Dyotropic rearrangement of α-lactone to β-lactone: a computational study of small-ring halolactonisation†
J. Grant
Buchanan
a,
Giuseppe D.
Ruggiero
a and
Ian H.
Williams
*ab
aDepartment of Chemistry, University of Bath, Bath, UK BA2 7AY
bDepartment of Chemistry, University of Bath, Bath, UK BA2 7AY. E-mail: i.h.williams@bath.ac.uk; Fax: +44 1225 386231; Tel: +44 1225 386625
First published on 13th November 2007
Abstract
Transition structures have been optimised using the B3LYP/6-31+G* density functional level method, in vacuum and in implicit (PCM) and explicit (DFT/MM) aqueous solvation, for the degenerate rearrangement of the α-lactone derived by the formal addition of Cl+ to acrylate anion and for the dyotropic rearrangement of this to the β-lactone. Despite being lower in energy than the α-lactone, there is no direct pathway to the β-lactone from the acrylate chloronium zwitterion, which is the transition structure for the degenerate rearrangement. This may be rationalised by consideration of the unfavorable angle of attack by the carboxylate nucleophile on the β-position; attack on the α-position involves a less unfavorable angle. Formation of the β-lactone may occur by means of a dyotropic rearrangement of the α-lactone. This involves a high energy barrier for the acrylate derived α-lactone, but dyotropic rearrangement of the β,β-dimethyl substituted α-lactone to the corresponding β-lactone involves a much lower barrier, estimated at about 46 kJ mol−1 in water, and is predicted to be a facile process.
Introduction
Halolactonisation is the well-known synthetic procedure whereby electrophilic halogenation (often iodination) of an unsaturated carboxylic acid yields a lactone product.1 The mechanism is generally considered to involve initial formation of an intermediate cyclic halonium ion which then undergoes intramolecular nucleophilic substitution with the carboxyl group attacking the halonium moiety in accordance with Baldwin's rules for ring closure.2 For example, Barnett and Sohn showed that the kinetic product of iodolactonisation of 2,2-dimethylbut-3-enoate (Scheme 1) is the γ-iodo-β-lactone, which at longer reaction times rearranges to the thermodynamic product β-iodo-γ-lactone.3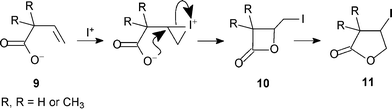 | ||
| Scheme 1 | ||
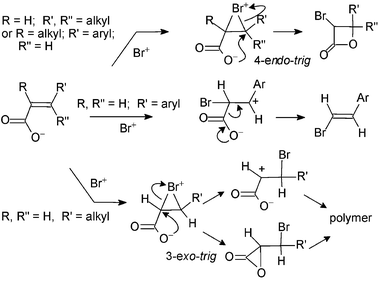
Rousseau and co-workers have reported the preparation of α-bromo-β-lactones by use of bis(collidine)bromine(I) hexafluorophosphate in dichloromethane as the electrophilic reagent with β-substituted α,β-unsaturated acids.4,5 With R = H and R′, R″ = alkyl or with R = alkyl, R′ = aryl, R″ = H (Scheme 2), Rousseau represents the process as a 4-endo-trigcyclisation with attack of the (undissociated) carboxyl group on a cyclic bromonium ion. However, since two molecules of collidine are present in the reagent it is more likely that the carboxylate form is involved. When R, R″ = H and R′ = aryl the intermediate is believed to be a resonance-stabilised benzylic cation which undergoes decarboxylation. With R, R″ = H and R′ = alkyl, polymeric products were reported which were considered to arise from the intermediacy of a highly reactive α-lactone.
 | ||
| Scheme 2 | ||
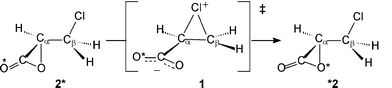
We have previously reported results of electronic structure calculations for the α-lactone produced by halide elimination from an α-halocarboxylate,6 and have estimated the strain energy of the three-membered ring of this smallest example of a cyclic ester.7 We have argued that, despite unusual electronic properties in the ring,8 this species is best described as an α-lactone, and have shown that it exists in an energy minimum in a hybrid quantum-mechanical/molecular-mechanical (QM/MM) model for explicit solvation in water.9 We have presented experimental evidence for the intermediacy of an α-lactone in the aqueous bromination of 2,3-dimethylmaleate and fumarate dianions.10 We have also communicated density-functional theoretical (DFT) results within the polarised continuum model (PCM) of aqueous solvation indicating that addition of Cl+ to acrylate anion leads directly to the chloromethyl-α-lactone 2.11 The cyclic chloronium species 1 is a transition structure (TS) for a degenerate rearrangement of 2 that exchanges the two oxygen atoms of the carboxylate group (Scheme 3, 2*→1→*2). Whereas in principle it could be imagined (Scheme 4) that the carboxylate nucleophile in 1 could have a choice to attack either Cα or Cβ leading to the α-lactone 2 and β-lactone 3, respectively, in practice this competition does not exist since 1 is not an intermediate.
 | ||
| Scheme 3 | ||
 | ||
| Scheme 4 | ||
This paper addresses primarily the question of how the thermodynamically more stable β-lactone 3 may be formed from the kinetically preferred α-lactone 2: we present results of calculations for a novel 1,2-transposition via TS 4. Although this type-1 dyotropic rearrangement12 of the acrylate-derived chloromethyloxiranone 2 involves a high barrier, the β,β-dimethyl substituted α-lactone 5 (formally derived from addition of Cl+ to senecioate anion) converts into the corresponding β-lactone 7via TS 6 in a low-energy rearrangement. We propose that β-lactones produced during electrophilic halogenation of the salts of simple α,β-unsaturated monocarboxylic acids dialkylated on Cβ are formed by dyotropic rearrangement of initially formed α-lactones. We also present a more complete discussion regarding the degenerate rearrangement of α-lactone 2via TS 1, including the effects of both implicit and explicit solvation by water.
Computational methods
The GAUSSIAN98 package of programs13 was used with the B3LYP/6-31+G* level14 of DFT and the polarized continuum model (PCM)15 for aqueous solvation (ε = 78.4). TSs were located by means of the EF algorithm and characterized by frequency calculations, and by intrinsic reaction coordinate calculations to verify the adjacent local minima. The PCM energies given in the Tables include both electrostatic and the non-electrostatic (surface-area dependent cavity) contributions to the free energy of the solvated species.Hybrid QM/MM calculations were performed using the CHARMM29b2 program16 as follows. The PCM-optimised TSs 1 and 4 were placed in the centre of a 15 Å-radius random sphere of TIP3P17water molecules; water molecules overlapping with the solute were removed, and a PCM Mulliken point charge was assigned to each solute atom. The water molecules were restrained by imposition of a stochastic boundary to mimic the effect of bulk solvent beyond the 15 Å-sphere. A sequence of initial energy minimisation using only the molecular mechanics (MM) energy function was performed in order to produce a reasonably well-packed arrangement of the 496 solvating water molecules around the solute species. Molecular dynamics calculations were performed with the solute atoms fixed throughout the simulations using CHARMM. The system was heated to 300 K over a period of 18 ps followed by 2 ps equilibration; it was then equilibrated for a further 20 ps. The equilibrated structure was taken as a starting point to initiate a TS search with the B3LYP/6-31+G* method for the QM solute, together with TIP3P solvent, using an interface with GAMESS-UK.18 The ab initioQM/MM stationary-point location and characterization were guided by means of GRACE,19 which uses a Newton–Raphson algorithm. The system was divided into two parts, the core and the environment; the Hessian matrix was calculated explicitly only for the core atoms, and stationary point location was carried out in the degrees of freedom of the core. Before each energy and gradient evaluation step for the core, the degrees of freedom of the environment were relaxed to maintain an approximately zero gradient and to minimise the potential energy. The core Hessian was then used to start an intrinsic reaction coordinate (IRC) calculation: the environment was continually relaxed while the core atoms followed the mass weighted reaction coordinate down from the saddle point to reactants (or products). A few QM/MM optimization steps were then used to lower the r.m.s. gradient to 0.005 kJ mol−1Å−1.
Results and discussion
A selection of key geometrical parameters for each optimised structure is presented in Table 1, along with relative energies and (for each TS) the imaginary frequency (expressed as a wavenumber) corresponding to the transition vector. Results are shown for species with R = H (Scheme 4) in vacuum, in PCM water and with explicit QM/MM solvation by TIP3P water, and with R = CH3 in vacuum and PCM water only.| Species | Rel. energy | ν ‡ | Cα–Cl | Cβ–Cl | Oα–Cα | Oα–Cβ | OαCαCl | OαCβCl |
|---|---|---|---|---|---|---|---|---|
| R = H | ||||||||
| α-Lactone 2 | 0 | 2.704 | 1.808 | 1.536 | 2.550 | 152.0 | 141.7 | |
| 0 | 2.703 | 1.808 | 1.537 | 2.762 | 152.0 | 141.7 | ||
| 0 | 2.732 | 1.885 | 1.635 | 2.618 | 146.8 | 136.9 | ||
| ‡degenerate1 | 171 | [73i] | 2.058 | 1.867 | 2.341 | 3.676 | 118.9 | 79.0 |
| 71 | [75i] | 1.988 | 1.882 | 2.352 | 3.683 | 112.1 | 78.5 | |
| 99 | 1.952 | 1.872 | 2.318 | 3.531 | 129.8 | 78.5 | ||
| ‡dyotropic4 | 177 | [484i] | 1.985 | 2.233 | 2.133 | 2.392 | 144.4 | 116.0 |
| 137 | [379i] | 1.872 | 2.278 | 2.224 | 2.445 | 138.7 | 108.5 | |
| 151 | 1.976 | 2.340 | 2.247 | 2.513 | 133.8 | 106.4 | ||
| β-Lactone 3 | −83 | 1.786 | 2.852 | 2.124 | 1.472 | 130.8 | 106.3 | |
| −92 | 1.800 | 2.842 | 2.122 | 1.489 | 126.3 | 103.5 | ||
| −87 | 1.717 | 2.807 | 2.145 | 1.479 | 129.5 | 105.1 | ||
| R = CH3 | ||||||||
|---|---|---|---|---|---|---|---|---|
| α-Lactone 5 | 0 | 2.644 | 1.851 | 1.544 | 2.587 | 156.4 | 134.8 | |
| 0 | 2.636 | 1.862 | 1.563 | 2.602 | 155.3 | 133.3 | ||
| ‡degenerate8 | 140 | [71i] | 1.965 | 2.014 | 2.401 | 3.029 | 108.2 | 86.9 |
| 64 | [55i] | 1.858 | 2.385 | 2.415 | 2.990 | 102.4 | 76.1 | |
| ‡dyotropic6 | 112 | [234i] | 1.952 | 2.307 | 2.176 | 2.686 | 151.9 | 106.4 |
| 46 | [45i] | 1.864 | 2.299 | 2.298 | 2.937 | 145.7 | 98.2 | |
| β-Lactone 7 | −82 | 1.787 | 2.901 | 2.121 | 1.499 | 133.8 | 105.1 | |
| −54 | 1.791 | 2.904 | 2.122 | 1.518 | 132.8 | 95.6 | ||
The energy difference between the α-lactone 2, chloromethyloxiranone, and the β-lactone 3, 3-chlorooxetan-2-one, in vacuum and in implicit or explicit water is similar in value to the energy difference between the isomeric α- and β-lactones derived from succinate,20 and reflects the difference in strain energy of the three- and four-membered rings. The Oα–Cα bond of 2 is unusually long, as noted previously for the parent oxiranone,8,9 and is further elongated by specific hydrogen-bonding interactions formed in the explicit QM/MM solvation model (Fig. 1) but not in PCM water.9 The Cβ–Cl bond is also significantly elongated by specific solvation of α-lactone 2. The Oα–Cβ and Cα–Cl bonds of the β-lactone 3 are of normal length and are not significantly affected by solvation.
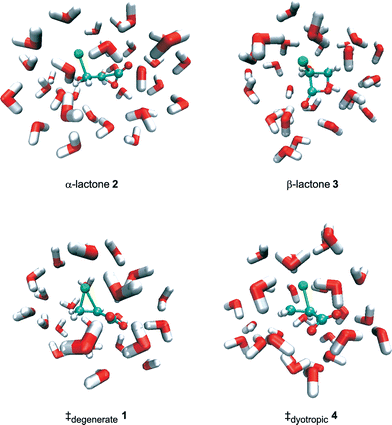 | ||
| Fig. 1 B3LYP/6-31+G*/TIP3P optimised structures with explicit aqueous solvation (only the closest of the 496 water molecules are shown for the sake of clarity). | ||
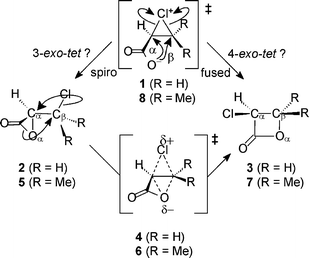
In discussing his rules for ring closure, Baldwin stated that ‘the rules for opening three membered rings to form cyclic structures seem to lie between those for tetrahedral and trigonal systems, generally preferring exo modes’.2 Regarding the two carbon atoms within the three-membered ring of the chloronium species 1 as being both tetrahedral, intramolecular nucleophilic attack by the carboxylate group would appear to be either a 3-exo-tet or 4-exo-tet process, depending upon whether it leads to the α- or the β-lactone (Scheme 4). Alternatively, consideration of the chloronium as involving only a loose association of Cl+ with a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond would suggest these same processes to be 3-exo-trig or 4-endo-trig, respectively. It is not at all clear that it is appropriate to consider the carbons in the chloronium ring as being tetrahedral: the sums of the CCC and two HCC angles about Cα and Cβ in 1 are 356.2 and 358.8°, respectively, indicating trigonal planar character. Moreover, the angles Cl–Cα–Cβ = 65.1° and Cl–Cβ–Cα = 71.1° deviate even more from the ideal tetrahedral angle. The relevant diagram in Baldwin's paper2 to illustrate these modes of ring closure, where the leaving group is part of a three-membered ring, does not employ the designations trig or tet. Instead, the modes labelled in our Scheme 4 as “3-exo-tet”or “4-exo-tet” are differentiated only as “exo” and “endo”, respectively. The latter usage is arguably at odds with the definition given earlier in the paper, which would apparently require the breaking bond to be endocyclic to the smallest ring formed in the process.21 An alternative way to distinguish the modes of ring closure is to adopt Danishefsky's terminology: formation of the α-lactone is “spiro” whereas formation of the β-lactone is “fused”, these terms describing the relationship of the making and breaking rings in the (putative) transition states.22
C double bond would suggest these same processes to be 3-exo-trig or 4-endo-trig, respectively. It is not at all clear that it is appropriate to consider the carbons in the chloronium ring as being tetrahedral: the sums of the CCC and two HCC angles about Cα and Cβ in 1 are 356.2 and 358.8°, respectively, indicating trigonal planar character. Moreover, the angles Cl–Cα–Cβ = 65.1° and Cl–Cβ–Cα = 71.1° deviate even more from the ideal tetrahedral angle. The relevant diagram in Baldwin's paper2 to illustrate these modes of ring closure, where the leaving group is part of a three-membered ring, does not employ the designations trig or tet. Instead, the modes labelled in our Scheme 4 as “3-exo-tet”or “4-exo-tet” are differentiated only as “exo” and “endo”, respectively. The latter usage is arguably at odds with the definition given earlier in the paper, which would apparently require the breaking bond to be endocyclic to the smallest ring formed in the process.21 An alternative way to distinguish the modes of ring closure is to adopt Danishefsky's terminology: formation of the α-lactone is “spiro” whereas formation of the β-lactone is “fused”, these terms describing the relationship of the making and breaking rings in the (putative) transition states.22
In practice, species 1 is not an energy minimum, either in vacuum or in implicit (PCM) or explicit (TIP3P) water, but a TS for a degenerate rearrangement (Scheme 3) of equivalent α-lactones 2 which serves to scramble the two oxygen atoms.11Fig. 2 shows how the carboxylate group in the cyclic chloronium species TS ‡degenerate may rotate in either direction to form an α-lactone with either one or the other oxygen atom. This unstable species 1 spontaneously collapses to the α-lactone 2 with an accompanying decrease in energy of 171, 71 or 99 kJ mol−1 in vacuum, PCM, or TIP3P water, respectively. All efforts to locate a TS leading from 1 directly to the β-lactone 3 have failed: this is not surprising, since a TS can only interconnect two valleys,23 even in a degenerate rearrangement. (A TS may interconnect with a third valley only by means of another saddle point of even higher energy.) Formation of the α-lactone by the 3-exo-tet or spiro mode of ring closure is evidently the sole favoured process. The TS ‡degenerate1 is zwitterionic in nature (dipole moment 9.0 D) and hence is more strongly solvated than 2 (dipole moment 2.9 D); thus the barrier to rearrangement in PCM water is much lower than in vacuum owing to this favourable solvation energy contribution. The Cα–Cl bond in 1 is consistently longer than the Cβ–Cl bond in all computational models. Although the barrier to degenerate rearrangement with implicit solvation is a little lower than with explicit solvation, nonetheless the PCM model provides overall similar results to the QM/MM model. While it is tempting to speculate upon the origin of geometric differences between structures optimised with implicit and explicit solvation methods, it should be noted that the stationary points in Table 1 for the QM/MM model each represent only one among many accessible solvent configurations with differing hydrogen-bonding patterns; a proper analysis must await a future study of ensemble-averaged structures.
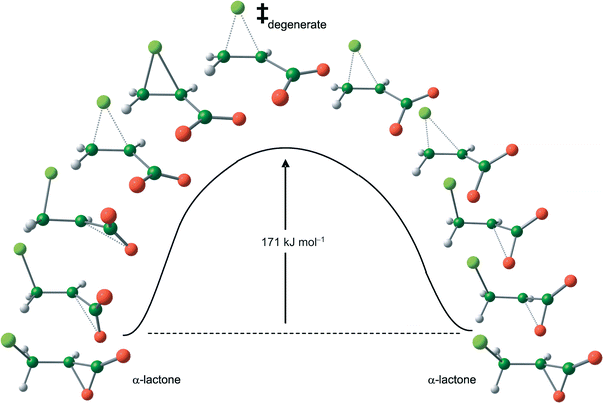 | ||
| Fig. 2 B3LYP/6-31+G* energy profile in vacuum with selected structures along the intrinsic reaction path for degenerate rearrangement of α-lactone 2. | ||
The β-lactone 3 can be formed from the α-lactone by a dyotropic rearrangement12 involving concerted intramolecular nucleophilic substitution at vicinal carbon atoms. The leaving group of one component is the nucleophile for the other, and vice versa. The key frontier-orbital interaction of each “SN2” (four electrons in three orbitals) component is between an nX lone-pair on the nucleophilic atom X (designated in Fig. 3 as “HOMO”, but not necessarily the true HOMO) and a σ*CY antibonding orbital involving the leaving group Y (designated in Fig. 3 as “LUMO”, but not necessarily the true LUMO); both components are important in the TS (Fig. 3) but in the absence of symmetry (X ≠ Y) these orbitals are not degenerate. The third orbital in each component (not shown in Fig. 3) is the σCY bonding orbital of the reactant which becomes an nY lone-pair in the product. The barrier for this process via TS ‡dyotropic4 (Fig. 4) is only slightly higher in vacuum than for the degenerate rearrangementvia‡degenerate1, but in implicit and explicit water is predicted to be some 50 or 60 kJ mol−1 higher, with a value of 151 kJ mol−1 in the QM/MM calculation. The TS 4 is rather dipolar in nature, with considerable chloronium carboxylate character (dipole moment 6.7 D), and so is more strongly solvated than either 2 or 3 (dipole moment 4.0 D). The Pauling bond orders for the Oα–Cα and Oα–Cβ bonds are both small (0.29 and 0.20, respectively), whereas those for the Cα–Cl and Cβ–Cl bonds are both substantial (0.89 and 0.45, respectively). This suggests that the two components of intramolecular nucleophilic substitution are concerted but not synchronous. It also suggests that the relative energy of the TSs ‡degenerate1 and ‡dyotropic4 is determined more by strain effects than by orbital symmetry considerations.
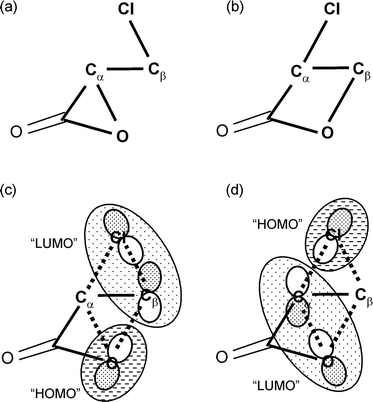 | ||
| Fig. 3 Connectivity for (a) α-lactone 2 and (b) β-lactone 3 and frontier molecular orbitals (c) and (d) of TS 4 for the dyotropic rearrangement. | ||
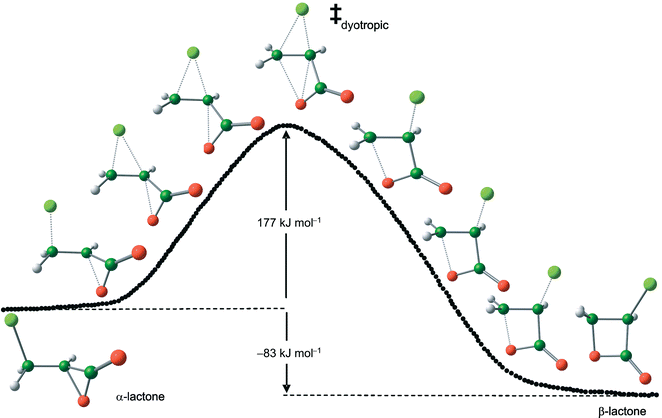 | ||
| Fig. 4 B3LYP/6-31+G* energy profile in vacuum with selected structures along the intrinsic reaction path for dyotropic rearrangement of α-lactone 2 to β-lactone 3. | ||
By what means may α-lactone 2 be generated in the first place? While this question is not the primary concern of the present study, and its detailed answer in the case of chloromethyloxiranone will be presented elsewhere, it is of interest in view of the fact that the chloronium carboxylate 1 is not an intermediate but a TS. In our recent B3LYP/6-31+G(d) study of aqueous bromination of alkene dicarboxylates,10b addition of Br2 to 2,3-dimethylfumarate dianion in PCM water was found to be exothermic by 100 kJ mol−1, yielding a structure in which the two bromine atoms were aligned perpendicularly to the plane of the fumarate moiety, but with a very elongated Br⋯Br distance; this intermediate was 35 kJ mol−1 higher in energy than the bromoalkyloxiranone and Br−. We suspect the existence of a TS in which electrophilic attack by molecular bromine on Cβ is coupled with departure of the bromide leaving group and with nucleophilic attack by the carboxylate group on Cα leading to the α-lactone and avoiding formation of the unfavourable cyclic bromonium dicarboxylate. This TS may be very difficult to locate and characterise within a PCM treatment of water, since an important component of the reaction coordinate is likely to be specific solvation of the incipient bromide anion.
Consider again the acrylate-derived chloronium zwitterion 1. Formally we may conceive of a “4-exo-tet” or “fused” intramolecular nucleophilic substitution (Scheme 4) leading to the β-lactone by means of ‡dyotropic4. In principle either of the two carboxylate O atoms could act as the nucleophile: the two possible angles OnuCβCl in 1 are 79 and 91° (Scheme 5) and this process is uphill in energy by 52 kJ mol−1 in TIP3P water. Alternatively 1 may undergo a “3-exo-tet” or “spiro” intramolecular nucleophilic substitution leading directly to the α-lactone 2: the two possible angles OnuCαCl in 1 are 103 and 112° (Scheme 5), and this process is downhill by 99 kJ mol−1 in TIP3P water. The favoured option is that involving the largest angle OnuCCl, and thus the least angle strain, just as in the case of chlorosuccinate.20
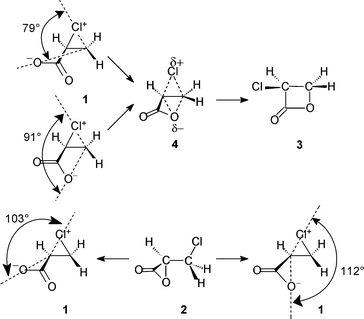 | ||
| Scheme 5 | ||
The chlorosuccinate dianion provides an interesting comparison because it involves competition between α- or β-lactone formation involving a choice of two carboxylate nucleophiles to attack a single carbon atom attached to a chloride leaving group. The β-lactone product is favoured because it involves the TS with atoms OnuCCl closer to collinearity than that for α-lactone formation (angle OnuCCl = 174 vs. 139°) and therefore involving less angle strain;20 the chloride leaving group is attached to a normal tetrahedral carbon atom (Fig. 5). In contrast, the carbon atoms in the three-membered ring of 1 are substantially distorted from tetrahedrality, each being “tied back” by approximately 40°. The OnuCCl angles made by the internal nucleophile with the two leaving groups are significantly affected by this distortion: the angle OnuCβCl is much closer to 90° than to 180°, whereas OnuCαCl becomes the larger angle. In consequence the α-lactone product is now kinetically favoured, as implied by the stereochemical result of aqueous bromination of the dianions of 2,3-dimethylmaleic and fumaric acids as recently determined10 by X-ray crystallography.
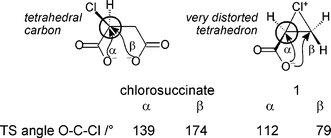 | ||
| Fig. 5 Comparison of angles of intramolecular nucleophilic attack in chlorosuccinate dianion and acrylate-derived chloronium zwitterion. | ||
Addition of Cl+ to the 3-methylbut-2-enoate (senecioate, β,β-dimethylacrylate) anion leads to α-lactone 5 (Scheme 4, R = CH3) as the first-formed intermediate. Both the Cα–Oα and Cβ–Cl bonds are longer than in the unsubstituted species 2, in vacuum and in PCM water. The β-lactone 7 has essentially the same Cα–Cl bond length as in 3 but a significantly longer Cβ–Oα bond, owing to the effect of its gem-dimethyl substitution. While the greater stability of 7 with respect to 5 in vacuum is the same as that of 3 relative to 2, in PCM water this difference is diminished, possibly due to enhanced polar character of the Cβ–Cl bond in 5 (dipole moment 3.4 D) than in 2. In TS 8 for the degenerate rearrangement of the α-lactone, Cβ–Cl is significantly longer than Cα–Cl, in contrast to the situation in TS 1; this suggests appreciably more carbocation character at Cβ in the gem-dimethyl substituted chloronium zwitterion (dipole moment 10.3 D). The same dissymmetry is found between the Cα–Cl and Cβ–Cl bonds in TS 6 for the dyotropic rearrangement of 5 to 7 (Fig. 6); moreover, both the Cα–Oα and Cβ–Oα bonds are longer in 6 than in 4, with that to Cβ being significantly the longer of the two. This suggests greater carbocation character at Cβ in TS 6 also (dipole moment 8.9 D). The Pauling bond orders for the Oα–Cα and Oα–Cβ bonds in 6 are both small (0.26 and 0.09, respectively) and smaller than in 4, whereas those for the Cα–Cl and Cβ–Cl bonds are both larger (0.90 and 0.44, respectively) and about the same as in 4. This suggests that the two components of intramolecular nucleophilic substitution are once again concerted but now even less synchronous.
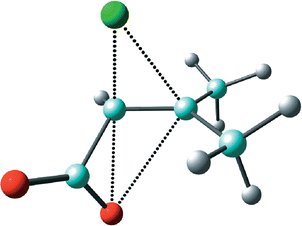 | ||
| Fig. 6 B3LYP/6-31+G* optimised transition structure ‡dyotropic6 for interconversion of α-lactone 5 and β-lactone 7. | ||
The barrier for degenerate rearrangementvia TS 8 is lower (140 vs. 171 kJ mol−1) than that via1 in vacuum, owing to the greater carbocation character at Cβ. In PCM water the barrier for each is considerably lower (64 vs. 71 kJ mol−1) with more similar values; the stabilising influence of solvation is more important than the substituent effect. The barrier for dyotropic rearrangementvia TS 6 is also lower (112 vs. 177 kJ mol−1) than that via4 in vacuum, presumably for the same reason. However, now there is both a significant substituent effect and a substantial solvation effect in PCM water, with a barrier of only 46 kJ mol−1via6 as compared with 137 kJ mol−1via4. In particular, the barrier for dyotropic rearrangement is 18 kJ mol−1 less than that for the degenerate rearrangement and easily surmountable at room temperature. While specific hydrogen-bonding interactions might affect the value of this barrier upwards (as for the unsubstituted TS 4) it is unlikely to alter the prediction of a facile dyotropic rearrangement for the less stable gem-dimethyl substituted α-lactone 5 to the more stable β-lactone 7.
Solas and Wolinsky reported the synthesis of α-chloro-β-lactones by treatment of certain α,β-unsaturated acids (including 3-methylbut-2-enoate) with hypochlorous acid in a two-phase system.24 In view of their use of excess 70% calcium hypochlorite as the reagent, we consider that under their experimental conditions it is the salt of the acid that reacts with Cl+. Consequently, in our view it is most likely that the initial kinetic product is an α-lactone 5 which then undergoes dyotropic rearrangement to β-lactone 7 as the thermodynamically preferred product.
As noted above, Homsi and Rousseau have reported the use of a solution of bis(collidine)bromine(I) hexafluorophosphate in dichloromethane to prepare α-bromo-β-lactones from α,β-unsaturated acids disubstituted in the β-position.4,5 In contrast to the mechanism shown in Scheme 2, we believe that an α-lactone is initially involved which rearranges into a β-lactone.
There is already some evidence for such a rearrangement in the regioisomeric β,γ-unsaturated acids (Scheme 6). Barnett and Sohn showed that iodolactonisation of carboxylate anions 9 afforded β-lactones 10 as the kinetic products.3 Under the reaction conditions these rearranged into the more stable γ-lactones 11. Since the reagents were still present, particularly iodide ion, there remains the possibility of an alternative mechanism for the rearrangement: besides dyotropy, γ-lactone might be produced slowly from the alkene formed by reversal of the fast initial step that led to β-lactone (Scheme 6). However, when thallium salts are used in a non-polar solvent the insoluble thallium iodide is produced and under these conditions rearrangement of β- to γ-lactone still takes place, thus ruling out the alternative mechanism; dyotropy provides a viable pathway.25 Most convincingly, Ganem isolated γ-bromo-β-lactones 12 from similar reactions and showed that they were converted thermally into the corresponding β-bromo-γ-lactones 13.26
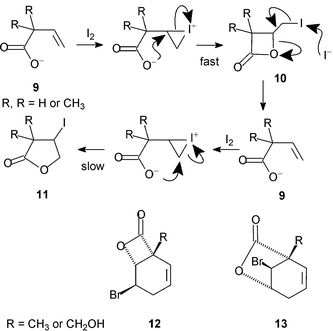 | ||
| Scheme 6 | ||
Conclusions
Transition structures have been optimised using the B3LYP/6-31+G* density functional level method, in vacuum and in implicit (PCM) and explicit (DFT/MM) aqueous solvation, for the degenerate rearrangement of the α-lactone 2 and for the dyotropic rearrangement of this to the β-lactone 3. Despite being lower in energy than the α-lactone, there is no direct pathway to the β-lactone from the acrylate chloronium zwitterion, which is the transition structure for the degenerate rearrangement. This may be rationalised by consideration of the unfavourable angle of attack by the carboxylate nucleophile on the β-position; attack on the α-position involves a less unfavourable angle. Formation of the β-lactone 3 may occur by means of a dyotropic rearrangement of the α-lactone 2 involving a high energy barrier, but the similar rearrangement of the β-dimethyl substituted α-lactone 5 to β-lactone 7 involves a much lower barrier, estimated at about 46 kJ mol−1 in water, and is predicted to be a facile process.Acknowledgements
We thank the EPSRC for a ROPA grant (GR/R94060/01) and the HEFCE/EPSRC JREI (GR/R55269) for computer equipment. I. H. W. thanks Prof. Phil Fuchs (Purdue University) for helpful discussion.References and notes
- S. Ranganathan, K. M. Muraleedharan, N. K. Vaish and N. Jayaraman, Tetrahedron, 2004, 60, 5273–5308 CrossRef CAS; M. D. Dowle and D. I. Davies, Chem. Soc. Rev., 1979, 8, 171–197 RSC.
- J. E. Baldwin, J. Chem. Soc., Chem. Commun., 1976, 734–736 RSC.
- W. E. Barnett and W. H. Sohn, Tetrahedron Lett., 1972, 1777–1780 CrossRef CAS; W. E. Barnett and W. H. Sohn, J. Chem. Soc., Chem. Commun., 1972, 472 Search PubMed.
- F. Homsi and G. Rousseau, J. Org. Chem., 1999, 64, 81–85 CrossRef CAS.
- S. Robin and G. Rousseau, Eur. J. Org. Chem., 2002, 3099–3114 CrossRef CAS.
- C. F. Rodriquez and I. H. Williams, J. Chem. Soc., Perkin Trans. 2, 1997, 953–957 RSC.
- C. F. Rodriquez and I. H. Williams, J. Chem. Soc., Perkin Trans. 2, 1997, 959–965 RSC.
- G. D. Ruggiero and I. H. Williams, J. Chem. Soc., Perkin Trans. 2, 2001, 733–737 RSC.
- J. G. Buchanan, M. H. Charlton, M. F. Mahon, J. J. Robinson, G. D. Ruggiero and I. H. Williams, J. Phys. Org. Chem., 2002, 15, 642–646 CrossRef CAS.
- (a) J. J. Robinson, J. G. Buchanan, M. H. Charlton, R. G. Kinsman, M. F. Mahon and I. H. Williams, Chem. Commun., 2001, 485–486 RSC; (b) N. Pirinççioğlu, J. J. Robinson, M. F. Mahon, J. G. Buchanan and I. H. Williams, Org. Biomol. Chem., 2007 10.1039/b711538e.
- G. D. Ruggiero and I. H. Williams, Chem. Commun., 2002, 732–733 RSC.
- M. Reetz, Angew. Chem., Int. Ed. Engl., 1972, 11, 129–130 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, Jr., R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, J. L. Andres, M. Head-Gordon, E. S. Replogle and J. A. Pople, GAUSSIAN 98 (Revision A.6), Gaussian, Inc., Pittsburgh, PA, 1998 Search PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS; A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS; P. J. Stevens, J. F. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef CAS; R. Cammi and J. Tomasi, J. Chem. Phys., 1994, 100, 7495–7502 CrossRef CAS.
- B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan and M. Karplus, J. Comput. Chem., 1983, 4, 187–217 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS . Note that CHARMM contains a modified TIP3P potential that includes intramolecular terms.
- GAMESS-UK/CHARMM is a coupling of GAMESS-UK 6.2 and CHARMM c29b2 by P. Sherwood, E. Billings, B. R. Brooks, H. L. Woodcock, M. Hodoscek, P. Sherwood, Y. S. Lees, H. F. Schaefer and B. R. Brooks, Theor. Chem. Acc., 2003, 109, 140–148 Search PubMed.
- V. Moliner, A. J. Turner and I. H. Williams, Chem. Commun., 1997, 1271–1272 RSC; A. J. Turner, V. Moliner and I. H. Williams, J. Phys. Chem. Chem. Phys., 1999, 1, 1323–1331 Search PubMed.
- J. G. Buchanan, R. A. Diggle, G. D. Ruggiero and I. H. Williams, Chem. Commun., 2006, 1106–1108 RSC.
- Professor Baldwin has employed the same “endo” label to designate intramolecular nucleophilic substitution at the distal position of an epoxide, leading to an alcohol with net ring expansion, in contrast to “exo” attack at the proximal position (personal communication to J. G. Buchanan, 9 January 1978).
- S. Danishefsky, J. Dynak, E. Hatch and M. Yamamoto, J. Am. Chem. Soc., 1973, 96, 1256–1259.
- J. N. Murrell and K. J. Laidler, Trans. Faraday Soc., 1968, 64, 371–377 RSC.
- D. Solas and J. Wolinsky, Synth. Commun., 1981, 11, 609–614 CAS.
- R. C. Cambie, R. C. Hayward, J. L. Roberts and P. S. Rutledge, J. Chem. Soc., Perkin Trans. 1, 1974, 1864–1867 RSC; R. C. Cambie, K. S. Ng, P. S. Rutledge and P. D. Woodgate, Aust. J. Chem., 1979, 32, 2793–2795 CAS.
- G. W. Holbert, L. B. Weiss and B. Ganem, Tetrahedron Lett., 1976, 4435–4438 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Cartesian coordinates and total energies for all optimised structures. See DOI: 10.1039/b714118a |
| This journal is © The Royal Society of Chemistry 2008 |