Synthetic nucleic acid secondary structures containing the four stereoisomers of 1,4-bis(thymine-1-yl)butane-2,3-diol†
Mikkel S.
Christensen
ab,
Andrew D.
Bond
b and
Poul
Nielsen
*ab
aNucleic Acid Center‡, University of Southern Denmark, 5230, Odense M, Denmark
bDepartment of Physics and Chemistry, University of Southern Denmark, 5230, Odense M, Denmark. E-mail: pon@ifk.sdu.dk; Fax: +45 66158780; Tel: +45 65502565
First published on 7th November 2007
Abstract
The four stereoisomers of the double-headed acyclic nucleoside 1,4-bis(thymine-1-yl)butane-2,3-diol were incorporated in the central position of four 13-mer oligonucleotides. The phosphoramidite building blocks were synthesized in four or six steps from either D- or L-2,3-O-isopropylidenethreitol. Two epimeric and fully deprotected double-headed nucleosides were analyzed by X-ray crystallography. The incorporation into oligonucleotides was hampered by steric hindrance and formation of a cyclic phosphate. The use of pyridinium chloride as the activator and a kinetic analysis based on 31P NMR of the coupling and detritylation processes led to improved yields of the oligonucleotides. In comparison with the (S)-GNA monomer, one of the four stereoisomers was found to show a similar destabilization of a DNA duplex, indicating that the additional base can be introduced without a thermal penalty. Another stereoisomer was found to induce a thermal stabilization of a DNA:RNA three-way junction. Thus, the stereochemistry of this acyclic double-headed nucleoside motif is important, indicating potential for the design of artificial nucleic acid secondary structures.
Introduction
Synthetic nucleic acid chemistry has been predominantly motivated by the potential of selective nucleic acid based therapeutics using gene silencing approaches.1,2 Nevertheless, new perspectives appear within nucleic acid nanobioscience and nanoscale engineering.3–5 The synthetic design is based on the predictable Watson–Crick base pairing capacity of nucleic acids. Simple duplexes built with canonical base pairing, however, span quite a restricted set of possible structure types. Nucleic acid secondary structures are envisioned to be among the structural elements that may form the foundation of bottom-up constructs within nanotechnology.5 Thus, within synthetic nucleic acid chemistry, there has been a drive to expand the structural diversity by mimicking natural secondary structures other than duplexes, such as bulges and three-way junctions, through application of synthetic nucleoside analogs.6,7 In this line of research, non-conventional nucleosides bearing two nucleobases (called double-headed nucleosides) have been synthesized, to give nucleic acids that provide an extra recognition possibility for either interstrand or intrastrand communication.8–11Nucleosides based on the natural 2′-deoxyribonucleoside structure but with an additional nucleobase attached in different positions have been studied (Fig. 1). Thus, we introduced the two double-headed nucleosides 1 and 2 with the additional thymine positioned in either the 2′- or the 5′-position,9,11 whereas Herdewijn and co-workers introduced 3 with either an additional thymine or an adenine in the 4′-position.10 Furthermore, a simpler acyclic double-headed nucleoside 4 has been studied.8 In our studies, oligodeoxynucleotides (ODNs) containing 1 or 2 were used to form bulged and three-way junction structures. In other studies, we have approached these structures with more complicated cyclic dinucleotides,12,13 but the simpler constructs were synthetically more appealing and, in fact, more promising. Thus, 1 incorporated in the branching point of a nucleic acid three-way junction increased the thermal stability of this significantly.9 The double-headed nucleosides 3 and 4 were mainly studied for developing interstrand communication between complementary nucleic acid sequences through base–base H-bond interaction on the duplex surface.8,10 On the other hand, a strong base–base stacking interaction in the minor groove was found for two complementary ODNs containing 2 in a zipper motif.11
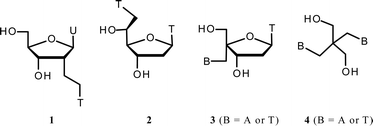 | ||
| Fig. 1 Double-headed nucleosides. U = uracil-1-yl, T = thymin-1-yl, A = adenin-9-yl. | ||
In this paper we wish to report our efforts towards simpler acyclic double-headed nucleoside building blocks mimicking the very simple (S)-GNA (glycerol nucleic acid) structure (Fig. 2) reported earlier by us14 and others,15,16 and elaborated recently by Meggers et al. to fully modified sequences forming a surprisingly stable antiparallel base pairing system.17,18 This system, which can be regarded as an acyclic analog of the TNA (threitol nucleic acid) reported by Eshenmoser and co-workers19 (Fig. 2), has also been shown to form stable cross-pairing with RNA. We decided to elaborate the simple glycerol skeleton to an erythrol/threitol system with two thymines, preparing and incorporating the four different stereoisomers of 1,4-bis(thymine-1-yl)butane-2,3-diol into ODNs (X, Fig. 2) in order to study their effect on DNA and RNA secondary structures. Due to the non-restricted rotation around the C2′–C3′ bond, the glycols are envisioned to conform better to these non-canonical structures than natural nucleosides or, possibly, better than the double-headed nucleosides with the natural 2′-deoxyribofuranose skeleton.
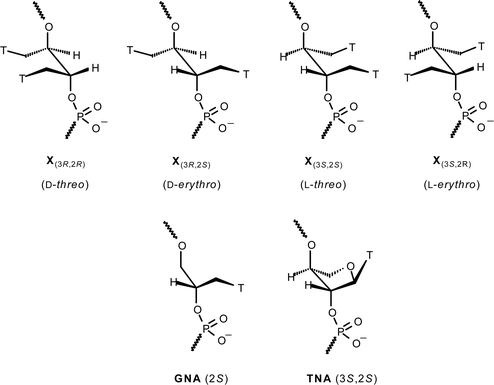 | ||
| Fig. 2 Incorporation of the four stereoisomers of 1,4-bis(thymine-1-yl)butane-2,3-diol into ODNs compared to GNA and TNA. T = thymin-1-yl. | ||
Results and discussion
Chemical synthesis
The four diastereoisomeric double-headed phosphoramidite building blocks were synthesised from the two commercially available enantiopure compounds D- and L-2,3-O-isopropylidene threitol, 5a and 5b, respectively (Scheme 1). A Mitsunobu reaction20 with N3-benzoylated thymine21,22 furnished the regioselective N1-alkylation of thymine. After chromatography, the compounds were taken directly to the next step and hydrolysed by a standard protocol to give the double-headed nucleosides 6a and 6b in a yield of 50 and 36% over the two steps, somewhat hampered by extensive chromatography well known for the Mitsunobu reaction.21 The polarity sense of the nucleosides was fixed by protecting one of the symmetry-equivalent hydroxy groups with a DMT group. Hence, selective DMT-protection by treatment with excess DMTCl in anhydrous pyridine afforded the two enantiopure compounds 7a and 7b constituting a divergence point between the threo- and erythro-configured stereoisomers. With the orientation set by the DMT protection, the projected threo-configured phosphoramidites 8a and 8b were immediately obtained after isolation of the DMT-protected glycols in 74 and 77% yield followed by a standard phosphitylation in 64 and 39% yield. To render an erythro-configuration, an inversion at the C2′ alcohol was required prior to phosphitylation. An approach involving the participation of the neighboring thymine was envisioned. To facilitate the enolic tautomer of the thymine base, compounds 7a and 7b were debenzoylated, and the compounds 9a and 9b were prepared in overall yields of 11 and 18% based on the starting materials 5a and 5b. A leaving group was introduced by mesylation in 41 and 50% yield to give 10a and 10b, and the configuration was subsequently inverted by treatment with aqueous NaOH in ethanol.23 The reaction presumably occurs via the anticipated intermediary O2-anhydro compound, which is subsequently hydrolysed. By analogy, Holy isolated the O2-anhydro compound obtained from the corresponding GNA-monomer 3-dimethoxytrityloxy-2-mesyloxy-1-(thymine-1-yl)propane treated with Et3N under anhydrous conditions.16 The erythritols 11a and 11b were obtained in 84 and 88% yield and finally phosphitylated to give the amidites 12a and 12b in 69 and 64% yield.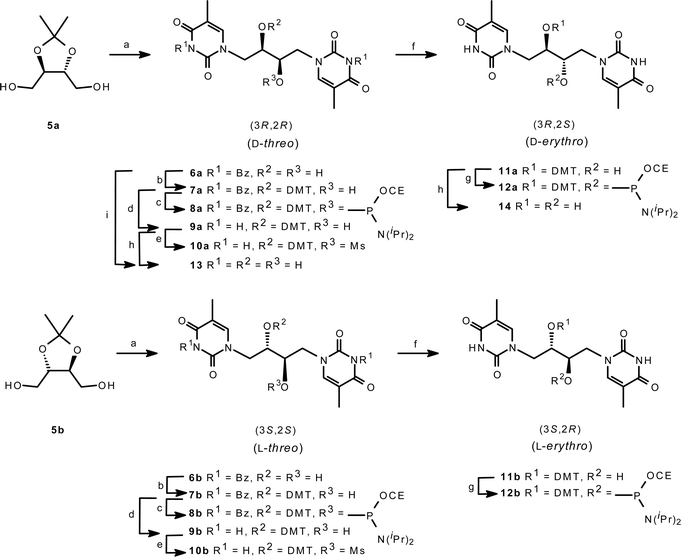 | ||
| Scheme 1 Reagents and conditions: (a) i. 3-N-benzoylthymine, Ph3P, DEAD, THF; ii. TFA–H2O (3 : 1) (6a 50% and 6b 36%); (b) DMTCl, pyr (7a 74% and 7b 77%); (c) CEOPClN(i-Pr)2, DIPEA, CH2Cl2 (8a 64% and 8b 39%); (d) aq. NaOH, dioxane (9a 11% from 5a and 9b 18% from 5b); (e) MsCl, Pyr (10a 41% and 10b 50%); (f) aq. NaOH, EtOH (11a 84% and 11b 88%); (g) CEOPClN(i-Pr)2, DIPEA, DCM (12a 69% and 12b 64%); (h) DCA–SiO2, DCM; (j) 80% aq. AcOH (42% from 5a). CE = 2-Cyanoethyl. DMT = 4,4′-Dimethoxytrityl. | ||
X-Ray crystallography, NMR spectroscopy and molecular modelling
Confirmation of the C2′ inversion was made possible by crystallization of the fully deprotected compounds 13 and 14 obtained from acidic hydrolysis on the analytical scale of compounds 9a and 11a, respectively. Compound 13 has also been prepared directly from 6a. Thus, the D-threo- and the meso-1,4-bis(thymin-1-yl)-2,3-butanediol, 13 and 14, respectively, were crystallized from a DMSO–water system to give the structures shown in Fig. 3. Both molecules were found to form an extended H-bonded network in the crystalline state. Both molecules assume the expected conformation based on gauche interactions between the electronegative substituents around the C1′–C2′ bond. On the other hand, only the threo-configured isomer 13 conforms to this theory for the C2′–C3′ bond as well, while the erythro-configured 14 assumes an antiperiplanar conformation for all its substituents. This could indicate that the anti relationship for the methylene (thymine) groups in this case is more important than the gauche interactions for the electronegative groups.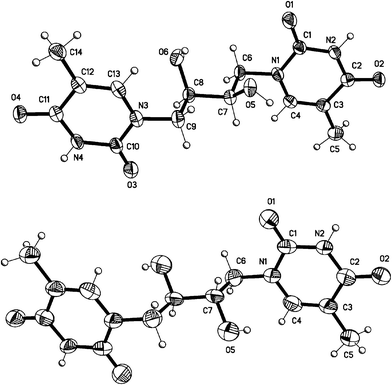 | ||
| Fig. 3 X-Ray crystal structures of the deprotected double-headed nucleosides 13 (top) and 14 (bottom). Displacement ellipsoids are shown at the 50% probability level for non-H atoms. In the meso-isomer 14, unlabelled atoms are related to labelled atoms by a crystallographic center of inversion. | ||
Conformational analysis based on the 1H NMR coupling constants from 13 in DMSO-d6 also indicates an average gauche (+) conformation around the C1′–C2′ bond (J = 2 Hz, 9 Hz and 13 Hz) (see ESI-Fig. 1). For 14, similar coupling constants also indicated this conformation (J ∼ 0 Hz, 8 Hz and 13 Hz). The alternative gauche (–) conformation with a similar coupling constant pattern seems less likely because of the unfavorable steric overlap from the butane moiety and the pyrimidine. It was not possible to discern the conformation around the C2′–C3′ bond from coupling constants. Nevertheless, the conformation around the C1′–C2′ bond discourages the possibility that the thymine bases stack intramolecularly in solution; a conformation that potentially could later impede its ability to fit properly into a duplex environment.
In order to investigate how well the structures adapt to a B-type DNA environment, models with the two isomers X(3S,2S) (L-threo) and X(3R,2S) (D-erythro) were investigated with a 0.5 ns MD-simulation (Fig. 4). Initially, the monomers in the conformations obtained from the X-ray analysis were used as starting points. Due to the 2(S)-configuration, the particular two isomers mimic the (S)-GNA carbon framework; moreover, the X(3R,2S) monomer assumes a conformation that resembles the quasi-2′,3′-trans-diaxial backbone of TNA with its 2′,3′-antiperiplanar backbone. The models in Fig. 4a and b indicate that the crystal state conformations are accommodated well in a B-DNA duplex with the extra base placed on the outside of the helix. Rotation around the C1′–C2′ bond is the only significant adaptation required to maintain base pairing. However, the P–P internucleotide distance fluctuates around 4.9 Å for X(3S,2S) and 5.9 Å for X(3R,2S) compared with 6.5 Å for the normal backbone. The short distance of 4.9 Å may be an incitement for the threo-configured X(3S,2S) to shift the conformation to a 2′,3′-antiperiplanar backbone. Indeed, a similar MD-simulation with X(3S,2S) in this conformation (Fig. 4c) and another with the C3′–C4′ bond also assuming an antiperiplanar conformation (Fig. 4d) give longer P–P internucleotide distances, around 5.3 Å and 5.9 Å, respectively.
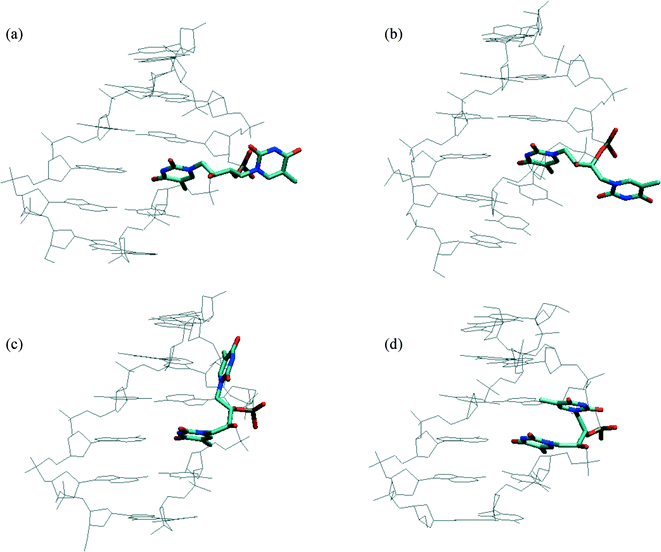 | ||
| Fig. 4 Modelling of two double-headed glycol nucleosides (highlighted) placed in a B-DNA environment. The average structures were obtained from the last 50 ps of 500 ps MD-simulations. (a) The D-erythro-configured isomer (X(3R,2S)) based on the X-ray structure of 14. (b) The L-threo-configured isomer (X(3S,2S)) based on the X-ray structure of 13 (inverted stereochemistry) (c) The same L-threo-configured isomer with an antiperiplanar conformation of the C2′–C3′-bond. (d) The same L-threo-configured isomer with an antiperiplanar conformation of both the C2′–C3′ and the C3′–C4′ bond. | ||
Synthesis of oligodeoxynucleotides
Table 1 presents the oligodeoxynucleotides (ODNs) prepared in this study. To investigate the effect of the four stereoisomeric double-headed nucleosides on DNA and RNA secondary structures, four ODNs incorporating any of the four building blocks at the target site were synthesized (ON1–ON4). An ODN that contains GNA–T allowing for a single base pairing was included in the study (ON5) to investigate the effect of the extra (thymine-1-yl)methyl moiety. Furthermore, an ODN containing just an ethylene glycol linker providing no possibility for base pairing was included (ON6).| ODN sequencesa | MW (found/calc.)b | |
|---|---|---|
| a X refers to the incorporation of 8a, 12a, 8b and 12b, respectively. T refers to the incorporation of the GNA–T phosphoramidite and E to the ethylene glycol phosphoramidite. b MALDI-MS data obtained in negative mode. | ||
| Ref. | 5′-GCTCACTCTCCCA | — |
| ON1 | 5′-GCTCACX(3R,2R)CTCCCA | 3925.7/3925.1 |
| ON2 | 5′-GCTCACX(3R,2S)CTCCCA | 3925.7/3926.1 |
| ON3 | 5′-GCTCACX(3S,2S)CTCCCA | 3925.7/3925.6 |
| ON4 | 5′-GCTCACX(3S,2R)CTCCCA | 3925.7/3926.1 |
| ON5 | 5′-GCTCACT(2S)CTCCCA | 3787.7/3788.5 |
| ON6 | 5′-GCTCACECTCCCA | 3649.7/3645.9 |
The six ODNs were synthesised on an automated DNA synthesiser employing phosphoramidite chemistry and standard coupling procedures with 4,5-dicyanoimidazole as the activator except for the non-conventional nucleosides, for which we used pyridinium chloride (pyrHCl) as the activator. This activator has been shown to give good coupling with ribofuranosyl phosphoramidites containing bulky protecting groups on the 2′-hydroxyl group,24 and in a preliminary experiment, 1H-tetrazole proved inefficient (see ESI-Fig. 2). With pyrHCl as activator, the incorporation of X = 8a into the short model ODN-sequence 5′-TTXTT was achieved with 47–58% efficiency with pyrHCl compared to 8–9% for 1H-tetrazole. For comparison, the synthesis of the corresponding n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) –1 oligomer 5′-TTTT was also performed. Yield assessment was based on analytical ion chromatography (IC) after spiking and correcting the extinction coefficient by the 4 : 6 ratio of thymine bases (see ESI-Fig. 3). Mass analysis of the product indicated that the major byproducts were n–1 oligomers missing thymidines [M – 304] for both sequences. There was no detection of an [M – 400] peak corresponding to an oligomer missing the modified nucleoside X. This indicates that elongation must be retarded only after the incorporation of the modification and also rules out insufficient capping. Thus, pyrHCl was found to be the best activator for these amidites, but the overall coupling yield was still around 50%. Thus, when it was attempted to synthesise a tract of four modifications flanked by unmodified 2′-deoxynucleotides, 5′-CGCT-XXXX-TGCG, with X = 8a using pyrHCl, the isolated yield of the modified oligonucleotide was <2%.
–1 oligomer 5′-TTTT was also performed. Yield assessment was based on analytical ion chromatography (IC) after spiking and correcting the extinction coefficient by the 4 : 6 ratio of thymine bases (see ESI-Fig. 3). Mass analysis of the product indicated that the major byproducts were n–1 oligomers missing thymidines [M – 304] for both sequences. There was no detection of an [M – 400] peak corresponding to an oligomer missing the modified nucleoside X. This indicates that elongation must be retarded only after the incorporation of the modification and also rules out insufficient capping. Thus, pyrHCl was found to be the best activator for these amidites, but the overall coupling yield was still around 50%. Thus, when it was attempted to synthesise a tract of four modifications flanked by unmodified 2′-deoxynucleotides, 5′-CGCT-XXXX-TGCG, with X = 8a using pyrHCl, the isolated yield of the modified oligonucleotide was <2%.
Recently, Zhou et al. reported that the elongation step in the phosphoramidite protocol of the 3-amino analogs of GNA was impaired by a cyclization from attack by the nucleophilic amino group on the P(III) level.25 We studied the similar cyclization as a potential explanation for the low coupling yields of the double-headed phosphoramidites. A 31P-NMR study of the coupling steps between 8b and 3′-O-tert-butyldimethylsilylthymidine did, however, indicate that the steps were fast and quantitative when pyrHCl was employed as activator and t-BuOOH as oxidizer, and indicated no formation of cyclic compounds at the P(III) oxidation state during the coupling step (see ESI-Fig. 4). This result is supported by the observed absence of [M – 400] masses for the aforementioned 5′-TTXTT synthesis. Therefore, the coupling with 8b as the amidite cannot be the reason for poor yields, indicating that the following detritylation or its subsequent coupling with an unmodified phosphoramidite is retarding the elongation.
To investigate the final detritylation reaction, the dinucleotide was resynthesized on a preparative scale and the detritylation step was studied using catalytic amounts (2%) of trichloroacetic acid in CD2Cl2 (Fig. 5). The reaction was very fast (t1/2 <2 min) as evident by 1H-NMR, and it can be concluded that the following coupling must be the weak step. However, the same experiment revealed, after longer reaction time, a new set of peaks appearing in the region of the 31P-NMR around δ = 15 ppm. This indicates a cyclisation to form a five-membered ring at the P(V) oxidation state26 (Fig. 5, see also ESI-Fig. 5). The reaction was conducted with both catalytic and stoichiometric amounts of trichloroacetic acid and in both cases found to have a t1/2 = 2 h independent of the acid concentration. Thus, while cyclisation reaction is easily avoided in the fast detritylation step, it could represent a plausible mechanism by which elongation is retarded in the subsequent coupling step, if the coupling itself is slower.
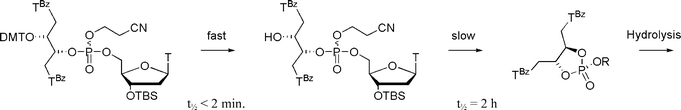 | ||
| Fig. 5 Detritylation of the dinucleotide obtained from 8a with trichloroacetic acid in CD2Cl2 at T = 24 °C. The reactions were monitored with 1H and 31P NMR spectroscopy at 200 MHz (see ESI). | ||
The poor coupling was only observed with the threo-configured phosphoramidites 8a and 8b. The erythro-configured amidites 12a and 12b along with the GNA–T amidite were coupled using pyrHCl as activator with good yields (>90%).
Hybridisation studies
The oligonucleotides were annealed with a set of DNA and RNA strands to give various structural motifs at the complementary site of the modification (Table 2). The formation of a duplex with a complementary DNA strand is seen to be destabilised by the replacement of a thymidine with a double-headed nucleoside as evident from the decrease in melting temperatures of –10 to –13 °C (entry 1). On the other hand, the destabilizations are smaller when compared to the insertion of an abasic moiety (ON6). Among the four different stereoisomers, the double-headed nucleoside in ON2 (containing the D-erythro-configuration, X(3R,2S), see Table 1) does not give any further destabilization, when compared to the GNA-nucleoside in ON5. Thus, in the duplex formed with ON2, the extra (thymin-1-yl)methyl moiety does not seem to affect the structure significantly. This is in contrast with ON3, which has the same C2′ configuration but with the C3′ configuration inverted (L-threo-configuration). The latter duplex is destabilised by about 2 °C compared with the former two, indicating that the orientation of the extra (thymin-1-yl)methyl moiety plays a role in the duplex stability.| Entry | Complements | T m/°C | ΔTm/°C | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Ref. | ON1 | ON2 | ON3 | ON4 | ON5 | ON6 | |||
| a Melting temperatures obtained from the maxima of the first derivatives of the melting curves (A260 vs. temperature) recorded in a medium salt buffer (Na2HPO4 (7.5 mM), NaCl (100 mM), EDTA (0.1 mM), pH 7.0)) using 1.0 µM concentrations of each strand. All Tm values are given as averages of double determinations. ΔTm values are the differences between the determined Tm values and the shown Tm values of the corresponding unmodified reference complexes. ND = Not determined. b Obtained by the addition of MgCl2. c Obtained in a high salt buffer. | |||||||||
| 1 | DNA | 3′-dCGAGTGAGAGGGT-5′ | 52 | –13 | –10 | –12 | –12 | –10 | –16 |
| 2 | DNA, with mismatch | 3′-dCGAGTGTGAGGGT-5′ | — | — | –14 | — | — | — | — |
| 3 | DNA, with mismatch | 3′-dCGAGTGGGAGGGT-5′ | — | — | –10 | — | — | — | — |
| 4 | DNA, with mismatch | 3′-dCGAGTGCGAGGGT-5′ | — | — | –24 | — | — | — | — |
| 5 | DNA, A-bulge | 3′-dCGAGTGAAGAGGGT-5′ | 53 | –19 | –15 | –18 | –16 | ND | –22 |
| 6 | DNA, AG-bulge | 3′-dCGAGTGAGAGAGGGT-5′ | 42 | –10 | –8 | –7 | –8 | ND | –13 |
| 7 | DNA hairpin (with 10 mM MgCl2) | 3′-dCGAGTGACGCGT4CGCGAGAGGGT-5′ | 32 | –1 | –2 | –2 | –1 | ND | –3 |
| 8 | DNA bulged hairpin (with 10 mM MgCl2) | 3′-dCGAGTGACCCGCGT4CGCGAGAGGGT-5′ | 33 | –6 | –5 | –6 | –5 | ND | –7 |
| 9 | RNA | 3′-rCGAGUGAGAGGGU-5′ | 59 | –11 | –8 | –11 | –11 | –5 | –14 |
| 10 | RNA, A-bulge | 3′-rCGAGUGAAGAGGGU-5′ | 60 | –18 | –14 | –15 | –16 | ND | –19 |
| 11 | RNA, AG-bulge | 3′-rCGAGUGAGAGAGGGU-5′ | 49 | –9 | –6 | –7 | –7 | ND | –10 |
| 12 | RNA hairpin | 3′-rCGAGUGACGCGU4CGCGAGAGGGU-5′ | 39 | –3 | –2 | –3 | +2 | ND | –4 |
| (with 5 mM MgCl2)b | 43 | –3 | –5 | –5 | +1 | ND | –4 | ||
| (with 10 mM MgCl2)b | 45 | –3 | –3 | –5 | –1 | ND | –4 | ||
| (with 1 M NaCl)c | 48 | –3 | –3 | –4 | +1 | ND | ND | ||
| (with 10 mM MgCl2 and 1 M NaCl)b,c | 48 | –2 | –3 | –4 | +1 | ND | ND | ||
The model studies (Fig. 4) give some explanation of these results. Considering the structural analogy with TNA–T, for which a crystal structure of its incorporation into a B–DNA duplex is available,27 the orientation of the methylene linker in ON3 would point towards the minor groove in an idealised mimic of the O3′–C3′–C2′–O2′ antiperiplanar conformation found for TNA–T. Thus, the modelling indicates that the extra base in ON3 will either align itself with the minor groove (Fig. 4c) or sit perpendicular to the groove (Fig. 4d) depending on the C3′–C4′ conformation. In contrast, the extra base in ON2 seems to be oriented towards the bulk solvent (Fig. 4a). This could indicate that the slightly larger destabilization observed in the ON3 duplex is partly attributed to a greater perturbation of the minor groove hydration from the presence of the extra thymine base.
A mismatch study (Table 2, entries 2–4) for ON2 shows large discrimination towards cytidine, but no discrimination towards guanosine and low discrimination towards thymidine. Nevertheless, this indicates that base-pairing is taking place between the double-headed nucleotide and the complementary nucleotide.
Despite the slightly smaller destabilization for ON2, the stereochemistry seems in general to be of little importance in the duplex. Insertion of an extra adenosine in the opposite strand (A-bulge) enhances the stereochemical differences and again favors ON2 over the other sequences (entry 5). However, an overall very large destabilization is evident. In the case of structure ON1, the destabilization approaches the result for the abasic analog in ON6.
Introducing an additional G bulge between the adenosines lowers the relative destabilization (entry 6). In this motif, ON3 forms the more stable hybrid followed by ON2 and ON4. However, the mismatch study for the ordinary duplex formed by ON2, showing no discrimination towards guanosine (entry 3), makes it impossible to discern whether or not an A or a G bulge is actually formed.
The next motif comprises a three-way junction with 10 mM MgCl2 added to stabilize the structure (entry 7).9 Here, only a slight destabilization by the unconventional nucleosides is observed. However, this is also evident for the oligonucleotide containing an abasic site at the junction (ON6), indicating that canonical base pairing is not an important structural factor in this motif. As indicated by Seemann and co-workers, it is probably not possible to form all canonical base pairs at the branch point in a DNA three-way junction.28 Hence, the DNA three-way junction is known to display a flexible structure, a trait which may be imparted by incorporating a dinucleotide bulge adjacent to the structural branch point.29,30 Therefore, we employed the same three-way motif with a CC bulge adjacent to the junction to render a less flexible motif (entry 8).11,13 The results suggest, however, that the double-headed modifications do not fit in the motif, as the destabilizations are larger and the differences between the abasic site and the double-headed nucleosides are again of minor importance.
The results obtained for the RNA based motifs are also given in Table 2. With an unbulged RNA complement (entry 9), ON2 forms a duplex, which is more destabilized by approx. –3 °C compared to the duplex formed by ON5 incorporating the GNA thymidine analog. Compared to the native duplex, however, ON2 is less destabilized in an RNA environment than in the DNA environment (–8 °C compared to –10 °C). This is not surprising as GNA has been shown to cross-pair with RNA but not with DNA.17 The effect, however, appears to be less pronounced for the other three stereoisomers in ON1, ON3 and ON4 and the abasic site in ON6. Again, it is particularly interesting that ON2 is observed to form more stable duplexes than ON3 with a difference of 3 °C. This is a clear indication that the orientation of the extra methyl linker moiety at C3′ is important for the accommodation of the extra thymine base in a duplex structure.
While the bulged RNA motifs display destabilized structures without significant stereochemical influence, similar to the DNA congener (entries 10–11), the stability of the RNA three-way junction is observed to be very dependent on the stereochemistry of the double-headed nucleosides (entry 12). Indeed, ON4 stabilizes this RNA motif by +2 °C without MgCl2 whereas the other oligonucleotides destabilize the motif by –2 °C to –3 °C. This property is seen to be true in both medium and high concentrations of NaCl. Although the observed stabilization for ON4 seems to level off with increasing concentration of MgCl2, the differences from the stereochemical relationship are almost constant. Although the stabilization is modest, the fairly large difference (which is dependent on stereochemistry) indicates that this motif is not unduly flexible and that it is possible to target and stabilize the secondary structure with a designed molecule. Indeed, ON4, which shows the best fit into the RNA three-way junction, incorporates a nucleoside that is the mirror image of the nucleoside incorporated into ON2, found to be the better oligonucleotide in simple duplex formation.
Discussion
The synthesis of the four stereoisomeric double-headed nucleosides was fairly simple. The two threo-configured phosphoramidites were obtained in only four steps from cheap commercially available starting materials, and the erythro-configured analogs demanded only two additional steps. On the other hand, the preparation of the latter was based on the so-called anhydro approach, which is only possible with the pyrimidine nucleobases. Therefore, the procedure would not be immediately useful for the preparation of erythro-configured purine analogs. In contrast to the relatively easy preparation of the phosphoramidites, their oligomerisation was a challenging task. The kinetic studies proved that pyrHCl is the preferred activator for these amidites and that the incorporation can be performed with reasonable efficiency. Nevertheless, steric problems in the coupling step and the ring-closure during the detritylation and subsequent coupling step hamper the incorporation of the threo-configured amidites so much that more than single incorporations in the ODNs were impossible. On the other hand, the erythro-configured amidites were more efficiently incorporated, and a direct study of the cooperativity between the double-headed acyclic nucleosides and their GNA counterparts can thus be performed in the near future.Compared to other double-headed nucleosides studied by us and others,8–11 the four stereoisomeric acyclic nucleosides of this study display the most pronounced destabilization of duplexes. In other words, the combination of conformational flexibility, a backbone that is shortened by one carbon atom, and an additional base is not optimal for duplex formation. It is not surprising, however, that an intact 2′-deoxyribose skeleton (as in 1–3) is preferred, as single incorporations of the GNA-monomers into duplexes are also known to induce significant destabilizations.14 For that reason, the effect of additional bases in fully modified GNA-sequences might give completely different results. The different stereochemistries give, however, some differences in the destabilization of duplexes, and the most “GNA-like” analog (as in ON2, Table 2), with the 2′(S)-configuration of GNA and the additional thymine pointing away from the duplex (Fig. 4a), shows almost the same thermal stability as the GNA-sequence (ON5). The study of a 5′-positioned (thymine-1-yl)methyl group using the double-headed nucleoside 2 has recently shown an intriguing interstrand base–base interaction in a DNA-zipper motif.11 A similar effect of the (thymine-1-yl)methyl group of the D-erythro-configured compound 14 in a corresponding GNA-zipper motif might be envisioned.
The study of three-way junctions has demonstrated that it is impossible to obtain large increases in thermal stability by simple and flexible acyclic double-headed nucleosides in the model system studied. On the other hand, the four different stereochemical configurations have demonstrated some influence, as one of them actually gives a slight stabilization of the DNA–RNA three-way junction. However, the three-way junction is not really very stable, and the stabilization obtained with the double-headed nucleoside 1 in the presence of Mg2+ was significantly more powerful. The TWJ stabilization obtained with one of the four stereoisomers indicates an important effect on TWJ structure of one or rather both of the nucleobases.
Conclusion
A set of four diastereomeric 1,4-bis(thymine-1-yl)butane-2,3-diols has been prepared and incorporated into oligonucleotides. In general, the nucleosides destabilize both DNA and RNA duplexes. However, not unexpectedly the nucleosides bearing an 2′(S)-configuration are marginally less destabilising. This is also true for bulged motifs. For the three-way junctions formed with DNA no proper indication of base pairing at the junction was observed. However, in the case of three-way junctions formed with RNA it is found that the stereochemistry of the nucleosides is important, as one of the nucleosides demonstrates a stabilization effect, while the others display a varying degree of destabilization in a consistent manner when varying the Mg2+ and the Na+ concentrations. In essence, these results demonstrate the possibility of targeting and stabilizing nucleic acid secondary structures with suitably designed nucleosides and oligonucleotides.Experimental
Reactions were carried out under an atmosphere of nitrogen when anhydrous solvents were used. Column chromatography was carried out using glass columns with silica gel 60 (0.040–0.063 mm). NMR spectra were recorded on Varian Gemini 2000 spectrometers. NMR spectra were recorded at 300 or 200 MHz for 1H NMR, 75 or 50 MHz for 13C NMR and 121.5 or 81 MHz for 31P NMR. The δ values are in ppm relative to tetramethylsilane as internal standard (for 1H and 13C NMR) and relative to 85% H3PO3 as external standard (for 31P NMR). Assignments of NMR spectra are based on 2D experiments and DEPT. HR MALDI and ESI mass spectra were recorded in positive-ion mode except for the oligonucleotides, which were recorded in negative-ion mode. Optical rotation was recorded on a Perkin Elmer Model 141 polarimeter.Preparation of (2R,3R)-1,4-bis(3-N-benzoylthymin-1-yl)butan-2,3-diol (6a)
3-N-Benzoylthymine (1.6 g, 6.9 mmol), 2,3-O-isopropylidene-D-threitol5a (0.52 g, 3.2 mmol) and triphenylphosphine (2.6 g, 9.9 mmol) were dissolved in anhydrous THF (50 mL) and the solution was stirred at room temperature. When all the compounds had dissolved, the solution was cooled to 0 °C and DEAD (1.5 mL, 10 mmol) was added over 15 minutes. The mixture was stirred for 16 h at room temperature and then concentrated under reduced pressure at 30 °C. The residue was purified by silica gel column chromatography (CHCl3–EtOH 99 : 1 v/v) to give a crude intermediate containing triphenylphosphine oxide. This intermediate was dissolved in a mixture of trifluoracetic acid and water (3 : 1, 20 mL) and the solution was stirred for 4.5 h. The reaction was quenched by the addition of a saturated aqueous solution of NaHCO3 (170 mL) and the mixture was extracted with CHCl3 (4 × 20 mL). The combined organic phases were dried (MgSO4) and the solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography (EtOAc–petrol ether 1 : 1 v/v, to remove triphenylphosphine oxide, and then CHCl3–EtOH 19 : 1 v/v). The residue was finally precipitated from CHCl3 by the slow addition of hexane to give the product 6a as a white powder (0.88 g, 50%). Rf 0.35 (CHCl3–EtOH 9 : 1); δH (300 MHz, DMSO-d6, Me4Si) 7.94–7.91 (4H, m, Ar H), 7.79–7.73 (2H, m, Ar H), 7.69 (2H, d, J = 0.9 Hz, H-6), 7.60–7.55 (4H, m, Ar H), 5.31 (2H, d, J = 5.7 Hz, OH), 3.93–3.65 (6H, m, H-1′, H-1″, H-2′), 1.83 (6H, d, J = 0.9 Hz, 2 × CH3), δC (75 MHz, DMSO-d6, Me4Si) 169.70 (C-4), 162.89 (PhC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 149.52 (C-2), 143.59 (C-6), 135.26, 131.17, 130.13, 129.34 (Ar C), 107.50 (C-5), 68.58 (CH), 51.04 (CH2), 11.76 (CH3); HR MALDI MS m/z 569.1667 ([M + Na]+, C28H26N4O8Na calcd 569.1643).
O), 149.52 (C-2), 143.59 (C-6), 135.26, 131.17, 130.13, 129.34 (Ar C), 107.50 (C-5), 68.58 (CH), 51.04 (CH2), 11.76 (CH3); HR MALDI MS m/z 569.1667 ([M + Na]+, C28H26N4O8Na calcd 569.1643).
Preparation of (2S,3S)-1,4-bis(3-N-benzoylthymin-1-yl)butan-2,3-diol (6b)
Same procedure as above. 2,3-O-Isopropylidene-L-threitol (0.888 g) was used to give the product 6b as a white powder (1.07 g, 36%). NMR data are identical to data for the enantiomer 6a; HR MALDI MS m/z 569.1641 [M + Na]+.Preparation of (2R,3R)-1,4-bis(3-N-benzoylthymin-1-yl)-3-(4,4-dimethoxytrityloxy)butan-2-ol (7a)
The diol 6a (0.77 g, 1.4 mmol) was co-evaporated with anhydrous pyridine (3 × 6 mL) and redissolved in anhydrous pyridine (9 mL). DMTCl (1.2 g, 3.5 mmol) was added and the mixture was stirred for 16 h. Another portion of DMTCl (0.59 g, 1.7 mmol) was added and the mixture was stirred for another 24 h. The reaction mixture was diluted with EtOAc (40 mL) and washed with water (150 mL). The water phase was extracted with EtOAc (40 mL) and the combined organic fractions were dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (CHCl3–petrol ether 1 : 1 v/v containing 0.2% Et3N) to give the product 7a as a powder binding 2.5 equivalents of Et3N (0.89 g, 74%). Rf 0.60 (CHCl3–EtOH 9 : 1); δH (300 MHz, DMSO-d6, Me4Si) 7.92–7.81 (4H, m, Ar H), 7.66–7.59 (2H, m Ar H), 7.51–7.40 (10H, m, Ar H), 7.33–7.23 (4H, m Ar H), 7.15 (1H, d, J = 1.2 Hz, H-6), 6.88–6.84 (4H, m, Ar H), 6.32 (1H, s, H-6), 4.23 (1H, d, J = 13.5 Hz), 3.98 (1H, m), 3.90-3.85 (2H, m), 3.78 (6H, s), 3.68 (1H, dd, J = 3.9, 13.5 Hz), 3.58 (1H, m), 3.29 (1H, dd, J = 3.0, 13.8 Hz), 1.86 (3H, s, CH3), 1.81 (3H, s, CH3). δC (75 MHz, DMSO-d6, Me4Si) 169.20, 168.56 (C-4), 157.85 (Ar C), 150.70, 146.57 (C-2), 143.15, 141.92 (C-6), 136.36, 136.14, 129.60, 127.43, 127.36, 126.19, 112.81 (Ar C), 111.67, 109.93 (C-5), 87.48 (C(Ar)3), 73.17, 69.75 (CH), 55.42 (OCH3), 51.04, 47.00 (CH2), 12.39, 12.36 (CH3).Preparation of (2S,3S)-1,4-bis(3-N-benzoylthymin-1-yl)-3-(4,4-dimethoxytrityloxy)butan-2-ol (7b)
Same procedure as above. Diol 6b (0.13 g) was used to give the product 7b as a powder binding 2.5 equivalents of Et3N (0.16 g, 77%). NMR data are identical to data for the enantiomer 7a.Preparation of (2R,3R)-1,4-bis(3-N-benzoylthymin-1-yl)-3-(4,4-dimethoxytrityloxy)but-2-oxy-β-cyanoethoxy-N,N-diisopropylaminophosphite (8a)
Compound 7a (1.75 g, 2.06 mmol) was dissolved in anhydrous CH2Cl2 (10 mL) and to the solution was added diisopropylethylamine (2.0 mL, 12 mmol) followed by chloro-(2-cyanoethoxy)(diisopropylamino)phosphane (1.0 g, 4.2 mmol). The mixture was stirred at room temperature for 1 h, diluted with EtOAc (50 mL) and washed with brine (4 × 25 mL). The organic phase was dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (EtOAc–Et3N 99 : 1 v/v). The residual compound was dissolved in CH2Cl2 (2 mL) and precipitated into hexane (20 mL) overnight at –20 °C to give the product 8a as a white powder (1.43 g, 64%). Rf 0.66 (EtOAc–Et3N 99 : 1); δP (121 MHz, CDCl3) 152.13, 149.73; HR MALDI MS m/z 1071.4007 ([M + Na]+, C58H62N6O11PNa calcd 1071.4028).Preparation of (2S,3S)-1,4-bis(3-N-benzoylthymin-1-yl)-3-(4,4-dimethoxytrityloxy)but-2-oxy-β-cyanoethoxy-N,N-diisopropylaminophosphite (8b)
Same procedure as above. Compound 7b (125 mg) was used to give the product 8b as a white powder (61 mg, 39%). δP (121 MHz, CDCl3) 152.15, 149.74; HR MALDI MS m/z 1071.4072 [M + Na]+.Preparation of (2R,3R)-1,4-bis(thymin-1-yl)-3-(4,4-dimethoxytrityloxy)butan-2-ol (9a)
3-N-Benzoylthymine (12.80 g, 55.6 mmol), 2,3-O-isopropylidene-D-threitol5a (4.03 g, 24.9 mmol) and triphenylphosphine (23.1 g, 88.2 mmol) were dissolved in anhydrous THF (400 mL). The solution was stirred at rt for 30 min, cooled to 0 °C and a 40% w/w solution of DEAD in toluene (40 mL, 87 mmol) added over 15 min. The mixture was stirred at rt for 16 h and concentrated under reduced pressure. The residue was dissolved in a mixture of trifluoroacetic acid and water (3 : 1 v/v, 100 mL) and stirred for 5 h. The mixture was diluted with water (300 mL) and neutralized by slow addition of Na2CO3 (53 g). The mixture was extracted with CHCl3 (4 × 100 mL), and the combined extracts were dried (MgSO4) and concentrated under reduced pressure. The residue was purified by column chromatography (CHCl3–EtOH 19 : 1 v/v). The residue was dried under vacuum for 16 h and then dissolved in anhydrous pyridine (100 mL). DMTCl (4.9 g, 15 mmol) was added and the mixture was stirred for 24 h; another portion of DMTCl (3.5 g, 10 mmol) was then added and the mixture stirred for another 24 h. The mixture was concentrated under reduced pressure, and the residue was suspended in a mixture of 1,4-dioxane (100 mL) and a 2 M aqueous solution of NaOH (100 mL). The mixture was stirred for 2 d and neutralized by the addition of neat AcOH (12 mL). Brine (100 mL) was added and the mixture was extracted with EtOAc (3 × 100 mL). The combined extracts were dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by column chromatography (EtOAc–petrol ether 1 : 1 v/v containing 0.2 vol% Et3N, then CH2Cl2–MeOH 19 : 1 v/v containing 0.2 vol% Et3N) to give the product 9a as a white powder (1.68 g, 11% over the four steps). Rf 0.36 (CHCl3–EtOH 9 : 1); δH (300 MHz, DMSO-d6, Me4Si) 11.15 (1H, s, NH) 11.12 (1H, s, NH), 7.38–7.08 (10H, m, Ar H and H-6), 6.76–6.69 (4H, Ar H), 5.48 (1H, d J = 4.8 Hz, OH), 4.01-3.81 (3H, m), 3.72 (6H, s, 2 × OCH3), 3.60-3.50 (1H, m), 3.21-3.17 (1H, m), 1.63 (3H, s, CH3), 1.48 (3H, s, CH3); δC (75 MHz, DMSO-d6, Me4Si) 164.27, 164.13 (C-4), 157.85 (Ar C), 150.70, 146.57 (C-2), 143.15, 141.92 (C-6), 136.36, 136.14, 129.60, 127.43, 127.36, 126.19, 112.81 (Ar C), 108.09, 106.96 (C-5), 86.57 (C(Ar)3), 72.41, 67.60 (CH), 54.81 (OCH3), 50.23, 47.59 (CH2), 11.49, 11.44 (CH3); HR ESI MS m/z 663.2768 ([M + Na]+, C35H36N4O8Na calcd 663.2453). [α]25D = +86 (c = 1.00 g mL–1, DMSO-d6).Preparation of (2S,3S)-1,4-bis(thymin-1-yl)-3-(4,4-dimethoxytrityloxy)butan-2-ol (9b)
Same procedure as above. 2,3-O-Isopropylidene-L-threitol5b (3.06 g) was used to give the product 9b as a white powder (2.14 g, 18%). NMR data are identical to data for the enantiomer 9a; HR ESI MS m/z 663.2472 [M + Na]+. [α]25D = –83 (c = 1.00 g mL–1, DMSO-d6).Preparation of (2R,3R)-1,4-bis(3-N-benzoylthymin-1-yl)-3-(4,4-dimethoxytrityloxy)but-2-yl methansulfonate (10a)
Compound 9a (1.38 g, 2.15 mmol) was dissolved in anhydrous pyridine (70 mL), and the reaction mixture was stirred at 0 °C. MsCl (0.50 mL, 6.4 mmol) was added slowly, and the reaction mixture was stirred for 3 h, another portion of MsCl (0.50 mL, 6.4 mmol) added, stirred for 4 h, then added another portion of MsCl (0.50 mL, 6.4 mmol) and stirred for 12 h. The mixture was quenched in water (100 mL) and extracted with CH2Cl2 (4 × 40 mL) The combined extracts were washed with brine (100 mL), dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by column chromatography (CH2Cl2–MeOH 98:2 v/v containing 0.2 vol% Et3N) to give a brown oil, which was dissolved in CH2Cl2 (3 mL) and precipitated into hexane (30 mL) to give the product 10a as a white powder (0.640 g, 41%). Rf 0.33 (CHCl3–EtOH 9 : 1); δH (300 MHz, DMSO-d6, Me4Si) 11.35 (1H, s, N-H), 11.06 (1H, s, N-H), 7.43–7.40 (2H, m, Ar H), 7.31–7.20 (8H, m, Ar H), 7.09 (1H, s, H-6), 6.86–6.80 (4H, m, Ar H), 4.61-4.57 (1H, m), 4.52 (1H, dd, J = 2.1, 14.7 Hz), 4.10-3.91 (3H, m), 3.74 (6H, s, 2 × OCH3), 3.68-3.63 (1H, m), 2.87 (3H, s, CH3SO3), 1.73 (3H, s, CH3), 1.56 (3H, s, CH3); δC (75 MHz, DMSO-d6, Me4Si) 164.19, 164.09 (C-4), 158.40, 158.38 (Ar), 150.94, 150.60 (C-2), 145.36 (Ar), 141.72, 141.61 (C-6), 135.10, 135.02, 130.50, 130.38, 127.93, 127.80, 126.93, 113.21, 113.09 (Ar), 108.54, 108.21 (C-5), 87.73 (C(Ar)3), 77.72, 70.01 (CH), 55.04, 54.92 (OCH3), 47.84, 45.71 (CH2), 38.08 (CH3SO3), 11.90, 11.71 (CH3); HR ESI MS m/z 741.2226 ([M + Na]+, C36H38N4O10SNa calcd 741.2201).Preparation of (2S,3S)-1,4-bis(3-N-benzoylthymin-1-yl)-3-(4,4-dimethoxytrityloxy)but-2-yl methansulfonate (10b)
Same procedure as above. Compound 9b (1.84 g) was used to give the product 10b as a white powder (1.04 g, 50%). NMR data are identical to data for the enantiomer 10a. HR ESI MS m/z 741.2231 [M + Na]+.Preparation of (2S,3R)-1,4-bis(thymin-1-yl)-3-(4,4-dimethoxytrityloxy)butan-2-ol (11a)
Compound 10a (0.421 g, 0.586 mmol) was dissolved in a mixture of EtOH (9 mL) and a 1 M aqueous solution of NaOH (9 mL) and the mixture stirred at reflux for 4 h. After cooling to room temperature, the mixture was neutralized with AcOH (approx. 0.5 mL) and diluted with water (20 mL). The mixture was extracted with CH2Cl2 (4 × 20 mL), and the combined extracts were dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by column chromatography (CH2Cl2–MeOH 98 : 2 v/v containing 0.2 vol% Et3N) to give the product 11a as a white powder (0.314 g, 84%). Rf 0.55 (CHCl3–EtOH 9 : 1); δH (200 MHz, DMSO-d6, Me4Si) 11.15 (1H, s, N-H), 11.12 (1H, s, N-H), 7.38–7.08 (10H, m, Ar H and H-6), 6.76–6.69 (4H, Ar H), 5.48 (1H, d, J = 6.6 Hz, OH), 4.01-3.81 (3H, m), 3.72 (6H, s, 2 × OCH3), 3.60-3.50 (1H, m), 3.21-3.17 (1H, m), 1.63 (3H, s, CH3), 1.48 (3H, s, CH3); δC (50 MHz, DMSO-d6, Me4Si) 164.27, 164.13 (C-4), 157.85 (Ar C), 150.70, 146.57 (C-2), 143.15, 141.92 (C-6), 136.36, 136.14, 129.60, 127.43, 127.36, 126.19, 112.81 (Ar C), 108.09, 106.96 (C-5), 86.57 (C(Ar)3), 72.41, 67.60 (CH), 54.81 (OCH3), 50.23, 47.59 (CH2), 11.49, 11.44 (CH3); HR ESI MS m/z 663.2473 ([M + Na]+, C35H36N4O8Na calcd 663.2453). [α]25D = +107 (c = 1.00 g mL–1, DMSO-d6).Preparation of (2R,3S)-1,4-bis(thymin-1-yl)-3-(4,4-dimethoxytrityloxy)butan-2-ol (11b)
Same procedure as above. Compound 10b (0.91 g) was used to give the product 11b as a white powder (0.71 g, 88%). NMR data are identical to data for the enantiomer 11a. HR ESI MS m/z 663.2442 [M + Na]+. [α]25D = –104 (c = 1.00 g mL–1, DMSO-d6).Preparation of (2S,3R)-1,4-bis(3-N-benzoylthymin-1-yl)-3-(4,4-dimethoxytrityloxy)but-2-oxy-β-cyanoethoxy-N,N-diisopropylaminophosphite (12a)
Compound 11a (0.311 g, 0.486 mmol) was dissolved in anhydrous CH2Cl2 (10 mL) and diisopropylethylamine (0.60 mL), and chloro-2-cyanoethoxydiisopropylaminophosphane (0.30 g, 1.3 mmol) was added. The mixture was stirred at room temperature for 1 h, transferred directly onto a column and purified by chromatography (EtOAc containing 0.2 vol% Et3N) to give the product 12a as a white powder (0.281 g, 69%). δP (121 MHz, CDCl3) 151.29, 151.17; HR ESI MS m/z 863.3486 ([M + Na]+, C44H53N6O9PNa calcd 863.3504).Preparation of (2R,3S)-1,4-bis(3-N-benzoylthymin-1-yl)-3-(4,4-dimethoxytrityloxy)but-2-oxy-β-cyanoethoxy-N,N-diisopropylaminophosphite (12b)
Same procedure as above. Compound 11b (0.59 g) was used to give the product 12b as a white powder (0.50 g, 64%). NMR data are identical to data for the enantiomer 12a. HR ESI MS m/z 863.3494 [M + Na]+.Synthesis of (2R,3R)-1,4-bis(thymin-1-yl)butane-2,3-diol (13)
A small amount of compound 9a was dissolved in a little CH2Cl2 and suspended with silica. A few drops of dichloroacetic acid were added and the mixture was stirred for 10 min and concentrated under reduced pressure. The powder was purified by chromatography on a Pasteur pipette column (CHCl3–EtOH 4 : 1 v/v) to give the crude product. This was dissolved in a small amount of DMSO in an open vial and placed in a closed beaker with water to give crystals after a few days. δH (300 MHz, DMSO-d6, Me4Si) 11.21 (2H, s, N-H), 7.41 (2H, s, H-6), 5.10 (2H, d, J = 6 Hz, OH), 3.80 (2H, dd, J = 2 Hz, 13 Hz, H-1′), 3.66 (2H, m, H-2′), 3.55 (2H, dd, J = 9 Hz, 13 Hz, H-1″), 1.75 (6H, s, CH3).Synthesis of meso-1,4-bis(thymin-1-yl)butan-2,3-diol (14)
Similar reaction procedure as above, but using 11a. The crude product was dissolved in a little DMSO and evaporated under reduced pressure at 100 °C until crystals appeared. Then, the crystal growth was allowed to proceed while slowly cooling down to room temperature over 5 h. δH (300 MHz, DMSO-d6, Me4Si) 11.19 (2H, s, N-H), 7.41 (2H, s, H-6), 5.25 (2H, br s, OH), 4.09 (2H, d, J = 13 Hz, H-1′), 3.55 (2H, m, H-2′), 3.35 (2H, dd, J = 8 Hz, 13 Hz, H-1″), 1.74 (6H, s, CH3).Synthesis of a model dinucleotide and 31P-NMR study of the detritylation step
A solution of 8a (0.66 g, 0.63 mmol) and 3′-O-(tert-butyldimethylsilyl)thymidine (0.27 g, 0.75 mmol) in anhydrous MeCN (2.0 mL) was stirred at room temperature and a 0.5 M solution of pyrHCl in MeCN (2.0 mL, 1 mmol) was added. The mixture was stirred for 20 h, and a 5.5 M solution of t-BuOOH in decane (0.2 mL, 1 mmol) was added. The mixture was stirred for 1 h and directly purified by column chromatography (CH2Cl2–MeOH 99 : 1 v/v containing 0.5 vol% pyridine). The residue was co-evaporated with a xylene mixture and dried under reduced pressure to give the dinucleotide product as a white compound (0.43 g, 57%). Rf 0.55 (CH2Cl2–MeOH 19 : 1); 31P-NMR (81 MHz, CDCl3) δ 0.03, –0.36. The kinetics were acquired as follows: the dinucleotide (0.191 g, 0.159 mmol) was dissolved in CD2Cl2 (0.50 mL) in an NMR tube. A solution of trichloroacetic acid (26 mg, 0.16 mmol)) in CD2Cl2 (234 mg) was added to initiate the reaction. Spectra recorded in 256 s were acquired in the spectral window from δ = 100 to –100 ppm.Oligonucleotide synthesis
The oligonucleotides were synthesized on an automated DNA synthesizer Expedite 8909 on a 0.2 µmol scale using CPG support and employing a standard phosphoramidite protocol except for the couplings involving the unconventional nucleosides. These were coupled manually by treating the CPG support with a 0.05 M mixture of the phosphoramidite in acetonitrile (100 µL) and a 0.5 M solution of activator (1H-tetrazole or pyridinium chloride) in acetonitrile (100 µL) for 10 minutes. Resuming the automatic cycle, the support was washed and capped according to the standard protocol and then again manually treated with a mixture of acetonitrile (300 µL) and a 5.5 M solution of t-BuOOH in decane (100 µL) for 5 minutes. The next elongation was conducted according to the standard protocol except for a prolonged coupling time of 15 minutes. The oligonucleotides were removed from the support and deprotected by treatment with a 32% w/w aqueous NH3 solution (1 mL) at 55 °C for 16 h. Subsequent purification was performed using reversed HPLC (column XTerra C18, 10 mm, 7.8 × 300 mm, with a gradient of buffers A and B (A, 90% 0.1 M NH4HCO3 + 10% CH3CN; B, 25% 0.1 M NH4HCO3 + 75% CH3CN), 1 mL min–1), and detritylation was performed using aqueous acetic acid. Precipitation with EtOH afforded the oligonucleotides.Thermal denaturation experiments
The thermal denaturation studies were conducted in a medium salt buffer containing Na2HPO4 (10 mM), NaCl (100 mM) and EDTA (0.1 mM), pH 7.0 with 1 µM concentrations of the two strands. The concentrations were determined assuming the unconventional double-headed nucleosides to have an extinction coefficient twice the normal thymidines. The hyperchromicity was measured as a function of time while increasing the temperature linearly between 10 and 80 °C at a time rate of 1 °C min–1. The melting temperature was then determined as the maximum of the first derivative of the hyperchromicity. Probes with both up and down curves were in general found to be identical.X-Ray crystallography
Single-crystal X-ray diffraction data for 13·DMSO·2H2O were collected at 180(2) K using a Bruker-Nonius X8APEX-II CCD diffractometer, with MoKα radiation (λ = 0.7107 Å). Incorporation of DMSO into the crystal allowed the absolute structure to be confirmed on the basis of the diffraction data. Crystals of 14·2DMSO were small and weakly diffracting, and diffraction data were collected at 150(2) K at Beamline I911-3 of the MAX-II storage ring at MAX-Lab, Lund, Sweden (λ = 0.7500 Å). On account of experimental constraints, data were collected using only a single 360° φ scan, resulting in ca. 88% completeness to a resolution of 1 Å. Data were corrected for beam decay during integration on the basis of ring-current values using the TWINSOLVE package.31 Both structures were solved and refined using SHELXTL.32 H atoms bonded to C atoms were placed geometrically and refined using a riding model. For 13, H atoms bonded to O atoms were located in difference Fourier maps and refined with restrained O–H distances and isotropic displacement parameters constrained to be 1.2 or 1.5 times the equivalent isotropic displacement parameter of the O atom to which they are bonded. For 14, the H atom of the unique hydroxyl group was placed so as to form a linear O–H⋯O hydrogen bond to the incorporated DMSO molecule, and refined as riding with an isotropic displacement parameter constrained to be 1.5 times the equivalent isotropic displacement parameter of the parent O atom. The data and resulting structure of 14·2DMSO are of comparatively low quality, but are sufficient to establish the meso-configuration.![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 5.224(2), b = 9.313(4), c = 12.558(6) Å, α = 101.76(3), β = 91.68(3), γ = 105.00(3)°, V = 575.6(4) Å3, Z = 1, ρcalcd = 1.427 g cm–3, µ(λ = 0.7500 Å) = 0.283 mm–1, 4.32 < 2θ < 22.03°; of 1231 measured reflections, 1047 were independent (Rint = 0.025) and 918 observed with I > 2σ(I); R1 = 0.117, wR2 = 0.316, GOF = 3.02 for 145 parameters, Δρmax/min = 0.90/–0.54. CCDC reference number 643525. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b713888a
, a = 5.224(2), b = 9.313(4), c = 12.558(6) Å, α = 101.76(3), β = 91.68(3), γ = 105.00(3)°, V = 575.6(4) Å3, Z = 1, ρcalcd = 1.427 g cm–3, µ(λ = 0.7500 Å) = 0.283 mm–1, 4.32 < 2θ < 22.03°; of 1231 measured reflections, 1047 were independent (Rint = 0.025) and 918 observed with I > 2σ(I); R1 = 0.117, wR2 = 0.316, GOF = 3.02 for 145 parameters, Δρmax/min = 0.90/–0.54. CCDC reference number 643525. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b713888a
Molecular modelling
MD-simulation was performed with the Amber99 force field with parameters as supplied with Amber7.33 The coordinates for the B-DNA were obtained from 1n1o.pdb, where one of the TNA monomers was replaced with the ordinary T monomer and the other replaced with the double-headed glycol nucleosides. The partial charges were derived using resp and the rhf/6-31g* wave function obtained from a GAMESS34 run using the nucleoside coordinates from a MMFF94 force field minimization of the 2′,3′-O-methyl analogs. The duplexes were neutralised with 10Na+ and placed in a 7.0 Å truncated octahedron with TIP3P waters. Using Sander with PBC and PME, the systems were then allowed to relax with 1000 minimisation steps using a 8 Å cutoff for the non-bonded potential terms and a 500 kcal mol–1 Å–2 restraint on the duplex. The hydrogens were then constrained using the SHAKE algorithm and the whole system relaxed by 2500 minimization steps. MD simulation was then initiated. While restraining the duplex with a 10 kcal mol–1 Å–2 force constant, the solvent environment was allowed to warm up to 298 K for 10 ps at constant volume using the Berendsen thermostat before the system was allowed to equilibrate unrestrained for 50 ps at 298 K and 1 atm (with time constants 1 ps and 2 ps, respectively). Finally, the dynamics of the system was simulated for 0.5 ns with time steps of 0.2 ps. The coordinates from the final 50 ps were used to produce an average structure of the final duplexes (Fig. 4).
Acknowledgements
The Danish National Research Foundation, The Danish Research Agency and Møllerens Fond are thanked for financial support of the project, and Mrs Birthe Haack is thanked for technical assistance. We are grateful to the Danish Natural Science Research Council and to Carlsbergfondet for provision of the X-ray equipment in Odense, and to MAX-Lab, University of Lund, Sweden, for synchrotron access. Assistance at MAX-Lab was provided by Yngve Cerenius, Krister Larsson and Christer Svensson.References
- C. J. Leumann, Bioorg. Med. Chem., 2002, 10, 841 CrossRef CAS.
- J. Kurreck, Eur. J. Biochem., 2003, 270, 1628 CAS.
- J. Wengel, Org. Biomol. Chem., 2004, 2, 277 RSC.
- B. Samori and G. Zuccheri, Angew. Chem., Int. Ed., 2005, 44, 1166 CrossRef CAS.
- N. C. Seeman and P. S. Lukeman, Rep. Prog. Phys., 2005, 68, 237 CrossRef CAS.
- A. Madder, R. Ehrl and R. Strömberg, ChemBioChem, 2003, 4, 1194 CrossRef CAS.
- K. Seio, T. Wada, K. Sakamoto, S. Yokoyama and M. Sekine, J. Org. Chem., 1998, 63, 1429 CrossRef CAS.
- W. Tongfei, M. Froeyen, G. Schepers, K. Mullens, J. Rozenski, R. Busson, A. Van Aerschot and P. Herdewijn, Org. Lett., 2004, 6, 51 CrossRef CAS.
- S. L. Pedersen and P. Nielsen, Org. Biomol. Chem., 2005, 3, 3570 RSC.
- W. Tongfei, K. Nauwelaerts, A. Van Aerschot, M. Froeyen, E. Lescrinier and P. Herdewijn, J. Org. Chem., 2006, 71, 5423 CrossRef CAS.
- M. S. Christensen, C. M. Madsen and P. Nielsen, Org. Biomol. Chem., 2007, 5, 1586 RSC.
- P. Børsting, K. E. Nielsen and P. Nielsen, Org. Biomol. Chem., 2005, 3, 2183 RSC.
- P. K. Sharma, B. H. Mikkelsen, M. S. Christensen, K. E. Nielsen, C. Kirchhoff, S. L. Pedersen, A. M. Sørensen, K. Østergaard, M. Petersen and P. Nielsen, Org. Biomol. Chem., 2006, 4, 2433 RSC.
- P. Nielsen, L. H. Dreiøe and J. Wengel, Bioorg. Med. Chem., 1995, 3, 19 CrossRef CAS.
- O. L. Acevedo and R. S. Andrews, Tetrahedron Lett., 1996, 37, 3931 CrossRef CAS.
- A. Holy, Collect. Czech. Chem. Commun., 1975, 40, 187 CAS.
- L. Zhang, A. Peritz and E. Meggers, J. Am. Chem. Soc., 2005, 127, 4174 CrossRef CAS.
- L. Zhang, A. E. Peritz, P. J. Carroll and E. Meggers, Synthesis, 2006, 4, 645.
- K.-U. Schöning, P. Scholz, S. Guntha, X. Wu, R. Krishnamurthy and A. Eschenmoser, Science, 2000, 290, 1347 CrossRef CAS.
- O. Mitsunobu, Synthesis, 1981, 1 CrossRef CAS.
- M. Frieden, M. Giraud, C. B. Reese and Q. Song, J. Chem. Soc., Perkin Trans. 1, 1998, 290, 2827 Search PubMed.
- A.-I. Hernández, J. Balzarini, A. Karlsson, M.-J. Camarasa and M.-J. Pérez-Pérez, J. Med. Chem., 2002, 45, 4254 CrossRef CAS.
- J. J. Fox and N. C. Miller, J. Org. Chem., 1963, 28, 936 CAS.
- M. Beier and W. Pfleiderer, Helv. Chim. Acta, 1999, 82, 879 CrossRef CAS.
- D. Zhou, I. M. Lagoja, J. Rozenski, R. Busson, A. Van Aerschot and P. Herdewijn, ChemBioChem, 2005, 6, 2298 CrossRef CAS.
- D. G. Gorenstein, Phosphorus-31 NMR: Principles and Applications, Academic Press, New York, 1984 Search PubMed.
- C. J. Wilds, Z. Wawrzak, R. Krishnamurthy and A. Eschenmoser, J. Am. Chem. Soc., 2002, 124, 13716 CrossRef CAS.
- R.-I. Ma, N. R. Kallenbach, R. D. Sheardy, M. L. Petrillo and N. C. Seeman, Nucleic Acids Res., 1986, 14, 9745 CrossRef CAS.
- N. B. Leontis, W. Kwok and J. S. Newman, Nucleic Acids Res., 1991, 19, 759 CrossRef CAS.
- B. Liu, N. B. Leontis and N. C. Seeman, Nanobiology, 1994, 3, 177 Search PubMed.
- C. Svensson, TWINSOLVE, University of Lund, Sweden, 2004 Search PubMed.
- G. M. Sheldrick, SHELXTL, Bruker AXS, Madison, Wisconsin, USA, 2000 Search PubMed.
- D. A. Case, D. A. Pearlman, J. W. Caldwell, T. E. Cheatham III, J. Wang, W. S. Ross, C. L. Simmerling, T. A. Darden, K. M. Merz, R. V. Stanton, A. L. Cheng, J. J. Vincent, M. Crowley, V. Tsui, H. Gohlke, R. J. Radmer, Y. Duan, J. Pitera, I. Massova, G. L. Seibel, U. C. Singh, P. K. Weiner and P. A. Kollman, AMBER7, University of California, San Francisco, 2002 Search PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, Jr., J. Comput. Chem., 1993, 14, 1347 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Data supporting the kinetic experiments on the phosphoramidite coupling sequences as well as 1H NMR analysis for 13 and 14; crystal structure data. See DOI: 10.1039/b713888a |
| ‡ The Nucleic Acid Center is funded by the Danish National Research Foundation for studies on nucleic acid chemical biology. |
| This journal is © The Royal Society of Chemistry 2008 |