Intramolecular direct arylation in an A,C-functionalized calix[4]arene†
Olaf G.
Barton
a,
B.
Neumann
b,
H.-G.
Stammler
b and
Jochen
Mattay
*a
aOrganische Chemie I, Universität Bielefeld, Universitätsstr. 25, 33501 Bielefeld, Germany. E-mail: oc1jm@uni-bielefeld.de; Fax: +49 521 106 6417; Tel: +49 521 106 2072
bAbteilung für Röntgenstrukturanalyse, Anorganische Chemie II, Universität Bielefeld, Universitätsstr. 25, 33501 Bielefeld, Germany
First published on 16th November 2007
Abstract
A recently developed efficient method for intramolecular direct arylation is employed on a doubly functionalized calix[4]arene fixed in the cone conformation. The reaction takes place in high yield leading to meta substituted calix[4]arenes. The functionalities are located at two opposite aromatic rings and the two possible diastereomers 1a and 1b are obtained in a 1 : 1 ratio. Full sets of data including crystal structures for both isomers are presented. The NMR data reveal that even at temperatures up to 120 °C both isomers are fixed in a flattened cone conformation with the substituted aromatic units pointing outwards.
Introduction
Phenolcalixarenes are well-established aromatic macrocycles.1 Their electron-rich aromatic systems provide good conditions for aromatic electrophilic substitution reactions with a regiochemistry governed by the para directing effect of the activating phenol group, whereas the ortho positions are blocked by the methylene moieties bridging the aromatic units. Therefore the vast majority of phenolcalixarenes functionalized at the wide rim are substituted at the para position. However, phenolcalixarenes functionalized at the meta position are rare. Of special interest are systems with fused rings because they should expand the calixarene cavity. So far only calix[4]arenes with the expansion of one of the four aromatic units have been described. These systems are inherently chiral, which explains the interest in developing their chemistry. The first system of that kind was synthesized in 1996 by transforming a monoformylcalix[4]arene into a naphthalene-containing calix[4]arene.2 Recently, inherently chiral calix[4]quinolines were prepared starting from monoaminocalix[4]arenes.3 A ring expansion employing metal coordination was achieved by the complexation of MnIII and UO2 with salen calix[4]arene ligands, which were generated from meta-formylated monohydroxycalix[4]arenes.4 Very recently, oxidative photocyclization on monostyrylcalix[4]arenes resulting in the formation of phenanthrene-containing calix[4]arenes was investigated.5In this paper, we present the first application of intramolecular direct arylation to a calixarene system. For this purpose, we adopted a procedure developed by Fagnou et al.6 As a substrate we used the doubly functionalized calix[4]arene 2 fixed in the cone conformation (Scheme 1). The reaction centres are located at two opposite aromatic rings, the remaining two aromatic rings bear no functional groups. Therefore ring closure could lead to two diastereomers 1a and 1b. In the achiral Cs-symmetric isomer 1a both newly formed rings reside near the same unfunctionalized aromatic unit B. In the C2-symmetric isomer 1b each of the newly formed rings points to a different unfunctionalized aromatic unit. 1b represents a racemic mixture of inherently chiral calixarenes . We were interested in the question of whether the first ring closure directs the orientation of the second ring closure. In that case one of the isomers 1a or 1b should be formed in excess.
 | ||
| Scheme 1 Synthesis of 1a and 1b; indices of protons according to calixarene nomenclature are given in the experimental section. | ||
Results and discussion
Synthesis
The synthetic route to 1a and 1b is outlined in Scheme 1. It starts with the dibromocalix[4]arene 3 which could be prepared according to reported protocols.7 Compound 3 is fixed in a cone conformation due to O-alkylation of the phenolic groups with propyl groups which are bulky enough to prevent inversion of the aromatics.8 Lithiation, arylboronate formation and oxidative carbon–boron bond cleavage led to the formation of dihydroxycalix[4]arene 4 in a yield of 85%. This transformation has been reported before, however without experimental data.9 Here, we present an experimental procedure with complete characterization. Alkylation of 4 at the phenol groups on the wide rim with benzyl bromide 5via a Williamson ether synthesis with Cs2CO3 in acetone produced dibenzyl ether 2 in a yield of 65%. Subsequent direct arylation using Fagnou’s protocol afforded the meta substituted calix[4]arenes 1a and 1b in a 1 : 1 ratio in 94% yield.6 These diastereomers were separated after careful multiple chromatography . In the first flash column chromatography (silica gel, 95% cyclohexane–5% ethyl acetate), most of the impurities were removed, a second flash column chromatography (75% chloroform–25% cyclohexane) yielded slightly impure 1b, a mixed fraction of 1a and 1b and a fraction containing pure 1a. A final purification step using HPLC (88% cyclohexane–6% chloroform–6% diethyl ether) gave pure 1b. The ratio of the diastereomers was gathered from the masses of the pure fractions and the 1H NMR spectrum of the mixed fraction.NMR studies
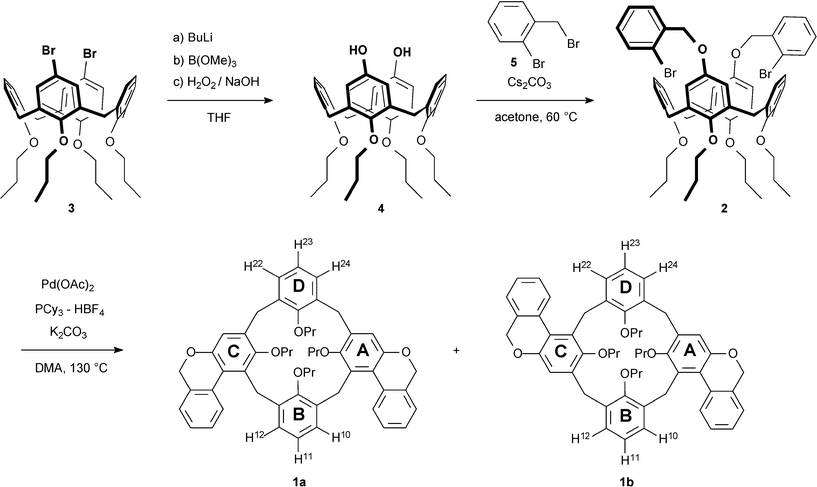
The two isomers isolated by column chromatography could be easily distinguished in 1H as well as 13C NMR spectroscopy. Isomer 1b was first eluted during the purification process. It is expected that its C2 symmetry generates two sets of signals for two sets of propoxy groups in a 1 : 1 ratio. In the 1H NMR spectrum two well separated triplet signals at 1.17 ppm and 0.94 ppm caused by protons of the terminal methyl groups are easily recognized (Fig. 1b). In contrast the Cs symmetric isomer 1a gives rise to three sets of signals for the three sets of propoxy groups in a 1 : 1 : 2 ratio. Again the proton signals of the terminal methyl groups at 1.15 ppm, 1.08 ppm, and 0.89 ppm can be found easily (Fig. 1a). Complete assignment of all proton and carbon signals is done on the basis of 1H–1H COSY, ROESY, HMQC and HMBC experiments. The assignment is facilitated by the fact that the signals of the two types of methylene bridges, close to the fused rings and at a distance from the fused rings, are different from each other. The latter ones show shifts which are common for calix[4]arenes in the cone conformation. In the 1H NMR spectrum of each isomer, two pairs of AB doublets (|2J| = 13–14 Hz) appear. In each case, one pair of doublets lies in the common region, whereas the other pair is noticeably shifted downfield with an extreme shift of the doublet for the equatorial protons. This is explained by the ring current effect of the closely fused aromatics. The corresponding 13C NMR signals are also shifted unusually and appear at a higher field (27.91 ppm for 1b and 27.38 ppm for 1a) than the signals of the bridges remote to the fused rings (31.33 ppm for 1b and 30.88 ppm for 1a), which again fall in the common region for calix[4]arenes adopting the cone conformation (Fig. 2).10 These tentative assignments are confirmed by HMBC experiments, which show coupling between the bridging carbons and the correct meta protons of the phenolic rings. Further support is a NOE contact between the strongly shifted equatorial protons and the bay region proton of the fused ring in each isomer.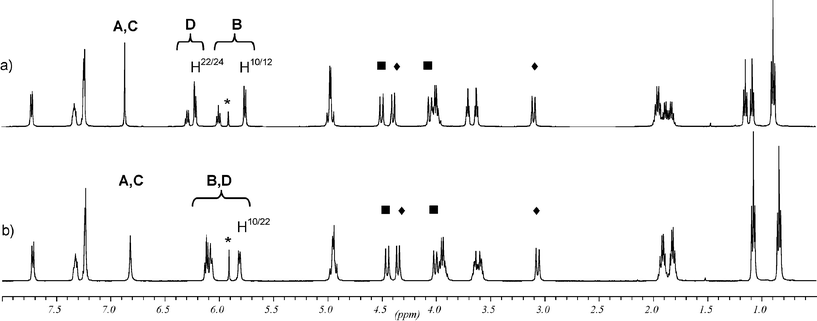 | ||
| Fig. 1 1H NMR spectra of (a) 1a in C2D2Cl4 at 55 °C and (b) 1b in C2D2Cl4 at rt, residual solvent signals are marked with an asterisk, signals for the two pairs of diastereomeric protons of the methylene bridges are marked with symbols (■: close to the annealed rings, ◆: distant to the annealed rings). | ||
 | ||
| Fig. 2 Partial 13C NMR spectra of (a) 1a in C2D2Cl4 at 55 °C and (b) 1b in CD2Cl2 at rt showing the signals for the propoxy groups. The symbols ◆ and ■ indicate signals from the methylene bridge carbons distant and close to the annealed rings, respectively. | ||
Due to the ring current effect of the fused benzene rings, the proton signals of the unfunctionalized aromatics which lie in proximity to the fused benzene rings are shifted upfield. In the case of the C2 symmetric isomer 1b, a highfield shifted doublet at 5.77 ppm can be assigned to the meta protons H10/22 near the fused benzene rings, whereas the signals of the other meta protons H12/24 overlap with resonances for the para protons H11/23 at 6.20–6.15 ppm. In the Cs symmetric isomer 1a signals for the protons of ring B, which is framed by the fused rings, emerge at higher field than the signals for the protons of ring D. The meta protons H10/12 on ring B resonate at 5.76 ppm compared to 6.22 ppm for the meta protons H22/24 on ring D.
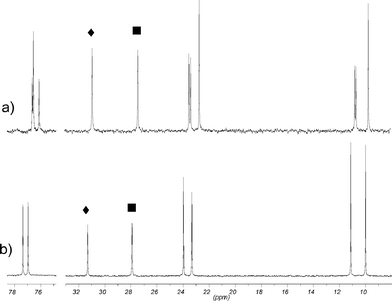
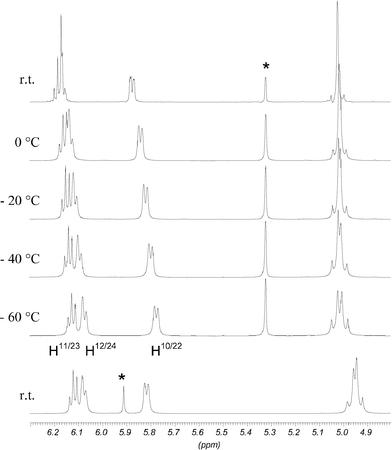
1H NMR experiments at low and high temperatures were carried out to examine the conformational dynamics of both isomers. In the range of −80 °C to room temperature, CD2Cl2 was used as the solvent and experiments from room temperature to 120 °C were conducted in C2D2Cl4. Surprisingly the spectra were basically unchanged from −80 °C to 120 °C. This means that the conformation of the calix[4]arene skeleton is fixed over the whole temperature range. A possible equilibrium between two flattened cone conformations can be ruled out. In that case, resonances for the protons experiencing ring current effects should be shifted markedly, because the shielding and deshielding is sensitive to geometrical changes. In fact all relevant signals stayed the same. A minor change regarding the meta protons H12/24 of rings B and D of the C2 symmetric isomer 1b should not be taken as a hint for any conformational dynamics. Their resonances at low temperature shifted by Δ = 0.11 ppm slightly more downfield than the signals did on average (Δ = 0.05 ppm). This led to a separation from the signals for the para protons H11/23. However, a separation was also seen at room temperature when C2D2Cl4 as solvent was applied (Fig. 3). Another example of small differences caused by the solvent are the resonances of the diastereotopic benzylic methylene protons of isomer 1a. From −80 °C to room temperature in CD2Cl2 they are isochronous, from room temperature to 55 °C in CDCl3 they are slightly split, whereas in C2D2Cl4 at room temperature as well as 120 °C they are clearly divided. All minor changes observed in the experiments at low temperature could be attributed to restrictions of local degrees of freedom. The only minor change displayed in the low temperature spectra of the Cs symmetric isomer 1a concerned the multiplet for the methylene group α to the methyl group of the propoxy chains on rings A and C. This multiplet became broader with new peaks appearing which indicates a splitting of two overlying multiplets consistent with two diastereotopic methylene protons. A similar but more pronounced change is observed in isomer 1b. Here, both methylene groups of the propoxy chains on rings A and C were affected. The multiplets of the diastereotopic protons became completely separated at −40 °C (Fig. 4). Additionally the signals of the diastereotopic benzylic methylene protons started to split up at low temperature (Fig. 3).
 | ||
| Fig. 3 Partial 1H NMR spectra of 1b at variable temperatures in CD2Cl2 (top) and at rt in C2D2Cl4 (bottom), residual solvent signals are marked with asterisks. | ||
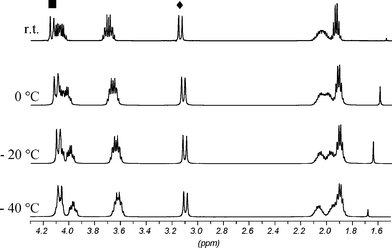 | ||
| Fig. 4 Partial 1H NMR spectra of 1b at variable temperatures in CD2Cl2; marked signals indicate equatorial methylene bridge protons close to (■) and remote from (◆) the annealed rings. | ||
X-Ray analysis
Crystals of isomers 1a and 1b suitable for single crystal X-ray structural analysis were grown from chloroform–ethanol and chloroform–dichloromethane–methanol. Fig. 5 shows the molecular structures in the solid state. No solvent molecules are included. The most prominent feature of both compounds is their flattened cone conformation in which the unsubstituted aromatic rings B and D are bent towards each other whereas the meta substituted phenolic rings A and C are directed away from each other. In case of isomer 1a, the unsubstituted ring D, which is most removed from the fused rings, is almost perpendicular to the plane defined by the four oxygen atoms of the propoxy groups (O4 plane) with a dihedral angle of 89.2°. The unsubstituted ring B lying between the fused rings is tilted towards ring D, its dihedral angle to the O4 plane is 76.1°. The substituent bearing phenolic rings A and C are flattened with dihedral angles of 136.1° and 151.3° to the O4 plane. In isomer 1b, both unsubstituted rings B and D are clearly tilted. The dihedral angles for rings A, B, C and D to the O4 plane are 127.9°, 77.7°, 149.8° and 77.3°, respectively. The phenolic rings A and C together with their substituents can be regarded as bridged biphenyl moieties to which axial chirality can be ascribed. For isomer 1a, the descriptor is Sa for ring A with an inter-ring torsion angle of 28.9° and Ra for ring C with an interplanar angle of 30.5°. The aromatic rings, which were attached to the calix[4]arene scaffold, are turned so that they stand steeper on the O4 plane than the phenolic rings. As a consequence, the bay region carbon atom lies under the plane of the corresponding phenolic ring and the carbon atom of the bridging methylene group is the upmost atom of the calixarene . In contrast, the attached aromatic rings of isomer 1b are arranged more flat to the O4 plane than the phenolic rings. Thus, the bay region carbon atom lies above the plane of the corresponding phenolic ring and the bridging methylene group points sidewards. The descriptor of axial chirality for the enantiomer shown in Fig. 5c and d is Ra for rings A and C with inter-ring torsion angles of 23.1° and 28.0°. An important result of the geometry of both isomers 1a and 1b in the solid state is the fact that there is no cavity large enough to include guest molecules. This can be illustrated by the short distance between the para carbon atoms of the upstanding rings A and C of 4.07 Å and 4.52 Å, respectively.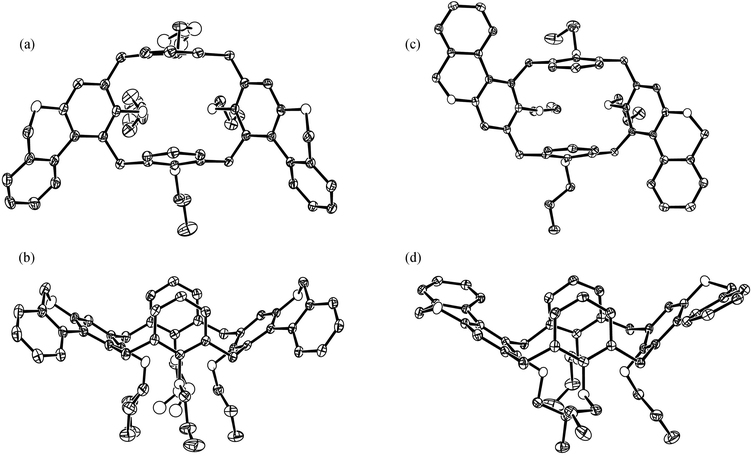 | ||
| Fig. 5 ORTEP view of 1a: (a) top view, (b) side view and 1b: (c) top view, (d) side view. Thermal ellipsoids are drawn at the 50% probability level, disorder in two propoxy groups of 1a is shown, hydrogen atoms are omitted for clarity. | ||
Intramolecular direct arylation in a fourfold functionalized calix[4]arene
The fixed flattened cone conformations in 1a and 1b raises the question of whether a fourfold arylation would be possible or sterically hindered. Therefore, we synthesised the appropriate probe molecule 6 (Scheme 2). It was treated under the same conditions that led successfully to the doubly meta substituted calix[4]arenes. Synthesis began from the known tetra iodinated calix[4]arene 7.11 It was transformed into the benzyl protected phenol derivative 8 under recently reported conditions for Ullmann reactions.12 The benzyl protecting groups were removed under standard conditions yielding the known tetrahydroxycalix[4]arene 9.13 Contrary to dihydroxycalix[4]arene 4, the subsequent alkylation with benzyl bromide 5 was incomplete in boiling acetone and had to be carried out in boiling acetonitrile to achieve completion. Tetrabenzyl ether 6 was subjected to direct arylation. The end of this reaction was indicated, like in the case of the A,C-functionalized calix[4]arene 2, by a colour change from yellow to gray. The crude product mixture and its TLC spots were examined by MALDI-ToF spectrometry. Masses corresponding to two, three and four times intramolecular arylation could be detected, as well as signals indicating one and two times hydrodehalogenation. Three different TLC spots include molecules with masses corresponding to fourfold intramolecular arylation. This is a hint that of the four possible isomers at least three were formed. However, none of the isomers could be isolated. Judging roughly from the HPLC trace these isomers constitute at most 5% of the product mixture.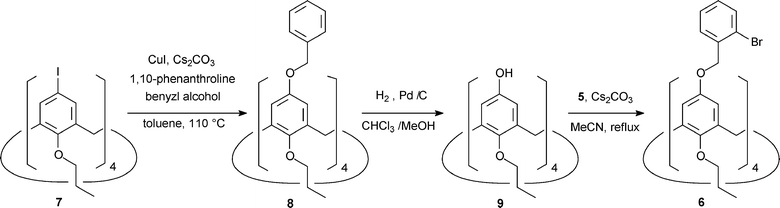 | ||
| Scheme 2 Synthesis of probe molecule 6. | ||
Conclusions
We examined the direct intramolecular arylation of an A,C-functionalized calix[4]arene with respect to the regioselectivity of the reaction. Both possible isomers were formed in a 1 : 1 ratio, with an overall yield of 94%. Therefore, no regioselectivity was observed. From NMR and X-ray analysis, we conclude that both isomers are fixed in a flattened cone conformation. This is a hint that an expansion of the calix[4]arene cavity cannot be achieved by annealing four arene rings in the described way. Accordingly, an experiment with the ABCD-functionalized calix[4]arene led to very poor results (<5% yield). We suppose that the steric crowding which is imposed by the first fused rings is the reason for a strongly diminished reactivity in further intramolecular arylations.‡Experimental
General
Melting points were measured in open capillaries with a Büchi B-540 melting point apparatus and are uncorrected. 1H and 13C NMR spectra were recorded on a Bruker DRX 500, chemical shifts were calibrated to the residual proton and carbon resonance of the solvent (CD2Cl2: δH = 5.32 ppm, δC = 53.80 ppm; CDCl3: δH = 7.24 ppm, δC = 77.00 ppm; C2D2Cl4: δH = 5.91 ppm, δC = 73.70 ppm). J values are given in Hz. MALDI-ToF mass spectra were obtained with a PerSeptive Biosystems Voyager-DE spectrometer using 2,5-dihydroxybenzoic acid as the matrix, a Bruker Esquire 3000 was used to record ESI mass spectra and exact masses were measured on a Bruker APEX III (FT-ICR). Elemental analyses were determined with a Perkin Elmer 240 instrument. X-Ray crystal structures were determined from data collected with a Nonius Kappa CCD area detector diffractometer, using graphite monochromatized Mo-Kα radiation. Analytical thin-layer chromatography was performed on silica gel 60 F254 (Merck), flash column chromatography on silica gel MN 60, 40–63 µm (Macherey-Nagel) and HPLC on a Nucleosil 100-7 silica gel column 250-20 mm (CS-Chromatographie). All reagents were reagent grade quality and used as received from commercial suppliers. Calix[4]arenes 3 and 7 were prepared according to the literature.7,115,17-Dihydroxy-25,26,27,28-tetra(1-propyloxy)calix[4]arene (4)
n-BuLi (1.6 M in hexanes, 2.00 mL, 3.20 mmol, 2.4 eq.) was added dropwise to a stirred solution of di-bromo compound 3 (1.00 g, 1.33 mmol) in THF (50 mL) under argon at −78 °C. The reaction mixture was stirred at that temperature for 20 min and then B(OMe)3 (0.75 mL, 6.7 mmol, 5 eq.) was added. The mixture was allowed to warm to room temperature and stirred for 18 h. Then the reaction flask was placed in an ice-water bath and the reaction mixture was treated with an ice-cold solution of 30% H2O2 (2.7 mL, 26 mmol, 20 eq.) in 3 N aqueous NaOH solution (4.4 mL, 13 mmol, 10 eq.). After stirring the mixture at room temperature for 6 h, H2O (100 mL) was added and the aqueous layer was extracted with a mixture of diethyl ether (100 mL) and dichloromethane (20 mL). The organic layer was washed with H2O (2 × 100 mL) and brine (100 mL) and dried over MgSO4. Chromatographic purification (flash column, silica gel, CHCl3–3% MeOH) and drying under high vacuum afforded a colourless oil (707 mg, 1.13 mmol, 85%). Found: C, 76.34; H, 7.73. C40H48O6 requires C, 76.89; H, 7.74%; δH(500 MHz, CDCl3) 7.02 (4 H, d, J 7.5), 6.81 (2 H, t, J 7.5), 6.29 (2 H, s), 5.53 (4 H, s), 4.39 (4 H, d, J 13.2), 3.96 (4 H, t, J 8.2), 3.61 (4 H, t, J 6.6), 3.06 (4 H, d, J 13.2), 1.94–1.80 (8 H, m), 1.09 (6 H, t, J 7.4), 0.84 (6 H, t, J 7.5); δC(125 MHz, CDCl3) 160.0, 149.8, 149.2, 136.7, 134.6, 128.9, 121.8, 114.2, 76.9, 76.4, 31.1, 23.4, 22.9, 10.8, 9.76; m/z (MALDI-ToF) 624 (M˙+), 647 ([M + Na]+), 663 ([M + K]+); ESI-HRMS: m/z calc. for C40H52NO6: 642.37891 ([M + NH4]+); found: 642.37820; for C40H48NaO6: 647.33431 ([M + Na]+); found: 647.33439.For crystallisation the oil was dissolved in dichloromethane. Under heating, MeOH was added and dichloromethane was allowed to evaporate. After cooling, colourless small needles were collected and dried under high vacuum (217 mg, yield not optimised; according to 1H NMR spectroscopy no solvent molecules included), mp 265 °C.
5,17-Di(2-bromo-benzyloxy)-25,26,27,28-tetra(1-propyloxy)calix[4]arene (2)
A mixture of dihydroxy-calixarene 4 (379 mg, 0.606 mmol), 2-bromobenzyl bromide (333 mg, 1.33 mmol) and caesium carbonate (433 mg, 1.33 mmol) in acetone (25 mL) was refluxed for 24 h. After cooling, the mixture was partitioned between water (100 mL) and diethyl ether (100 mL). The organic layer was washed with water (two times 100 mL) and brine (100 mL), dried over MgSO4 and concentrated in vacuo. Chromatographic purification (flash column, silica gel, 50% cyclohexane–50% chloroform) and drying under high vacuum afforded a colourless solid (378 mg, 0.393 mmol, 65%). Mp 58 °C; found: C, 67.32; H, 6.19. C54H58Br2O6 requires C, 67.36; H, 6.07; Br, 16.60; O, 9.97%; δH(500 MHz, CDCl3) 7.47 (2 H, dd, J 1.1, 7.9), 7.38 (2 H, dd, J 1.2, 7.5), 7.19 (2 H, dt, J 1.0, 7.5), 7.06 (2 H, dt, J 1.7, 7.6), 6.66 (4 H, d, J 7.2), 6.61–6.58 (2 H, m), 6.26 (4 H, s), 4.76 (4 H, s), 4.43 (4 H, d, J 13.3), 3.86 (4 H, t, J 7.5), 3.78 (4 H, t, J 7.44), 3.10 (4 H, d, J 13.3), 1.97–1.87 (8 H, m), 1.01–0.97 (12 H, m); δC(125 MHz, CDCl3) 156.6, 153.0, 150.9, 137.0, 135.7, 135.0, 132.2, 128.6, 128.2, 127.3, 122.1, 121.8, 114.5, 76.8, 76.7, 69.7, 31.2, 23.21, 23.19, 10.4, 10.3; m/z (MALDI-ToF) 983 ([M + Na]+), 999 ([M + K]+), 1093 ([M + Cs]+).Isomers 1a and 1b
A mixture of calixarene 2 (190 mg, 0.197 mmol) and potassium carbonate (109 mg, 0.789 mmol) in N,N-dimethylacetamide (DMA, 2 mL) in a screw-cap vial equipped with a magnetic stirrer was carefully degassed and argon-saturated by two freeze-and-pump cycles. After the addition of Pd(OAc)2 (4.4 mg, 0.020 mmol) and PCy3–HBF4 (14.5 mg, 0.0394 mmol), two more degassing cycles were carried out. The reaction mixture was heated to 130 °C for 24 h. At the end of the reaction the colour had changed from yellow to grey. After cooling to room temperature, the mixture was partitioned between water (100 mL) and diethyl ether (100 mL)–dichloromethane (20 mL). The organic layer was washed with water (100 mL) and brine (100 mL), dried over MgSO4 and concentrated in vacuo. The first chromatographic purification (flash column, silica gel, 95% cyclohexane–5% ethyl acetate) gave a mixture of three compounds. Collection of the last band of a second chromatography (flash column, silica gel, 75% chloroform–25% cyclohexane) and drying under high vacuum afforded isomer 1a as a colourless solid (53.2 mg), crystals suitable for single crystal X-ray structural analysis were grown from chloroform–ethanol. A mixed fraction of 1a and 1b was concentrated in vacuo and dried under high vacuum, the ratio of isomers was determined from 1H NMR spectroscopy (22.5 mg 1a and 3.0 mg 1b). Purification of the residual material by HPLC (88% cyclohexane–6% chloroform–6% diethyl ether) and drying under high vacuum yielded isomer 1b (70.4 mg), and crystals suitable for single crystal X-ray structural analysis were grown from chloroform–dichloromethane–methanol.Isomer 1a (75.7 mg, 0.0944 mmol, 48%)
Mp 282 °C (decomp.); found: C, 80.68; H, 7.09. C54H56O6 requires C, 80.97; H, 7.05%; δH(500 MHz, CDCl3) 7.72 (2 H, d, J 7.5, H6′/13′), 7.38–7.34 (2 H, m, H5′/12′), 7.31–7.26 (4 H, m, H3′/10′, H4′/11′), 6.88 (2 H, s, H4/18), 6.33–6.31 (1 H, m, H23), 6.26 (2 H, d, J 7.5, H22/24), 6.00 (1 H, t, J 7.5, H11), 5.73 (2 H, d, J 7.5, H10/12), 5.01 (2 H, d, J 13.0, H1′/8′), 4.99 (2 H, d, J 13.0, H1′/8′), 4.53 (2 H, d, J 14.4, H8ax./14ax.), 4.42 (2 H, d, J 13.3, H2ax./20ax.), 4.08 (2 H, d, J 13.6, H8eq./14eq.), 4.05 (4 H, m, O–CH2 (A/C)), 3.71 (2 H, t, J 6.6, O–CH2 (B)), 3.64 (2 H, t, J 6.6, O–CH2 (D)), 3.14 (2 H, d, J 13.4, H2eq./20eq.), 2.07–1.97 (4 H, m, CH2 (A/C)), 1.96–1.91 (2 H, m, CH2 (B)), 1.90–1.84 (2 H, m, CH2 (D)), 1.19 (3 H, t, J 7.5, CH3 (B)), 1.11 (3 H, t, J 7.5, CH3 (D)), 0.91 (6 H, t, J 7.5, CH3 (A/C)); δC(125 MHz, C2D2Cl4, 55 °C) 155.0 (C27), 154.9 (C25), 154.3 (C26/28), 150.6 (C5/17), 137.7 (C3/19), 134.3 (C7/15), 133.6 (C9/13, C2′/9′), 132.3 (C1/21), 130.8 (C7′/14′), 127.8 (C5′/12′), 127.1 (C22/24), 126.4 (C4′/11′), 126.3 (C10/12, C6′/13′), 124.9 (C3′/10′), 122.7 (C6/16), 122.4 (C11), 122.2 (C23), 116.5 (C4/18), 76.6 (O–CH2 (D)), 76.5 (O–CH2 (A/C)), 76.1 (O–CH2 (B)), 69.3 (C1′/8′), 30.9 (C2/20), 27.4 (C8/14), 23.5 (CH2 (B)), 23.3 (CH2 (D)), 22.7 (CH2 (A/C)), 10.8 (CH3 (B)), 10.7 (CH3 (D)), 9.7 (CH3 (A/C)); m/z (ESI, CHCl3–MeOH) 801.4 ([M + H]+).
Isomer 1b (73.4 mg, 0.0916 mmol, 46%)
Mp 282 °C (decomp.); found: C, 80.86; H, 7.13. C54H56O6 requires C, 80.97; H, 7.05%; δH(500 MHz, CD2Cl2) 7.77 (2 H, d, J 7.8, H6′/13′), 7.42–7.37 (2 H, m, H5′/12′), 7.31–7.30 (4H, m, H3′/10′, H4′/11′), 6.88 (2 H, s, H4/16), 6.20–6.15 (4 H, m. H11/23, H12/24), 5.87 (2 H, dd, J 2.2, 6.9, H10/22), 5.036 (2 H, d, J 12.7, H1′/8′), 5.020 (2 H, d, J 12.7, H1′/8′), 4.55 (2 H, d, J 14.1, H8ax./20ax.), 4.45 (2 H, d, J 13.5, H2ax./14ax.), 4.14 (2 H, d, J 14.3, H8eq./20eq.), 4.13–4.03 (4 H, m, O–CH2 (A/C)), 3.74–3.65 (4 H, m, O–CH2 (B/D)), 3.14 (2 H, d, J 13.6, H2eq./14eq.), 2.12–1.98 (4 H, m, CH2 (A/C)), 1.97–1.90 (4 H, m, CH2 (B/D)), 1.17 (6 H, t, J 7.4, CH3 (B/D)), 0.94 (6 H, t, J 7.5, CH3 (A/C)); δC(125 MHz, C2D2Cl4) 155.5 (C25/27), 155.1 (C26/28), 151.3 (C5/17), 138.3 (C3/15), 134.9 (C7/19), 134.33 (C9/21), 134.28 (C2′/9′), 132.9 (C1/13), 131.4 (C7′/14′), 128.2 (C5′/12′), 127.2 (C12/24), 127.0 (C10/22, C4′/11′), 126.9 (C6′/13′), 125.3 (C3′/10′), 123.3 (C6/18), 122.8 (C11/23), 117.0 (C4/16), 77.4 (O–CH2 (A/C)), 77.0 (O–CH2 (B/D)), 69.8 (C1′/8′), 31.3 (C2/14), 27.9 (C8/20), 24.0 (CH2 (B/D)), 23.3 (CH2 (A/C)), 11.1 (CH3 (B/D)), 10.0 (CH3 (A/C)); m/z (ESI, CHCl3–MeOH) 801.4 ([M + H]+).
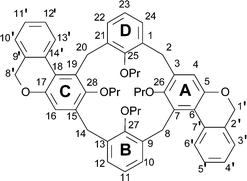
5,11,17,23-Tetrabenzyloxy-25,26,27,28-tetra(1-propyloxy)calix[4]arene (8)
Tetraiodocalix[4]arene 7 (547 mg, 0.500 mmol) was combined with copper(I) iodide (38 mg, 0.20 mmol), 1,10-phenanthroline (76 mg, 0.42 mmol), caesium carbonate (1.30 g, 4.00 mmol), benzyl alcohol (4.33 g, 40.0 mmol) and toluene (4.1 mL). The reaction tube was sealed and the mixture was stirred at 110 °C for 48 h. The resulting suspension was cooled to room temperature and filtered through a pad of silica gel, eluting with diethyl ether. The filtrate was concentrated in vacuo. Purification of the residue by flash chromatography (silica gel, 95–90% cyclohexane, 5–10% ethyl acetate) and subsequent trituration with dichloromethane–methanol provided a colourless solid (173 mg, 0.170 mmol, 34%). Mp 151 °C; found: C, 80.22; H, 7.25; C68H72O8 requires C, 80.28; H, 7.13%; δH(500 MHz, CDCl3) 7.33–7.27 (16 H, m), 7.24–7.21 (4 H, m), 6.30 (8 H, s), 4.74 (8 H, s), 4.41 (4 H, d, J 13.2), 3.77 (8 H, t, J 7.5), 3.03 (4 H, d, J 13.2), 1.94–1.87 (8 H, m), 0.97 (12 H, t, J 7.5); δC(125 MHz, CDCl3) 153.5, 150.8, 137.8, 135.6, 128.4, 127.6, 127.3, 114.2, 76.8, 70.2, 31.4, 23.1, 10.3; m/z (ESI, CHCl3–MeOH) 1017.4 ([M + H]+).5,11,17,23-Tetra(2-bromo-benzyloxy)-25,26,27,28-tetra(1-propyloxy)calix[4]arene (6)
A solution of calix[4]arene 8 (171 mg, 0.168 mmol) in CHCl3–MeOH (1 : 1, 50 mL) was subjected to hydrogenolysis over 10% palladium (134 mg) on activated carbon and under a high pressure of H2 (2 bar) for 20 h. The mixture was filtered over celite, the solid was washed with CHCl3–MeOH (1 : 1) and the combined filtrate was concentrated under reduced pressure to afford 5,11,17,23-tetrahydroxy-25,26,27,28-tetra(1-propyloxy)calix[4]arene9 which was used in the following step without further purification. A mixture of crude tetrahydroxycalix[4]arene 9, 2-bromobenzyl bromide (253 mg, 1.01 mmol) and caesium carbonate (331 mg, 1.01 mmol) in acetonitrile (25 mL) was refluxed for 24 h. After cooling, the mixture was partitioned between water (100 mL) and diethyl ether (100 mL). The organic layer was washed with water (two times 100 mL) and brine (100 mL), dried over MgSO4 and concentrated in vacuo. Purification by flash column chromatography (silica gel, 95% cyclohexane–5% ethyl acetate), HPLC (80% chloroform–20% cyclohexane) and drying under high vacuum afforded a colourless oil (138 mg, 0.103 mmol, 61% over two steps). Found: C, 61.27; H, 5.25; C68H68Br4O8 requires C, 61.28; H, 5.14%; δH(500 MHz, CDCl3) 7.45 (4 H, dd, J 1.3, 8.0), 7.42 (4 H, dd, J 1.5, 7.6), 7.21 (4 H, dt, J 1.3, 7.5), 7.04 (4 H, dt, J 1.8, 7.7), 6.32 (8 H, s), 4.77 (8 H, s), 4.41 (4 H, d, J 13.2), 3.77 (8 H, t, J 7.2), 3.05 (4 H, d, J 13.2), 1.94–1.87 (8 H, m), 0.97 (12 H, t, J 7.5); δC(125 MHz, CDCl3) 153.2, 150.9, 137.0, 135.6, 132.2, 128.6, 128.5, 127.3, 121.7, 114.3, 76.9, 69.7, 31.4, 23.2, 10.3; m/z (ESI, CHCl3–MeOH) 1333 [M + H]+.Direct intramolecular arylation of 6
A mixture of calixarene 6 (138 mg, 0.104 mmol) and potassium carbonate (115 mg, 0.828 mmol) in N,N-dimethylacetamide (DMA, 2.8 mL) in a screw-cap vial equipped with a magnetic stirrer was carefully degassed and argon-saturated by two freeze-and-pump cycles. After the addition of Pd(OAc)2 (4.7 mg, 0.021 mmol) and PCy3–HBF4 (15.3 mg, 0.0414 mmol), two more degassing cycles were carried out. The reaction mixture was heated to 130 °C. After 24 h, its colour had changed from yellow to brown and after a further 12 h to black. Subsequently it was cooled to room temperature and the mixture was partitioned between water (100 mL) and diethyl ether (100 mL)–dichloromethane (20 mL). The organic layer was washed with water (100 mL) and brine (100 mL), dried over MgSO4 and concentrated in vacuo.Crystal structure determination§
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 011 reflections measured, 9820 unique (Rint = 0.056) which were used in all calculations. The final wR(F2) was 0.1447 (all data). CCDC 657956.
011 reflections measured, 9820 unique (Rint = 0.056) which were used in all calculations. The final wR(F2) was 0.1447 (all data). CCDC 657956.
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 2, µ(Mo-Kα) = 0.080 mm−1, 59242 reflections measured, 12396 unique (Rint = 0.068) which were used in all calculations. The final wR(F2) was 0.1271 (all data). CCDC 657957.
, Z = 2, µ(Mo-Kα) = 0.080 mm−1, 59242 reflections measured, 12396 unique (Rint = 0.068) which were used in all calculations. The final wR(F2) was 0.1271 (all data). CCDC 657957.
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG). We thank Bayer Industry Services for the gift of chemicals. O. G. B. thanks Dr. Tina Kasper for discussing the manuscript.References
- M. Vysotsky, C. Schmidt and V. Böhmer, Chirality in Calixarenes and Calixarene Assemblies in Advances in Supramolecular Chemistry, ed. G. W. Gockel, JAI Press, Stamford, 2001, vol. 7, pp. 139–233 Search PubMed; C. D. Gutsche, Calixarenes Revisited, The Royal Society of Chemistry, Cambridge, 1998 Search PubMed; L. Mandolini and R. Ungaro, Calixarenes in Action, Imperial College Press, London, 2000 Search PubMed.
- A. Ikeda, M. Yoshimura, P. Lhotak and S. Shinkai, J. Chem. Soc., Perkin Trans. 1, 1996, 1945 RSC.
- R. Miao, Q. Zheng, C. Chen and Z. Huang, J. Org. Chem., 2005, 70, 7662 CrossRef CAS.
- M. E. Amato, F. P. Ballistreri, A. Pappalardo, G. A. Tomaselli, R. M. Toscano and D. J. Williams, Eur. J. Org. Chem., 2005, 3562 CrossRef CAS.
- M. Mastalerz, W. Hüggenberg and G. Dyker, Eur. J. Org. Chem., 2006, 3977 CrossRef CAS.
- L. Campeau, M. Parisien, A. Jean and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 581 CrossRef CAS; L. Campeau, D. R. Stuart and K. Fagnou, Aldrichimica Acta, 2007, 40, 35.
- A. Casnati, M. Fochi, P. Minari, A. Pochini, M. Reggiani, R. Ungaro and D. N. Reinhoudt, Gazz. Chim. Ital., 1996, 126, 99 CAS; R. H. Vreekamp, W. Verboom and D. N. Reinhoudt, Recl. Trav. Chim. Pays-Bas, 1996, 115, 363 CAS.
- K. Iwamoto, K. Araki and S. Shinkai, J. Org. Chem., 1991, 56, 4955 CrossRef CAS.
- K. Araki and H. Hajashida, Chem. Lett., 2000, 29, 20 CrossRef.
- C. Jaime, J. de Mendoza, P. Prados, P. M. Nieto and C. Sanchez, J. Org. Chem., 1991, 56, 3372 CrossRef CAS.
- A. Arduini, G. Giorgi, A. Pochini, A. Secci and F. Ugozzoli, J. Org. Chem., 2001, 66, 8302 CrossRef CAS.
- M. Wolter, G. Nordmann, G. E. Job and S. L. Buchwald, Org. Lett., 2002, 4, 973 CrossRef CAS.
- A. Arduini, L. Mirone, D. Paganuzzi, A. Pinalli and A. Pochini, Tetrahedron, 1996, 52, 6011 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra of all isolated compounds. See DOI: 10.1039/b713357j |
| ‡ In a preliminary experiment similar to ref. 5, we conducted oxidative photocyclization under standard diluted conditions with an ABCD styryl functionalized calix[4]arene. Mass analysis of the crude product mixture indicated that the reaction took place on only two positions. This result further supports the assumption that building up rings with a phenanthrene connectivity comprising the calix[4]arene para and meta positions is strongly sterically hindered. |
| § CCDC 657956 and 657957. For crystallographic data in CIF or other electronic format, see DOI: 10.1039/b713357j |
| This journal is © The Royal Society of Chemistry 2008 |