevant nucleic acid sequences.6
Experimental
NMR spectra were recorded on a Bruker DPX 300 operating at 300 MHz (1H) and 75.5 MHz (13C) or a Bruker DPX 400 operating at 400 MHz (1H) and 100.6 MHz (13C). Chemical shifts in 1H and 13C NMR spectra are expressed in ppm relative to tetramethylsilane and were internally referenced to the residual solvent signal. Chemical shift assignments for 1H and 13C spectra were assisted with COSY, DEPT, HMQC and HMBC experiments. The splitting patterns for NMR spectra are designated as follows: s (singlet), d (doublet), t (triplet), q (quartet), dd (doublet of doublets), ddd (doublet of doublet of doublets), m (multiplet) and br (broad). Mass spectra were obtained using electrospray (ES) on a MassLynx orthogonal accelerated-TOF mass spectrometer with samples introduced from a Waters 7240 sample injector. MALDI mass spectra were obtained on a Micromass TOF Spec 2e or a Shimadzu AXIMI-CF+ using α-cyano-4-hydroxycinnamic acid as matrix. Infrared spectra were recorded on a Nicolet Nexus 670 FT-IR spectrometer with samples prepared as a thin film on KBr discs or as Nujol mulls. UV measurements were carried out on a Varian Cary 400 spectrometer with cell-transport accessories with samples. Molar extinction coefficients (ε) were calculated from Beer–Lambert law from a sample solution of known concentration. Optical rotations were measured at 25 °C with an Optical Activity AA-1000 polarimeter. Melting points were determined with an electrothermal capillary apparatus and are uncorrected. X-Ray crystallographic analysis was collected on a Nonius κCCD diffractometer. Thin layer chromatography was performed on Fluka silica gel (60 F254) coated on aluminium plates. TLC plates were visualised by UV (254 nm) and/or developed using potassium permanganate, vanillin or Ehrlichs reagent and ninhydrin. Flash column chromatography was performed on silica gel LC 60A purchased from Fluorochem Ltd. Chemicals were purchased from Aldrich Chemical Company, Acros Organics and Lancaster Synthesis Ltd and were used without further purification unless otherwise noted. Solvents were purified and dried where necessary; THF was distilled from sodium with benzophenone as indicator under nitrogen. Dichloromethane was distilled from CaH2. DMF, 1,4-dioxane, DIEA and pyridine were purchased anhydrous from Aldrich Chemical Company or Acros Organics and used without further purification. Deionised water was used throughout. For reactions requiring anhydrous conditions, glassware was flame dried under vacuum and cooled under a positive pressure of nitrogen.
(2R,4R)-2-Ethoxycarbonyl-4-hydroxy-N-(benzyloxycarbonyl)pyrrolidine (5)
To a solution of pyrrolidine hydrochloride salt 4 (61.9 g, 0.316 mmol) in 1 : 1 water–1,4-dioxane (465 mL), was added triethylamine (110 mL, 0.783 mmol), followed by benzylchloroformate (66.4 mL, 0.472 mmol) dropwise. The reaction mixture was stirred at 50 °C for 2h then at room temperature for 18 h. Solvent was removed under reduced pressure, water (300 mL) was added and the product extracted with Et2O (4 × 500 mL). The organic fractions were combined and dried over MgSO4, MgSO4 was removed by filtration and solvent was removed under reduced pressure. The product was purified by flash chromatography (3 : 1 EtOAc–hexanes, Rf 0.6) to afford benzyloxycarbonyl-protected product 5 (84.4 g, 91%) as a colourless oil. [α]D + 19.5° (c = 1, CHCl3); νmax(KBr)/cm−1 3428 br (OH), 1754, 1712, (CO); 1H NMR (400 MHz, CDCl3) δ 1.14 and 1.31 (3H, 2 × t, J 7.1 Hz, CH2CH3 rotamers), 2.12 (1H, dd, J 14.1 Hz, 6.7 Hz, Ha3), 2.29–2.40 (1H, m, Hb3), 3.59 and 3.63 (1H, 2 × dd, J 11.9 Hz, 4.1 Hz, Ha5 rotamers), 3.73 and 3.78 (1H, 2 × d, J 11.9 Hz, Hb5 rotamers), 4.09 and 4.26 (2H, 2 × q, J 7.1 Hz, CH2CH3 rotamers), 4.36–4.43 (2H, m, H2 and H4), 5.06–5.21 (2H, m, benzyl CH2), 7.32–7.36 (5H, m, benzyl aromatic); 13C NMR (75.5 MHz, CDCl3) δ 13.4 and 13.5 (CH2CH3 rotamers), 37.3 and 38.1 (C3 rotamers), 54.4 and 54.7 (C5 rotamers), 57.3 and 57.6 (C4 rotamers), 60.8 and 60.9 (CH2CH3 rotamers), 66.5 and 66.6 (benzyl CH2 rotamers), 68.8 and 69.7 (C2 rotamers), 127.1, 127.2, 127.4, 127.8 and 127.9 (benzyl aromatic CH), 135.8 and 136.0, (benzyl ipso-C rotamers), 153.9 and 154.4 (CO2Bn rotamers), 172.7 and 172.8 (CO2Et rotamers); m/z (ES) 332 ([M+K]+ 70%); HRMS m/z (ES) 332.0895, calculated for C15H19O5NK 332.0895.
(2R,4R)-2-Ethoxycarbonyl-4-[(tert-butyl)dimethylsilyloxy]-N-(benzyloxycarbonyl)pyrrolidine (6)
To a solution of alcohol 5 (84.0 g, 0.286 mmol) in anhydrous DMF (110 mL) was added tert-butyldimethylsilylchloride (TBDMS–Cl) (64.3 g, 0.427 mmol), imidazole (38.1 g, 0.56 mmol) and DIEA (50 mL, 0.303 mmol) under nitrogen. The reaction mixture was stirred at room temperature under nitrogen for 18 h. The solvent volume was reduced in vacuo and water (200 mL) was added. The product was extracted with CH2Cl2 (4 × 300 mL) and the organic fractions were combined and dried over MgSO4, MgSO4 was removed by filtration and solvent was removed under reduced pressure. The product was purified by flash chromatography (3 : 1 hexanes–EtOAc, Rf 0.4) to afford TBDMS-protected alcohol 6 (99.6 g, 85%) as a colourless oil. Found: C 61.59; H 8.43, N 3.31, Calculated for C19H31O4NSi; C 61.88, H 8.16, N 3.44%; [α]D + 44.9° (c = 1, CHCl3); νmax(KBr)/cm−1 1750, 1708 (CO); 1H NMR (400 MHz, CDCl3) δ 0.00 and 0.01 (6H, 2 × s Si(CH3)2, rotamers), 0.81 (9H, s, SiC(CH3)3), 1.11 and 1.22 (2 × t, J 7.1 Hz, CH2CH3, rotamers), 2.07–2.15 (1H, m, Ha3), 2.22–2.31 (1H, m, Hb3), 3.36 and 3.40 (1H, 2 × dd, J 11.3 Hz, 2.6 Ha5, rotamers), 3.62 and 3.66 (1H, 2 × dd, J 11.3, 5.3 Hz, Hb5, rotamers), 4.03 and 4.13 (1H, 2 × q, J 7.1 Hz, CH2CH3, rotamers), 4.04 and 4.14 (1H, 2 × q, J 7.1 Hz, CH2CH3, rotamers), 4.31–4.35 (1H, m, H4), 4.36 and 4.42 (1H, 2 × dd, J 8.8, 3.7 Hz, H2 rotamers), 5.05 and 5.09 (1H, d, J 12.4 Hz, benzyl CH2, rotamers), 5.13 and 5.15 (1H, d, J 12.4 Hz, benzyl CH2, rotamers), 7.23–7.36 (5H, m, benzyl aromatic); 13C NMR (100.6 MHz, CDCl3) δ −5.5 and −5.4 (Si(CH3)2 rotamers), 13.6 and 13.7 (CH2CH3, rotamers), 17.4 (SiC(CH3)3), 25.2 (SiC(CH3)3), 38.3 and 39.2 (C3, rotamers), 54.3 and 54.7 (C5, rotamers), 57.3 and 57.6 (C2 rotamers), 60.5 (CH2CH3), 66.5 (benzyl CH2), 69.4 and 70.2 (C4 rotamers), 127.3, 127.4, 127.5 and 127.5, (benzyl aromatic, rotamers), 127.9 and 128.0 (benzyl aromatic, rotamers), 136.2 and 136.3 (benzyl ipso-C, rotamers), 154.0 and 154.3 (CO2Bn rotamers); m/z (ES) 446 ([M + K]+ 100%), 408 ([M + H]+ 40%); HRMS m/z (ES), 446.1768 calculated for C19H31O4NSiNa 446.1760.
(2R,4R)-2-Hydroxymethyl-4-[(tert-butyl)dimethylsilyloxy]-N-(benzyloxycarbonyl)pyrrolidine (7)
To a solution of ethyl ester 7 (99.0 g, 0.243 mmol) in anhydrous THF at 0 °C under nitrogen was added LiBH4 portionwise. The reaction mixture was allowed to warm to room temperature and stirred under nitrogen for 18 h. The reaction mixture was cooled to −10 °C and quenched by dropwise addition of 1 : 1 water–sat. aq. K2CO3 (200 mL), this mixture was stirred at room temperature for 24 h. Water (200 mL) was added and the product extracted with EtOAc (4 × 500 mL). The organic fractions were combined and dried over MgSO4, MgSO4 was removed by filtration and solvent was removed under reduced pressure. The product was purified by flash chromatography (2 : 1 hexanes–EtOAc, Rf 0.3) to afford alcohol 7 (62.2 g, 70%) as a colourless oil. [α]D + 10.7° (c = 1, CHCl3); νmax(KBr)/cm−1 3428 br (OH), 1762, 1708 (CO); 1H NMR (400 MHz, CDCl3) δ 0.08 and 0.12 (6H, 2 × s, Si(CH3)2 rotamers), 0.88 and 0.89 (9H, 2 × s, SiC(CH3)3 rotamers), 1.70 and 1.91 (1H, 2 × d, J 13.7 Hz, Ha3 rotamers), 2.10–2.31 (1H, m, Hb3), 3.45 and 3.37 (1H, 2 × d, J 11.7 Hz, Ha5 rotamers), 3.60–3.68 (1H, m, Hb5), 3.78–3.88 (2H, m, Ha6 and Hb6), 4.06–4.14 (1H, m, H2), 4.34 and 4.40 (1H, 2 × br s, H4 rotamers), 5.09–5,19 (2H, m, benzyl CH2), 7.29–7.37 (5H, m, benzyl aromatic); 13C NMR (75.5 MHz, CDCl3) δ −5.3 (Si(CH3)2), 17.6 (SiC(CH3)3), 25.2 and 25.4 (SiC(CH3)3 rotamers), 37.5 and 38.1 (C3 rotamers), 55.3 and 56.1 (C5 rotamers), 59.5 (C2), 66.1 (benzyl CH2), 66.7 and 66.8 (C6 rotamers), 70.0 and 70.2 (C4 rotamers), 126.5, 126.8, 127.6, 127.7, 128.0 and 128.2 (benzyl aromatic CH, rotamers), 136.2 (benzyl ipso-C) 154.6 and 156.2 (CO2Bn rotamers); m/z (ES) 388 ([M + Na]+ 50%); HRMS m/z (ES) 388.1915, calculated for C19H31O4NSiNa 388.1915.
(2S,4R)-2-Cyanomethyl-4-[(tert-butyl)dimethylsilyloxy]-N-(benzyloxycarbonyl)pyrrolidine (9)
To a solution of alcohol 7 (5.25 g, 14.36 mmol) in anhydrous CH2Cl2 (20 mL) at 0 °C, under nitrogen was added DIEA (3.8 mL, 23.0 mmol) followed by methanesulfonyl chloride (1.3 mL, 17.0 mmol) dropwise. The reaction mixture was allowed to warm to room temperature and stirred under nitrogen for 3 h. The reaction was quenched by addition of sat. NaHCO3 (aq) (25 mL). Water (25 mL) was added and the crude product was extracted with CH2Cl2 (4 × 100 mL). The organic fractions were combined and dried over MgSO4, MgSO4 was removed by filtration and solvent was removed under reduced pressure. The crude methanesulfonate 8 was dried under reduced pressure and dissolved in anhydrous DMF (40 mL). NaCN (3.55 g, 72.4 mmol) was added under nitrogen and the suspension stirred at 75 °C, under nitrogen for 20 h. Water (150 mL) was added to the reaction mixture and the product extracted with Et2O (4 × 250 mL). The aqueous layer was drained into a solution of NaOCl. The organic fractions were combined, washed with water and dried over MgSO4, MgSO4 was removed by filtration and solvent was removed under reduced pressure. Flash chromatography (hexanes–EtOAc 2 : 1 Rf 0.7) afforded nitrile 9 (3.65 g, 68%) as a pale yellow oil. [α]D + 21.3° (c = 2, CHCl3); νmax(KBr)/cm−1 2253 (CN), 1707 (CO); 1H NMR (400 MHz, CDCl3) δ 0.07 and 0.10 (6H, 2 × s, Si(CH3)2 rotamers), 0.89 (9H, s, SiC(CH3)3), 2.04–2.09 (1H, m, Ha3), 2.13–2.20 (1H, m, Hb3), 2.85–3.10 (2H, m, Ha6Hb6), 3.39 and 3.34 (1H, 2 × d, J 11.6 Hz, Ha5 rotamers), 3.55 and 3.60 (1H, 2 × dd, J 11.6, 4.5 Hz, Hb5 rotamers), 4.16–4.21 (1H, m, H2), 4.41 (1H, br s, H4), 5.08–5.18 (2H, m, benzyl CH2), 7.36–7.37 (5H, m, benzyl aromatic); 13C NMR (75.5 MHz, CDCl3) δ −5.1 and −5.0 (Si(CH3)2 rotamers), 17.8 (SiC(CH3)3, 22.3 and 23.2 (C6 rotamers), 25.6 (SiC(CH3)3, 38.2 and 39.0 (C3, rotamers), 53.5 and 54.0 (C2 rotamers), 55.6 and 56.2 (C5 rotamers), 67.0 and 67.3 (benzyl CH2 rotamers), 70.4 and 71.1 (C4 rotamers), 117.9 and 118.0 (CN rotamers), 127.8, 128.0, 128.1, 128.2, 128.4 and 128.6 (benzyl aromatic CH rotamers), 136.0 and 136.3 (benzyl ipso-C rotamers), 154.2 and 154.6 (CO2Bn rotamers); m/z (ES) 375 ([M + H]+ 100%); HRMS m/z (ES) 375.2100, calculated for C20H31O3N2Si 375.2098.
(2S,4R)-2-[2-(tert-Butoxycarbonylamino)ethyl]-4-[(tert-butyl)dimethylsilyloxy]-N-(benzyloxycarbonyl)pyrrolidine (11)
To a solution of nitrile 9 (230 mg, 0.66 mmol) in CH3OH (4 mL) at room temperature was added CoCl2·6H2O (315 mg, 1.32 mmol), followed by portionwise addition of NaBH4 (250 mg, 6.60 mmol). The solution was stirred at room temperature for 4 h. EtOAc (20 mL) was added to the reaction mixture and the resulting black precipitate removed by filtration. Water (20 mL) was added to the filtrate and the crude amine extracted with EtOAc (4 × 100 mL). The organic fractions were combined and dried over MgSO4, MgSO4 was removed by filtration and solvent was removed under reduced pressure. The crude amine 10 was dried under reduced pressure and dissolved in 1 : 1 H2O–1,4-dioxane (0.9 mL). To this solution was added triethylamine (200 µL, 1.43 mmol) and di-tert-butyl-dicarbonate (Boc anhydride) (220 mg, 1.01 mmol). The reaction mixture was stirred at room temperature for 18 h and the product was extracted with Et2O (5 × 100 mL). The organic fractions were combined and dried over MgSO4. MgSO4 was removed by filtration and solvent was removed under reduced pressure. Purification by flash chromatography (3 : 1 hexanes–EtOAc, Rf 0.7) afforded the Boc-protected product 11 (195 mg, 62%) as a colourless oil. [α]D −16.6° (c = 1, CHCl3); νmax(KBr)/cm−1 3357 (NH), 1710, 1685 (CO); 1H NMR (400 MHz, CDCl3) δ 0.05 (6H, s, Si(CH3)2), 0.86 (9H, s SiC(CH3)3), 1.43 (9H, s, C(CH3)3), 1.72–1.82 (2H, m, Ha3 and Ha6), 1.95–2.17 (2H, m, Hb3 and Hb6), 2.92–3.00 (1H, m, Ha8), 3.28 (1H, d, J 11.7 Hz, Ha5), 3.34–3.40 (1H, m, Hb8), 3.67 (1H, dd, J 11.7 Hz, 5.2 Hz, Hb5 rotamers), 3.93 and 4.06 (1H, 2 × d, J 7.0 Hz, H2 rotamers), 4.36 (1H. br s, H4), 5.07–5.17 (2H, m, benzyl CH2), 7.34–7.38 (5H, m, benzyl aromatic); 13C NMR (100.6 MHz, CDCl3) δ −5.0 (Si(CH3)2), 17.8 (SiC(CH3)3, 25.6 (SiC(CH3)3, 28.4 (C(CH3)3), 35.4 (C6), 37.5 (C8), 39.6 and 39.7 (C3 rotamers), 54.6 and 54.8 (C2 rotamers), 55.3 (C5), 66.7 and 67.0 (benzyl CH2 rotamers), 70.5 and 71.3 (C4 rotamers), 78.6 and 78.9 (C(CH3)3 rotamers), 127.7, 127.9, 128.1, 128.4 and 128.6, (benzyl aromatic CH rotamers), 136.4 and 136.7 (benzyl ipso-C rotamers), 155.6 (CO2Bn), 156.1 (CO2tBu); m/z (ES) 479 ([M + H]+ 100%); HRMS m/z (ES) 479.2929, calculated for C25H43O5N2Si 479.2936.
(2S,4R)-2-[2-(tert-Butoxycarbonylamino)ethyl]-4-[(tert-butyl)dimethylsilyloxy]-N-(methoxycarbonylmethyl)pyrrolidine (13)
A solution of 11 (12.84 g, 26.8 mmol) in anhydrous CH3OH (350 mL) was degassed with nitrogen before being added to 10% palladium on carbon (1.50 g) under a nitrogen atmosphere. Hydrogen was bubbled through the reaction mixture for 5 minutes and the reaction mixture was then stirred under a hydrogen atmosphere for 18 h. Palladium on carbon was removed by filtration and solvent was removed under reduced pressure. The crude amine 12 was dried and then dissolved in anhydrous CH2Cl2 (40 mL) under nitrogen. The solution was cooled to 0 °C and DIEA (9.8 mL, 59.1 mmol) was added followed by dropwise addition of methyl bromoacetate (4.9 mL, 53.1 mmol). The reaction mixture was allowed to warm to room temperature and stirred under nitrogen for 18 h. Solvent was removed under reduced pressure and flash chromatography (4 : 1 hexanes–EtOAc, Rf 0.5) afforded methyl ester 13 (7.21 g, 65%) as a pale yellow oil. [α]D −11.5° (c = 1, CHCl3); νmax(KBr)/cm−1 3360 (NH), 1751, 1710, 1688 (CO); 1H NMR (400 MHz, CDCl3) δ 0.03 (6H, s, Si(CH3)2), 0.86 (9H, s, SiC(CH3)3), 1.42 (9H, s, C(CH3)3), 1.55–1.75 (3H, m, Ha3, Ha6 and Hb6), 2.23 (1H, ddd, J 13,4, 7.3, 6.1 Hz, Hb3), 2.63 (1H, dd, J 9.7, 5.8 Hz, Ha5), 2.75–2.81 (1H, m, H2), 3.06 (1H, dd, J 9.7 1.6, Hz, Hb5), 3.14–3.27 (3H, m, Ha7, Ha8 and Hb8), 3.55 (1H, m, J 16.7 Hz, Hb7), 3.70 (3H, s, OCH3), 4.30–4.35 (1H, m, H4), 5.30 (1H, br s, NH); 13C NMR (100.6 MHz, CDCl3) δ −4.8 (Si(CH3)2), 18.1 (SiC(CH3)3, 25.8 (SiC(CH3)3, 28.4 (C(CH3)3), 32.2 (C6), 37.3 (C8), 40.3 (C3), 51.5 (OCH3), 53.7 (C7), 60.5 (C2), 62.3 (C5), 70.5 (C4), 78.7 (C(CH3)3), 156.0 (CO2tBu), 171.2 (CO2CH3); m/z (ES) 417 ([M + H]+ 100%); HRMS m/z (ES) 417.2787, calculated for C20H41O5N2Si 417.2779.
(2S,4R)-2-[2-(tert-Butoxycarbonylamino)ethyl]-4-hydroxy-N-(methoxycarbonylmethyl)pyrrolidine (14)
To a solution of 13 (6.51 g, 15.64 mmol) in anhydrous THF (65 mL) was added tetrabutylammonium fluoride (TBAF) (14.00 g, 44.37 mmol) under nitrogen. The reaction mixture was stirred at room temperature for 4 h. Solvent was removed under reduced pressure and flash chromatography (EtOAc, Rf 0.2) afforded alcohol 14 (3.88 g, 82%) as a pale yellow oil. [α]D + 28.5 (c = 1, CHCl3); νmax(KBr)/cm−1 3360 br (OH), 1740, 1684 (CO); 1H NMR (400 MHz, CDCl3) δ 1.42 (9H, s, (C(CH3)3), 1.56–1.66 (2H, m, Ha3 and Ha6), 1.74–1.79 (1H, m, Hb6), 2.30–2.37 (1H, m, Hb3), 2.54 (1H, dd, J 9.8 4.4 Hz, Ha5), 2.61–2.64 (1H, m, H2), 3.10–3.18 (4H, m, Hb5, Ha7, Ha8 and Hb8), 3.56 (1H, d, J 16.8 Hz, Hb7), 3.70 (3H, s, OCH3), 4.25 (1H, br s, H4); 13C NMR (100.6 MHz, CDCl3) δ 28.3 (C(CH3)3), 32.8 (C6), 37.3 (C8), 40.2 (C3), 51.6 (OCH3), 53.4 (C7), 60.5 (C2), 62.6 (C5), 69.6 (C4), 78.9 (C(CH3)3), 156.0 (CO2tBu), 171.4 (CO2CH3); m/z (ES) 325 ([M + Na]+ 100%), 303 ([M + H]+ 85%); HRMS m/z (ES) 303.1913, calculated for C14H27O5N2 303.1914.
(2S,4S)-2-[2-(tert-Butoxycarbonylamino)ethyl]-4-formyloxy-N-(methoxycarbonylmethyl)pyrrolidine (15)
To a solution of alcohol 14 (3.20 g, 10.59 mmol) in anhydrous THF (50 mL) under nitrogen, was added triphenylphosphine (3.60 g, 13.73 mmol). The solution was cooled to −20 °C and anhydrous formic acid (560 µL, 13.78 mmol) was added followed by dropwise addition of DIAD (2.75 mL, 13.97 mmol). The reaction mixture was allowed to warm to room temperature and stirred under nitrogen for 18 h. Triphenylphosphine (1.80 g, 6.87 mmol) was added and the reaction mixture was cooled to −20 °C, anhydrous formic acid (260 µL, 6.89 mmol) was added followed by dropwise addition of DIAD (1.38 mL, 6.99 mmol). The reaction mixture was allowed to warm to room temperature and stirred under nitrogen for 3 h. Solvent was removed under reduced pressure and flash chromatography (1 : 1 hexanes–EtOAcRf 0.3) afforded formyl ester 15 (2.45 g, 70%) as a pale yellow oil. [α]D−10.2° (c = 1, CHCl3); νmax(KBr)/cm−1 3336 (NH), 1739, 1720, 1685 (CO); 1H NMR (400 MHz, CDCl3) δ 1.43 (9H, s, (C(CH3)3), 1.50–1.59 (1H, m, Ha6), 1.75–1.82 (1H, m, Hb6), 1.92 (1H, ddd, J 13.6, 6.7, 2.1 Hz, Ha3), 2.04 (1H, ddd, J 13.6, 6.4, 2.2 Hz, Hb3), 2.54 (1H, dd, J 11.1, 3.6 Hz, Ha5), 2.92–2.98 (1H, m, H2), 3.09–3.19 (2H, m, Ha8 and Hb8), 3.22 (1H, d, J 16.7 Hz, Ha7), 3.60 (1H, d, J 16.7 Hz, Hb7), 3.69 (1H, dd, J 11.1, 6.3 Hz, Hb5), 3.72 (3H, s, OCH3), 4.95 (1H, br s, NH), 5.26–5.31 (1H, m, H4), 8.00 (1H, s, OCHO); 13C NMR (100.6 MHz, CDCl3) δ 28.4 (C(CH3)3), 32.4 (C6), 37.3 (C8), 37.4 (C3), 51.8 (OCH3), 54.0 (C7), 59.5 (C5), 59.9 (C2), 72.5 (C4), 79.1 (C(CH3)3), 155.9 (CO2tBu), 160.5 (OCHO), 171.0 (CO2CH3); m/z (ES) 331 ([M + H]+ 100%), 353 ([M + Na+] 40%); HRMS m/z (ES) 331.1864, calculated for C15H27O6N2 331.1864.
(2S,4S)-2-[2-(tert-Butoxycarbonylamino)ethyl]-4-hydroxy-N-(methoxycarbonylmethyl)pyrrolidine (16)
To a solution of formyl ester 15 (2.21 g, 6.68 mmol) in anhydrous CH3OH (15 mL) under nitrogen, was added anhydrous sodium methoxide (90 mg, 1.67 mmol). The reaction mixture was stirred at room temperature for 2 h. Anhydrous sodium methoxide (45 mg, 0.84 mmol) was added and the reaction mixture stirred for a further 3 h. Solvent was removed under reduced pressure and flash chromatography (EtOAcRf 0.2) afforded 4Salcohol 16 (1.82 g, 90%) as a pale yellow oil. [α]D + 31.4° (c = 1, CHCl3); νmax(KBr)/cm−1 3362 br (OH), 1744, 1690 (CO); 1H NMR (400 MHz, CDCl3) δ 1.39 (1H, s, (C(CH3)3), 1.44–1.51 (1H, m, Ha6), 1.63–1.75 (2H, m, Ha3 and Hb6), 1.93 (1H, dd, J 13.0, 6.1 Hz, Hb3), 2.53 (1H, d, J 10.8 Hz, Ha5), 3.01–3.13 (3H, m, H2, Ha8 and Hb8), 3.35 (1H, d, J 17.6 Hz, Ha7), 3.48 (1H, dd, J 10.8, 5.2 Hz, Hb5), 3.55 (1H, d, J 17.6 Hz, Hb7), 3.69 (3H, s, OCH3), 4.26–4.28 (1H, m, H4); 13C NMR (100.6 MHz, CDCl3) δ 28.2 (C(CH3)3), 32.7 (C6), 37.3 (C8), 40.5 (C3), 51.6 (OCH3), 53.2 (C7), 59.1 (C2), 61.8 (C5), 69.9 (C4), 78.8 (C(CH3)3), 155.9 (CO2tBu), 172.2 (CO2CH3); m/z (ES) 303 ([M + H]+ 100%); HRMS m/z (ES) 303.1914, calculated for C14H27O5N2 303.1914.
(2′S,4′R)-2-[2-(tert-Butoxycarbonylamino)ethyl]-4-(N3-benzoylthymin-1-yl)-N-(methoxycarbonylmethyl)pyrrolidine (17)
To a solution of alcohol 17 (400 mg, 1.32 mmol) in anhydrous THF (50 mL) under nitrogen, was added N3-benzoylthymine (370 mg, 1.61 mmol) and triphenylphosphine (420 mg, 1.60 mmol). The mixture was cooled to −20 °C and DIAD (360 µL, 1.83 mmol) was added dropwise. The reaction mixture was allowed to warm to room temperature and stirred under nitrogen for 18 h. Solvent was removed under reduced pressure and flash chromatography (1 : 1 hexanes–EtOAcRf 0.4) afforded thyminyl derivative 17 (453 mg, 67%) as a white foam. Found: C 60.28; H 6.76, N 10.36, Calculated for C26H34O7N4; C 60.69, H 6.66, N 10.89%; [α]D−49.3° (c = 0.5, CHCl3); νmax(KBr)/cm−1 3373 (NH), 1746, 1698, 1652 (CO); λmax (CH3OH)/nm 252 (ε/dm3mol−1 cm−1 1.6 × 104); 1H NMR (400 MHz, CDCl3) δ 1.42 (9H, s, C(CH3)3), 1.48–1.55 (1H, m, Ha3′), 1.60 (1H, dd, J 13.9, 7.2 Hz, Ha6′), 1.83–1.91 (1H, m, Hb6′), 1.99 (3H, s, thymine CH3), 2.56–2.66 (3H, m, H2′, Hb3′ and Ha5′), 2.99 (1H, d, J 17.2 Hz, Ha7′), 3.09–3.18 (2H, m, HaHb8′), 3.33 (1H, d, J 11.1 Hz, Hb5′), 3.69 (1H, d, J 17.2 Hz, Hb7′), 3.74 (3H, s, O–CH3), 4.85 (1H, br s, NH), 5.00–5.04 (1H, m, H4′), 7.47 (2H, t, J 7.5 Hz, Bz meta-H), 7.62 (1H, t, J 7.5 Hz, Bz para-H), 7.90 (2H, d, J 7.5 Hz, Bz ortho-H), 8.09 (1H, s, H6); 13C NMR (100.6 MHz, CDCl3) δ 12.7 (thymine CH3), 28.3 (C(CH3) 3), 32.6 (C6′), 37.2 (C8′), 38.7 (C3′), 51.8 (O-CH3), 51.9 (C4′), 52.7 (C7′), 58.7 (C5′), 60.7 (C2′), 79.3 (C(CH3)3), 111.3 (C5), 129.0 (Bz meta-C), 130.3 (Bz ortho-C), 131.6 (Bz ipso-C), 134.8 (Bz para-C), 137.7 (C6), 149.9 (C2), 155.8 (CO2tBu), 162.8 (C4), 169.2 (Bz CO), 170.9 (CO2CH3); m/z (ES) 537 ([M + Na]+ 100%), 515 ([M + H]+ 60%); HRMS m/z (ES) 515.2514, calculated for C26H35O7N4 515.2500.
(2′S,4′R)-2-[2-(tert-Butoxycarbonylamino)ethyl]-4-(thymin-1-yl)pyrrolidine-1-yl-acetic acid (18)
To a solution of methyl ester 17 (400 mg, 0.78 mmol) in THF (4 mL) was added 1 M aqueous NaOH (2.4 mL, 2.4 mmol) and the reaction mixture was stirred at room temperature for 18 h. THF was removed under a stream of nitrogen and the pH of the remaining aqueous solution was adjusted to 7 by addition of 0.1 M aqueous HCl. Water was removed under reduced pressure and the resulting white residue was submitted to column chromatography (7 : 3 EtOAc–CH3OHRf 0.2) followed by reversed phase chromatography (BondElut C18, H2O–CH3CN 9 : 1), the product was lyophilised to afford acid 18 (220 mg, 71%) as a white powder. [α]D + 5.2° (c = 1, CH3OH); νmax(KBr)/cm−1 3353 br (OH), 1720, 1680, 1651 (CO); λmax (CH3OH)/nm 267 (ε/dm3 mol-1 cm-1 1.27 × 104); 1H NMR (400 MHz, CD3OD) δ 1.35 (9H, s, (C(CH3)3), 1.72–1.77 (1H, m, Ha6′), 1.80 (3H, s, thymine CH3), 1.98–2.08 (2H, m, Ha3′ and Hb6′), 2.74–2.81 (1H, m, Hb3′), 3.03–3.08 (2H, m, Ha8′ and Hb8′), 3.28–3.38 (2H, m, H2′ and Ha5′), 3.43 (1H, d, J 16.2 Hz, Ha7), 3.74 (1H, d, J 16.2 Hz, Hb7′), 3.91 (1H, d, J 12.6 Hz, Hb5′), 4.68–4.75 (1H, m, H4′), 7.47 (1H, s, H6); 13C NMR (100.6 MHz, CD3OD) δ 12.4 (thymine CH3), 28.8 (C(CH3) 3), 31.9 (C6′), 36.6 (C3′), 38.2 (C8′), 56.2 (C7′), 58.5 (C4′), 60.2 (C5′), 66.5 (C2′), 80.3 (C(CH3)3), 111.6 (C5), 142.7 (C6), 153.2 (C2), 158.5 (CO2tBu), 166.5 (C4), 171.2 (CO2H); m/z (ES) 397 ([M + H]+ 100%), 419 ([M + Na]+ 50%); HRMS m/z (ES) 397.2094, calculated for C18H29O6N4 397.2082.
(2R,4R)-2-(Azidomethyl)-4-hydroxy-N-(methylpropanoate)pyrrolidine (20)
To a suspension of the azide hydrochloric salt 19 (4.17 g, 23.34 mmol) in anhydrous CH2Cl2, was added DIEA (1 mL, 8.22 g, 63.57 mmol) at 0 °C under nitrogen and the suspension stirred until dissolution. To the solution was added methyl acrylate (4.25 mL, 4.06 g, 47.19 mmol) dropwise, and the reaction mixture was stirred at 0 °C for 30 min. The reaction mixture was allowed to warm to room temperature and stirred for a further 18 h under nitrogen. Solvent was removed under reduced pressure and the crude product purified by flash chromatography (1 : 1 hexanes–EtOAc, Rf 0.2 EtOAc) to afford methyl ester 20 (4.52 g, 85%) as a pale yellow oil. [α]D +57.7 (c = 1, CHCl3); νmax(BaF)/cm−1 3401 br (OH), 2104 (N3), 1727 (CO); 1H NMR (400 MHz; CDCl3) δ 1.65 (1H, dd, J 14.3, 4.6 Hz, Ha3), 2.20–2.27 (1H, m, Hb3), 2.30 (1H dd, J 9.8, 4.0 Hz Ha5), 2.45–2.57 (3H, m, Ha7, Hb7 and Ha8), 2.65–2.71 (1H, m, H2), 2.83 (1H, s, OH), 3.05–3.17 (2H, m, Hb5 and Hb8), 3.29 (1H, dd, J 12.4, 4.4 Hz, Ha6), 3.46 (1H, dd, J 12.4, 3.2 Hz, Hb6), 3.65 (3H, s, OCH3) 4.17 (1H, s, H4); 13C NMR (100.6 MHz; CDCl3) δ 33.5 (C7), 38.1 (C3), 48.1 (C8), 51.6 (OCH3), 53.8 (C6), 61.8 (C5), 61.9 (C2), 70.1 (C4), 172.6 (CO2CH3); m/z (ES) 251 ([M + Na]+ 100%), 229 ([M + H]+ 55%); HRMS m/z (ES) 251.1118, calculated for C9H16N5O2 251.1115.
(2R,4R)-2-[(tert-Butoxycarbonyl)aminomethyl]-4-hydroxy-N-(methylpropanoate)pyrrolidine (21)
To a solution of the methyl ester azide20 (522 mg, 2.28 mmol) in THF (10 mL) was added a 1 M solution of trimethylphosphine in THF (3.43 mL, 3.43 mmol) and water (42 µL, 2.33 mmol). The solution was stirred until all the starting material had been consumed, as determined by TLC (ca. 1.5 h). The solution was cooled to −20 °C and 2-(tert-butoxycarbonyloxyimino)-2-phenylacetonitrile (Boc-ON) (1.41 g, 5.74 mmol) was added, the reaction mixture was stirred at −20 °C for 15 minutes then allowed to warmed to room temperature and stirred for a further hour. Solvent was removed under reduced pressure and the crude product was purified by flash chromatography (1 : 1 hexanes–EtOAc, Rf 0.1 EtOAc) to afford the Boc-protected product 21 (619 mg, 89%) as a pale yellow oil. [α]D +63.2 (c = 1, CHCl3); νmax(BaF)/cm−1 3389 br (OH) 1742 and 1692 (CO); 1H NMR (400 MHz, CDCl3) δ 1.42 (9H, s, C(CH3)3), 1.60 (1H, dd, J 14.3, 5.9 Hz, Ha3), 2.16–2.26 (1H, m, Hb3), 2.28 (1H, dd, J 9.8, 4.1 Hz, Ha5), 2.37–2.43 (1H, m, Ha8), 2.46–2.53 (2H, m, Ha7 and Hb7), 2.59 (1H, br s, OH), 3.07–3.17 (3H, m, Hb5, H2 and Ha6), 3.32–3.37 (1H, m, Hb6), 3.69 (3H, s, OCH3), 4.20 (1H, t, J 4.4 Hz, H4), 5.20 (1H, br s, NH); 13C NMR (100.6 MHz; CDCl3) δ 28.4 (C(CH3)3), 33.5 (C7), 38.0 (C3), 40.9 (C6), 48.2 (C8), 51.7 (OCH3), 61.9 (C2), 62.0 (C5), 69.9 (C4), 79.1 (C(CH3)3), 156.5 (CO2tBu), 173.0 (CO2CH3); m/z (ES) 325 ([M + Na]+ 100%), 303 ([M + H]+ 90%; HRMS m/z (ES) 325.1725, calculated for C14H26N2O5Na 325.1734.
(2R,4S)-2-[(tert-Butoxycarbonyl)aminomethyl]-4-formyloxy-N-(methylpropanoate)pyrrolidine (22)
To a solution of alcohol 21 (2.01 g, 6.68 mmol) in THF (30 mL), was added triphenylphosphine (2.25 g, 8.3 mmol) and anhydrous formic acid (0.32 mL, 8.48 mmol) under nitrogen. The solution was cooled to −20 °C and DIAD (1.7 mL, 8.57 mmol) was added dropwise. The reaction mixture was allowed to warm to room temperature and stirred under nitrogen for 18 h. Solvent was removed under reduced pressure and the crude product was purified by flash chromatography (5 : 1 hexanes–EtOAc, Rf 0.5 EtOAc) to afford the formyl ester 22 (1.44 g, 65%) as a pale yellow oil. [α]D +57.7 (c = 1, CHCl3); νmax(BaF)/cm−1 2976 (CHO), 1723 (CO); 1H NMR (400 MHz, CDCl3) δ 1.40 (9H, s, C(CH3)3), 1.82–1.96 (2H, m, Ha3, Hb3), 2.23 (1H, dd, J 10.6, 3.7 Hz, Ha5), 2.41–2.46 (3H, m, Ha8, Ha7, Hb7), 2.82 (1H, br s, H2), 3.08–3.15 (2H, m, Hb8, Hb5), 3.31–3.37 (1H, m, Ha6), 3.56 (1H, dd, J 10.6, 6.1 Hz Hb6), 3.67 (3H, s, OCH3), 5.04, 5.06 (1H, 2 × s, NH rotomers), 5.14–5.19 (1H, m, H4), 7.96 (1H, s, CHO); 13C NMR (100.6 MHz; CDCl3) δ 28.3 (C(CH3)3), 33.5 (C7), 34.5 (C3), 39.9 (C6), 48.7 (C8), 51.7 (OCH3), 59.0 (C5), 61.2 (C2), 72.3 (C4), 78.9 (C(CH3)3), 156.3 (CO2tBu), 160.5 (CHO), 172.8 (CO2CH3); m/z (ES) 353 ([M + Na]+ 100%), 331 ([M + H]+ 40%); HRMS m/z (ES) 353.1690, calculated for C15H26N2O6Na 330.1783.
(2R,4S)-2-[(tert-Butoxycarbonyl)aminomethyl]-4-hydroxy-N-(methylpropanoate)pyrrolidine (23)
To a solution of formyl ester 22 (1.39 g, 4.19 mmol) in anhydrous CH3OH (15 mL) was added sodium methoxide (34 mg, 0.62 mmol) under nitrogen at room temperature. The reaction mixture was stirred until no starting material was left (ca. 2 h), as shown by TLC. Solvent was removed under reduced pressure and the crude product was purified by flash chromatography (1 : 1 hexane–EtOAc, Rf 0.1 EtOAc) to afford the S-alcohol 23 (1.03 g, 82%) as a pale yellow oil. [α]D +111.7 (c = 1, CHCl3); νmax(KBr)/cm−1 3389 br (OH) 1737 and 1688 (CO); 1H NMR (400 MHz, CDCl3) δ 1.40 (9H, s, C(CH3)3), 1.70–1.75 (1H, m, Ha3), 1.78–1.90 (1H, m, Hb3), 2.20 (1H, dd, J 9.85, 4.67 Hz, Ha5), 2.40–2.51 (3H, m, Ha8, Ha7 and Hb7), 2.86 (1H, br s, H2), 3.07–3.13 (2H, m, Hb5 and Hb8), 3.25–3.30 (1H, m, Ha6), 3.40 (1H, dd, J 9.72, 5.81 Hz, Hb6), 3.66 (3H, s, OCH3), 4.30 (1H, br s, H4), 5.06 (1H, d, J 6.57, NH); 13C NMR (100.6 MHz; CDCl3) δ 28.3 (C(CH3)3), 33.7 (C7), 37.9 (C3), 40.4 (C6), 49.1 (C8), 51.7 (OCH3), 61.3 (C2), 61.7 (C5), 69.6 (C4), 78.9 (C(CH3)3), 156.4 (CO2tBu), 173.0 (CO2CH3); m/z (ES) 325 ([M + Na]+ 100%), 303 ([M + H]+ 75%); HRMS m/z (ES) 303.1916, calculated for C14H27N2O5 303.1914.
(2′R,4′R)-2-[(tert-Butoxycarbonyl)aminomethyl]-4-(N3-benzoylthymin-1-yl)-N-(methylpropanoate)pyrrolidine (24)
To the S-alcohol 23 (475 mg, 1.57 mmol) in anhydrous THF (20 mL) under nitrogen was added triphenylphosphine (550 mg, 2.04 mmol) and N3-benzoylthymine (462 mg, 2.00 mmol). The suspension was cooled to 0 °C and DIAD (462 µL, 2.00 mmol) was added dropwise and the reaction mixture was stirred for 5 min. The reaction mixture was allowed to warm to room temperature and stirred for 18 h under nitrogen. Solvent was removed under reduced pressure and the crude product was purified by flash chromatography (1 : 1 hexane–EtOAc, Rf 0.1 EtOAc) to afford the thyminyl derivative 24 (516 mg, 64%) as a white foam. [α]D +68.1 (c = 1, CHCl3); νmax(KBr)/cm−1 1746, 1699 and 1652 (CO); 1H NMR (400 MHz; CDCl3) δ 1.46 (9H, s, C(CH3)3), 1.63–1.70 (1H, m, Ha3′), 2.00 (3H, s, thymine CH3), 2.23–2.30 (1H, m, Ha8′), 2.52–2.60 (5H, m, Ha7′, Hb7′, Hb3′, Ha5′ and H2′), 3.19–3.32 (3H, m, Ha6′, Hb8′ and Hb5′), 3.59 (1H, dd, J 14.2, 9.6 Hz, Hb6′), 3.77 (3H, s, OCH3), 5.14 (1H, br s, H4′), 5.37 (1H, d, J 7.1 Hz, NH), 7.47 (2H, t, J 7.6 Hz, Bz meta-H), 7.62 (1H, t, J 7.6 Hz, Bz para-H), 7.79 (1H, s, H6), 7.89 (2H, d, J 7.6 Hz, Bz ortho-H); 13C NMR (100.6 MHz; CDCl3) δ 12.7 (thymine CH3), 28.3 (C(CH3)3), 33.2 (C7′), 35.5 (C3′), 39.0 (C6′), 47.2 (C8′), 51.0 (C4′), 51.8 (OCH3), 59.0 (C5′), 63.2 (C2′), 79.4 (C(CH3)3), 111.0 (C5), 129.1 (Bz ortho-C), 130.4 (Bz meta-C), 131.5 (Bz ipso-C), 134.9 (Bz para-C), 137.5 (C6), 149.8 (C2), 156.2 (CO2tBu), 162.7 (C4), 169.1 (benzamide CO) 173.2 (CO2CH3); m/z (ES) 515 ([M + H]+ 100%); HRMS m/z (ES) 515.2504, calculated for C26H35N4O7 515.2500.
(2′R,4′R)-2-(tert-Butoxycarbonylamino-methyl)-4-(thymin-1-yl)pyrrolidine-1-yl propanoic acid (25)
To a solution of methyl ester 24 (314.7 mg, 0.611 mmol) in THF (5 mL) was added 1 M aqueous NaOH (1.85 mL, 1.85 mmol). The reaction mixture was stirred at room temperature for 3 h. THF was removed under a stream of nitrogen and the aqueous solution was adjusted to pH 7 by addition of 0.1 M aqueous HCl. Water was removed under reduced pressure and the resulting white residue was submitted to column chromatography (EtOAc–CH3OH 7 : 3 Rf 0.2) followed by reversed phase chromatography (BondElut C18, H2O–CH3CN 9 : 1), the product was lyophilised to afford acid 25 (295 mg, 69%), as a white solid. [α]D + 60.3 (c = 1, CH3OH); νmax(KBr)/cm−1 1701, 1696, 1685, 1680 and 1675 (CO); 1H NMR (400 MHz; CD3OD) δ 1.31 (9H, s, C(CH3)3), 1.52–1.61 (1H, m, Ha3′), 1.84 (3H, s, thymine CH3), 2.32–2.39 (1H, m, Ha8′), 2.42–2.54 (3H, m, Ha7′, Hb3′ and Hb7′), 2.61–2.70 (2H, m, Ha5′ and H2′), 3.12–3.21 (1H, m, Ha6′), 3.25–3.42 (3H, m, Hb8′, Hb5′and Hb6′), 4.84 (1H, s, H4′), 7.76 (1H, s, H6). 13C NMR (100.6 MHz; CD3OD) δ 12.7 (thymine CH3), 28.7 (C(CH3)3), 34.7 (C7′), 36.6 (C3′), 40.8 (C6′), 50.4 (C8′), 54.6 (C4′), 58.9 (C5′), 65.7 (C2′), 80.3 (C(CH3)3), 111.5 (C5), 140.8 (C6), 153.0 (C2), 158.7 (CO2tBu), 166.5 (C4), 177.6 (CO2H); m/z (ES) 419 ([M + Na]+ 100%), 397 ([M + H]+ 10%); HRMS m/z (ES) 419.1909, calculated for C17H26O6N4Na 419.1901.
POM oligomer synthesis
All experiments were carried out in solid-phase synthesis vessels purchased from Kinesis and fitted a with porosity-3 frit. Resin was agitated by rotation of the vessel and reagents were removed by suction filtration through a Buchner flask. MBHA resin LL (100–200 mesh) (loading of 0.62 mmol/g), Boc–Lys–(2-Cl–Z)–OH and HBTU were purchased from Novabiochem. Fresh bottles of anhydrous solvents from Acros Organics were used for each POM oligomer synthesised. All other chemicals used in solid-phase work were obtained at the highest purity grade from Aldrich Chemical Company or Acros Organics and were used without further purification. Reagents used for the Kaiser test were prepared according to literature.17
General procedure for solid-phase synthesis
Into a 1 mL solid-phase synthesis vessel was weighed MBHA resin (5 equiv.). Washing of the resin was carried out 3 times with DMF (all washings use 1 mL per 25 µmol resin loading, in all cases performed with rotation of vessel for 30 s each time, after which solvent was removed through a Buchner flask under reduced pressure) and 3 times with CH2Cl2. The resin was swelled in CH2Cl2. Washing of the resin was carried out 3 times with DMF for 30 s each time, once with 5% piperidine–DMF for 4 minutes and 3 times with DMF–CH2Cl2 (1 : 1). In a separate small vial, Boc-POM–(T)–OH (1 equiv.), HBTU (0.95 equiv.) and DIEA (1.1 equiv.) in DMF–pyridine (3 : 1) (monomer concentration of 0.1 M) were allowed to activate for 3 min. The mixture was then added to the resin. Coupling was allowed to proceed with agitation for 6 h. The coupling reagent was removed and the resin washed 2 times with DMF for 30 s each time. The resin was treated with freshly prepared acetic anhydride–collidine–DMF (1 : 1 : 8) (1 mL per 25 µmol) with agitation for 15 min. The acetylating reagent was removed by vacuum suction and resin washed with DMF (3 times for 30 s each time), complete reaction was indicated by negative Kaiser test. The resin was then washed with 5% piperidine–DMF (once for 4 minutes) and DMF–CH2Cl2 (1 : 1) (3 times for 30 s each time). Deprotection of the resin-bound Boc-protected POM oligomer was accomplished using TFA–m-cresol (1 mL per 25 µmol) 4 times for 4 minutes each time. The resin was washed with DMF–CH2Cl2 (1 : 1) (3 times for 30 s each time) and deprotection was indicated by a positive Kaiser test. The resin was then washed with pyridine (2 times for 30 s each time). Subsequent coupling employed Boc-POM(T)–OH (5 equiv.), HBTU (4.75 equiv.), DIEA (5.5 equiv.) and coupling times of 2 h. In the case of lysine, Boc-Lys-(2-Cl-Z)–OH (6 equiv.), HBTU (5.7 equiv.), and DIEA (6.6 equiv.) were used. Capping after subsequent couplings was carried out for 5 min. The coupling–capping–deprotection sequence was repeated until the desired oligomer was obtained. Deprotection of Cbz-protected nucleobases and cleavage of the oligomer from the resin was achieved by the ‘Low–high TFMSA’ method. During ‘low TFMSA’ the resin was treated with a solution of (TFA–DMS–m-cresol (1 : 3 : 1)) and a solution of (TFA–TFMSA (9 : 1)) (each 1 mL per 20 µmol resin loading) each separately cooled to 0 °C before being added to resin and agitated for 1 h. The cleavage mixture was removed by vacuum suction. ‘High TFMSA’ was carried by treating the resin with a solution of TFMSA–TFA–m-cresol (1 : 8 : 1) (1 mL per 10 µmol resin loading) cooled to 0 °C before being added to resin and agitated for 1 h. The cleavage mixture was removed by vacuum suction. The cleavage solutions were separately concentrated under a stream of nitrogen to ∼50 µL and the oligomer was precipitated from the cleavage mixtures by addition of a ten-fold excess of anhydrous diethyl ether. The mixture was subject to centrifugation (10 min, 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 4 °C) and the resulting pellet was redissolved in formic acid and diluted again with anhydrous diethyl ether. The centrifugation process was repeated a further three times. After the final time the pellets were dissolved in water and lyophilised to give crude POM oligomers as off-white powders. The oligomers were then purified by semi-preparative reversed-phase HPLC on a C18 column (Phenomenex Gemini 5 µ C18, 250 × 10 mm) with a typical gradient of 0–10% acetonitrile with 0.1% HCO2H–0.1% aqueous HCO2H. Fractions collected were evaporated and lyophilised to give pure product as a white powder. Product purity was verified by analytical reversed-phase HPLC (Phenomenex Gemini 5 µ C18, 150 × 4.6 mm) and oligomers were characterised by MALDI-TOF mass spectrometry.
000 rpm, 4 °C) and the resulting pellet was redissolved in formic acid and diluted again with anhydrous diethyl ether. The centrifugation process was repeated a further three times. After the final time the pellets were dissolved in water and lyophilised to give crude POM oligomers as off-white powders. The oligomers were then purified by semi-preparative reversed-phase HPLC on a C18 column (Phenomenex Gemini 5 µ C18, 250 × 10 mm) with a typical gradient of 0–10% acetonitrile with 0.1% HCO2H–0.1% aqueous HCO2H. Fractions collected were evaporated and lyophilised to give pure product as a white powder. Product purity was verified by analytical reversed-phase HPLC (Phenomenex Gemini 5 µ C18, 150 × 4.6 mm) and oligomers were characterised by MALDI-TOF mass spectrometry.
POM Lys–(T)8–NH2 (27)
Retention time on analytical HPLC was 29 min, using a Phenomenex Gemini 5 µ C18 150 × 4.6 mm analytical column. Solvent A was H2O with 0.1% HCO2H and solvent B was acetonitrile with 0.1% HCO2H. The flow rate was 1 mL min−1 with 100% A for 9 min followed by a gradient from 100% A changing to 90% A with 10% B over 52 min. m/z MALDI-TOF MS 2259 ([M + H]+ 100%, C102H144N35O25 requires m/z, 2259.1).
bePOM I Lys–(T)8–NH2 (28)
Retention time on analytical HPLC was 33 min, using a Phenomenex Gemini 5 µ C18 150 × 4.6 mm analytical column. Solvent A was H2O with 0.1% HCO2H and solvent B was acetonitrile with 0.1% HCO2H. The flow rate was 1 mL min−1 with 100% A for 9 min followed by a gradient from 100% A changing to 90% A with 10% B over 52 min. m/z MALDI-TOF MS 2371 ([M + H]+ 100%, C110H160N35O25 requires m/z, 2371.2).
bePOM II Lys–(T)8–NH2 (29)
Retention time on analytical HPLC was 21 min, using a Phenomenex Gemini 5 µ C18 150 × 4.6 mm analytical column. Solvent A was H2O with 0.1% HCO2H and solvent B was acetonitrile with 0.1% HCO2H. The flow rate was 1 mL min−1 with 100% A for 9 min followed by a gradient from 100% A changing to 97% A with 3% B over 45 min. m/z MALDI-TOF MS 2372.1 ([M + H]+ 100%, C110H159N35O25 requires m/z, 2371.2).
Thermal denaturation experiments
UV melting plots of absorbance versus temperature were measured at 260 nm on a Varian Cary 400 Scan UV-visible spectrophotometer fitted with a 6 × 6 Peltier thermostatable multicell holder connected to a temperature-controller module. Experiments were performed in double-beam mode and controlled by an interfaced Dell OptiPlex GX150 computer. Denaturation experiments were performed in 10 mm path length 4 mm path width self-masking semi-micro quartz cells fitted with a Teflon stopper. Concentrations of POM oligomers, oligonucleotides and polynucleotides were measured spectrophotometrically at 80 °C from molar extinction coefficients of nucleotidyl units calculated from the literature.
Buffers were prepared as double-concentrated stock solutions and diluted to the appropriate concentrations during sample preparation. All appropriate equipment were autoclaved before use. Sterile nuclease, protease and DEPC-free deionised water was used throughout. All samples were stored at −20 °C. Oligonucleotides were purchased from Sigma-Genosys or sigma Proligo. PNA monomers were purchased from Applied Biosystems. Each thermal denaturation experiment consists of 3 ramps and an averaging time of 1 s was used throughout. Data was collected every 1 °C for the first ramp and 0.1 °C for subsequent part of the experiment. Samples were initially heated at a rate of 5 °C min−1 to 93 °C to dissociate all strands. After 1 min, samples were cooled at 0.2 °C min−1 to 15 °C and after a holding time of 1 min were heated at 0.2 °C min−1 to 93 °C. All Tm values were obtained from the maxima of first derivative curves calculated from Varian Thermal software using a filter size of 97 and smoothed every 0.3 °C.
Circular dichroism experiments
CD spectra were recorded on a JASCO J-715 spectropolarimeter. The CD spectra of the POM–DNA complexes and the relevant single strands were recorded in 10 mM potassium phosphate buffer, 0.12 M KCl at pH 7.0 unless otherwise stated. The CD spectra were recorded as an accumulation of 10 scans from 320 to 180 nm using a 0.5 cm cell, a resolution of 0.1 nm, band-width of 1.0 nm, sensitivity of 2 m deg, response of 2 seconds and a scan speed of 50 nm min−1.
Acknowledgements
This work was funded by the BBSRC (research grant 36/B15998) and EPSRC (PhD studentships to R. J. W. and N. M. B)
References
-
(a) J. Micklefield, Curr. Med. Chem., 2001, 8, 1157 CAS;
(b) H.-J. Hsu, M.-R. Liang, C.-T. Chen and B.-C. Chung, Nature, 2006, 439, 480 CrossRef CAS;
(c) I. A. Sheptopalov, S. Sinha and J. K. Chen, Nat. Chem. Biol., 2007, 3, 650 CrossRef;
(d) X. Tang, S. Maegawa, E. S. Weinberg and I. J. Dmochowski, J. Am. Chem. Soc., 2007, 129, 11000 CrossRef.
-
(a) B. Vester and J. Wengel, Biochemistry, 2004, 43, 13233 CrossRef CAS;
(b) P. E. Nielsen, Curr. Opin. Biotechnol., 2001, 12, 16 CrossRef CAS;
(c) M. Petersen and J. Wengel, Trends Biotechnol., 2003, 21, 74 CrossRef CAS.
-
(a) J. Wengel, Org. Biomol. Chem., 2004, 2, 277 RSC;
(b) P.-S. Ng and D. E. Bergstrom, Nano Lett., 2005, 5, 107 CrossRef;
(c) P. S. Lukeman, A. C. Mittal and N. C. Seeman, Chem. Commun., 2004, 1694 RSC.
-
(a) G. K. Mittapalli, K. R. Reddy, H. Xiong, O. Munoz, B. Han, F. De Riccardis, R. Krishnamurthy and A. Eschenmoser, Angew. Chem., Int. Ed., 2007, 46, 2470 CrossRef CAS;
(b) G. K. Mittapalli, Y. M. Osornio, M. A. Guerrero, K. R. Reddy, R. Krishnamurthy and A. Eschenmoser, Angew. Chem., Int. Ed., 2007, 46, 2478 CrossRef CAS.
-
(a) D. T. Hickman, P. M. King, M. A. Cooper, J. M. Slater and J. Micklefield, Chem. Commun., 2000, 2251 RSC;
(b) D. T. Hickman, T. H. S. Tan, J. Morral, P. M. King, M. A. Cooper and J. Micklefield, Org. Biomol. Chem., 2003, 1, 3277 RSC;
(c) T. H. S. Tan, D. T. Hickman, J. Morral, I. G. Beadham and J. Micklefield, Chem. Commun., 2004, 516 RSC;
(d) T. H. S. Tan, R. J. Worthington, R. G. Pritchard, J. Morral and J. Micklefield, Org. Biomol. Chem., 2007, 5, 239 RSC.
- R. J. Worthington, A. P. O'Rourke, J. Morral, T. H. S. Tan and J. Micklefield, Org. Biomol. Chem., 2007, 5, 249 RSC.
- P. E. Nielsen, M. Egholm, R. H. Berg and O. Buchardt, Science, 1991, 254, 1497 CrossRef CAS.
- Z. Huang and S. A. Benner, J. Org. Chem., 2002, 67, 3996 CrossRef CAS.
- K.-U. Schöning, P. Scholz, S. Guntha, X. Wu, R. Krishnamurthy and A. Eschenmoser, Science, 2000, 290, 1347 CrossRef CAS.
-
(a) C. J. Wilds, G. Minasov, F. Natt, P. von Matt, K.-H. Altmann and M. Egli, Nucleotides, Nucleosides & Nucleic Acids, 2001, 20, 991 Search PubMed;
(b) A. Lauritsen and J. Wengel, Chem. Commun., 2002, 530 RSC;
(c) N. Katagiri, Y. Morishita, I. Oosawa and M. Yamaguchi, Tetrahedron Lett., 1999, 40, 6835 CrossRef CAS;
(d) T. Govindaraju and V. A. Kumar, Chem. Commun., 2005, 495 RSC.
- G. L. Baker, S. J. Fritschel, J. R. Stille and J. K. Stille, J. Org. Chem., 1981, 46, 2954 CrossRef CAS.
- T. Satoh and S. Suzuki, Tetrahedron Lett., 1969, 52, 4555 CrossRef.
- O. Mitsunobu, Synthesis, 1981, 1, 1 CrossRef.
- K. A. Cruickshank, J. Jiricny and C. B. Reese, Tetrahedron Lett., 1984, 25, 681 CrossRef CAS.
- X. Uriza, F. Urpi, C. Viladomat and J. Vilarrasa, Tetrahedron Lett., 1998, 39, 9101 CrossRef.
- L. Christensen, R. Fitzpatrick, B. Gildea, K. H. Petersen, H. F. Hansen, T. Koch, M. Egholm, O. Buchardt, P. E. Nielsen, J. Coull and R. H. Berg, J. Pept. Sci., 1995, 3, 175.
- E. Kaiser, R. L. Colescott, C. D. Bossinger and P. I. Cook, Anal. Biochem., 1970, 34, 595 CrossRef CAS.
- H. T. Steely, D. M. Gray and R. L. Ratliff, Nucleic Acids Res., 1986, 14, 10071 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: HPLC traces, UV melting curves, and CD spectra. See DOI: 10.1039/b714580m |
|
| This journal is © The Royal Society of Chemistry 2008 |
Click here to see how this site uses Cookies. View our privacy policy here.