Metallo-nucleosides: synthesis and biological evaluation of hexacarbonyl dicobalt 5-alkynyl-2′-deoxyuridines†‡
Craig D.
Sergeant
a,
Ingo
Ott
*b,
Adam
Sniady
a,
Srinivasarao
Meneni
a,
Ronald
Gust
b,
Arnold L.
Rheingold
c and
Roman
Dembinski
*a
aDepartment of Chemistry and Center for Biomedical Research, Oakland University, 2200 N. Squirrel Rd., Rochester, Michigan 48309-4477, USA. E-mail: dembinsk@oakland.edu
bInstitute of Pharmacy, Free University of Berlin, Königin Luise Str. 2 + 4, 14195 Berlin, Germany. E-mail: ottingo@zedat.fu-berlin.de
cDepartment of Chemistry and Biochemistry, University of California, San Diego, La Jolla, California 92093-0358, USA
First published on 26th November 2007
Abstract
Reactions of 5-alkynyl-2′-deoxyuridines with dicobalt octacarbonyl Co2(CO)8 in THF at room temperature gave hexacarbonyl dicobalt nucleoside complexes (77–93%). The metallo-nucleosides were characterized, including an X-ray structure of a 1-cyclohexanol derivative. In crystalline form, the Co–Co bond is perpendicular to the plane of the uracil base, which is found in the anti position. The level of growth inhibition of MCF-7 and MDA-MB-231 human breast cancer cell lines was examined and compared to results obtained with the alkynyl nucleoside precursors. The cobalt compounds displayed good antiproliferative activities with IC50 values in the range of 5–50 µM. Interestingly, the coordination of the dicobalt carbonyl moiety to 5-alkynyl-2′-deoxyuridines led to a significant increase in the cytotoxic potency for alkyl/aryl substituents at the non-nucleoside side of the alkyne , but in the case of hydrogen (terminal alkyne ) or a silyl group, a decrease of the cytotoxic effect was observed. As demonstrated using examples for an active and a low active target compound, the cytotoxicity was significantly influenced by the uptake into the tumor cells and the biodistribution into the nuclei.
Introduction
Bioorganometallic chemistry provides new tools for control of biological interactions.1–8 In particular, organometallic compounds may offer innovative solutions for medicinal chemistry. Traditionally, platinum compounds have forged a path in this area, principally for cancer treatment. Yet, more recently, attention has encircled other transition metals and their carbonyl derivatives. For example, rhenium and technetium complexes (1, Fig. 1) show potential for breast cancer imaging and radiodiagnostics.9,10 Biological assays contrived with the aid of organometallic bioconjugates and IR spectroscopy (carbonyl metalloimmunoassay) have been elaborated.11 One current exciting development includes protein kinase inhibition by ruthenium complexes (such as 2, Fig. 1).12 The medicinal potential of metal carbonyl derivatives has been reviewed not long ago;3,13 their antitumor activity has also been noted.8 An example includes a triosmium cluster (3, Fig. 1), which exhibits anti-telomerase activity on semi-purified enzymes in a cell-free assay but is inactive towards the breast cancer MCF-7 cell line.14 Recently, a nucleoside –iron carbonyl complex (4, Fig. 1) has been reported as bestowing a significant apoptosis-inducing activity against BJAB tumor cells.15,16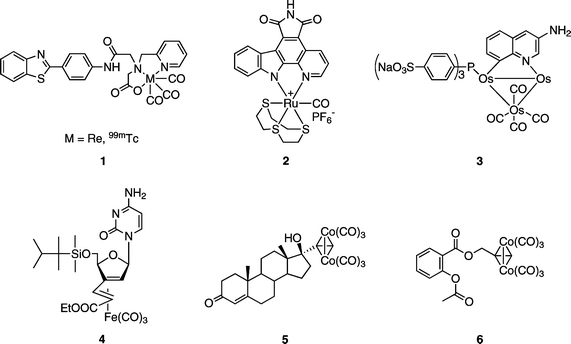 | ||
| Fig. 1 Structures of representative metal–carbonyl complexes investigated for medicinal potential. | ||
Hexacarbonyl dicobalt complexes of acetylenes ,17 in addition to material science, inorganic/organometallic or synthetic chemistry, are employed in medicinal-related investigations.11,18 A biochemical study of a cobalt derivative of 17α-ethynyltestosterone (5, Fig. 1) showed that the compound is still recognized by the androgen receptor even when the relative binding affinity is quite low (0.5%).19 The cytotoxicity of dicobalt hexacarbonyl complexes has also been reported.20–24 In particular, an aspirin cobalt carbonyl derivative (6, Fig. 1) was discovered to be antitumor active in vitro against MCF-7 and MDA-MB-231 human mammary tumor cells.22,23 In cell culture experiments hexacarbonyl[2-acetoxy-(2-propynyl)benzoate]dicobalt 6 has been found to be more active than cisplatin at each tested concentration. It has been demonstrated that the presence of the cobalt carbonyl is essential to achieve the cytotoxic effect, as the alkynyl precursor does not exhibit any activity. More systematic structure–activity relationship studies confirmed the aspirin derivative 6 as the lead compound and suggested a mode of action in which cyclooxygenase inhibition plays a major role.24
Modified nucleosides have already acquired an important role as therapeutic agents.25–29 Cytotoxic nucleoside analogues were among the first chemotherapeutic agents to be introduced for the medical treatment of cancer. This family of compounds has grown to include a variety of purine and pyrimidine nucleoside derivatives with activity in both solid tumors and hematological malignancies. These agents behave as antimetabolites, compete with physiologic nucleosides , and consequently, interact with a large number of intracellular targets to induce cytotoxicity.25 Potent biological properties have arisen by substitution at the 5-position of the uracil base.
Interest in the use of the ethynyl (acetylenic) fragment for modification of nucleoside bases has resulted in a great number of applications for 5-alkynyl uridines.30,31 Considering the antitumor activity of 5-alkynyl-2′-deoxyuridines (7)30 and the high activity of hexacarbonyl dicobalt species,20–24 a combination of both structural features in new target compounds was pursued. This strategy was further motivated by the significant apoptosis inducing properties of iron nucleoside analogues.15 Similar to the results obtained with aspirin-derived cobalt carbonyl complexes, the activity depends on the presence of the iron carbonyl moiety suggesting that metal carbonyls are useful functional groups for the modification or inducement of biological activity.
However, up to this date only one alkynyl nucleoside has been converted into its hexacarbonyl dicobalt complex, and its biological evaluation has not been pursued.32 In our ongoing interest in synthetic transformations of alkynyl-modified nucleosides 33 we have communicated a conversion of 5-(p-tolylethynyl)-2′-deoxyuridine (7d) into its hexacarbonyl dicobalt derivative (8d).32 Here, this methodology was extended to a series of 5-alkynyl-2′-deoxyuridines, for which significant antiproliferative properties have been recently reported.30 The preparation and structural characterization of the corresponding Co2(CO)6 complexes is described as well as the biological evaluation concerning cytotoxicity and uptake into the tumor cells and nuclei.
Chemistry
The series of 5-alkynyl-2′-deoxyuridines 7a–h (with the following groups: an alkyl, a cycloalkyl, a cycloalkanol, three alkylphenyls, a trialkylsilyl, and a terminal alkyne ) was synthesized, starting from 5-iodo-2′-deoxyuridine. Isolation in good yields (76–94%) was carried out according to an improved, frequently chromatography -free, larger scale protocol.30,34 Conversion to the respective cobalt carbonyl derivatives 8 came next. As illustrated in Scheme 1, the alkynyl nucleosides 7a–h were treated with Co2(CO)8 (1.2 equiv.) in THF at room temperature (22 °C) for approx. 1 h. Quantitative formation of 8 was observed by 1H NMR or TLC. Isolation was accomplished by open-air standard silica gel column chromatography and gave brown or dark red nucleosides 8a–h in high yield (77–93%). The functional groups of these nucleosides did not require any protection. The presence of a branched carbon next to the carbon–carbon triple bond did not affect the yield of 8c significantly (77%). This was not surprising, since synthesis of simple cobalt complexes with a 1-ethynylcyclohexan-1-ol motif have been reported with comparable yields.35 Structures for 8a–h are depicted in Table 1.| Compound | R | Yield (%) | IC50/µM | |
|---|---|---|---|---|
| MCF-7 | MDA-MB-231 | |||
| a Forms CHCl3 solvate. b Ref. 24. | ||||
| 8a |
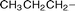
|
87 | 19.5 (±3.5) | 29.1 (±3.6) |
| 8b |

|
93 | 12.6 (±1.4) | 36.9 (±1.3) |
| 8c |
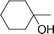
|
77 | 32.2 (±4.0) | 47.3 (±3.3) |
| 8d |
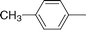
|
92 | 13.3 (±0.2) | 22.4 (±0.6) |
| 8e |
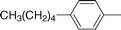
|
80 | 8.5 (±3.0) | 8.6 (±0.5) |
| 8f |
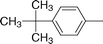
|
86 | 6.8 (±1.0) | 10.6 (±0.6) |
| 8g |
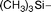
|
84a | 10.9 (±2.0) | 6.8 (±3.2) |
| 8h |
|
87 | 6.7 (±4.6) | 19.4 (±4.5) |
| Cisplatin b | — | — | 2.0 (±0.3) | 4.0 (±1.5) |
| 5-Fluorouracil b | — | — | 4.8 (±0.6) | 9.6 (±0.3) |
 | ||
| Scheme 1 Synthesis of dicobalt hexacarbonyl 5-alkynyl-2′-deoxyuridines 8. | ||
The dicobalt complexes were characterized by 1H and 13C NMR, IR, and MS. The characteristic NMR (acetone-d6) chemical shifts for 8a–h include the 1H signal of H-6 (8.29–8.57 ppm) and 13C signals of C-5 (111.9–113.7, which reflects a downfield shift as compared to the alkynyl precursors at 97.5–99.6), C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C (76.6–112.6), and CoCO (200.5–201.1 ppm).36 Tables comparing all 1H and 13C NMR chemical shifts for 8a–h are provided in the ESI† whereas a detailed characterization and similar comparison for 7a–h is available in our earlier report.30 IR spectra showed a CO stretching vibrational pattern characteristic for dicobalt hexacarbonyl alkynes (2096–2091, 2056–2050, and 2027–2021 cm−1). Absorptions attributable to the nucleoside core were also observed (usually three bands between 1702–1603 cm−1). Mass spectra for 8a–h exhibited intense molecular ions and a sequential fragmentation with a loss of consecutive carbonyl groups.37 Complexes 8 are obtained as an amorphous powder that can be stored for months in the freezer (−10 °C) under a nitrogen atmosphere without noticeable decomposition, as confirmed by 1H NMR. They are more susceptible to degradation, while in organic solvents in an ambient atmosphere for an extended time (weeks), however, no significant decomposition was noticed by 1H NMR when an acetone-d6 solution of 8f was stored in the freezer for 6 months.
C (76.6–112.6), and CoCO (200.5–201.1 ppm).36 Tables comparing all 1H and 13C NMR chemical shifts for 8a–h are provided in the ESI† whereas a detailed characterization and similar comparison for 7a–h is available in our earlier report.30 IR spectra showed a CO stretching vibrational pattern characteristic for dicobalt hexacarbonyl alkynes (2096–2091, 2056–2050, and 2027–2021 cm−1). Absorptions attributable to the nucleoside core were also observed (usually three bands between 1702–1603 cm−1). Mass spectra for 8a–h exhibited intense molecular ions and a sequential fragmentation with a loss of consecutive carbonyl groups.37 Complexes 8 are obtained as an amorphous powder that can be stored for months in the freezer (−10 °C) under a nitrogen atmosphere without noticeable decomposition, as confirmed by 1H NMR. They are more susceptible to degradation, while in organic solvents in an ambient atmosphere for an extended time (weeks), however, no significant decomposition was noticed by 1H NMR when an acetone-d6 solution of 8f was stored in the freezer for 6 months.
A molecular structure of a representative nucleoside was confirmed by X-ray crystallography.‡ Efforts to obtain diffraction quality crystals have only been successful in the case of 8c so far, by evaporation of a methanol–chloroform solution. Fig. 2 illustrates the molecular structure of the cyclohexanol-substituted nucleoside cobalt complex. It should first be noted that formation of a cobalt complex changes the position of the R group relative to the base since C–CC angles in cobalt complexes are of approximately 140–150° with the carbon atoms of the C–CC–C unit located in one plane. The C2 carbonyl group of 8c adopted an anti orientation towards the ribose ring in the crystalline form: the glycosidic bond torsion angle χ (O4′–C1′–N1–C2) is −126.7(2)°. The dihedral angle C(5)–C(7)–C(8)–C(9) of −4.21(7)° confirmed coplanarity of the alkyne and attached carbons. The dicobalt carbonyl unit is located syn to the ribose ring, with the Co–Co bond perpendicular to the uracil plane. The cyclohexanol ring adopted a staggered conformation across the C(8)–C(9) bond with O(9) of the hydroxyl group anti to the O(4′) of the ribose. In relation to the cyclohexanol ring, the Co2(CO)6 unit in 8c occupies the equatorial location. However, it is likely that the alkyne of non-coordinated 7c prefers an axial position in the chair conformation, which has been changed (with a ring flip) after incorporation of cobalt carbonyl, similar to observations during conformational studies of 1-ethynylcyclohexanols.35
![An ORTEP view of the 8c with the atom-labeling scheme. Thermal ellipsoids at the 50% probability level. Selected interatomic distances [Å]: C(5)–C(7) 1.456(4); C(7)–C(8) 1.347(4); C(8)–C(9) 1.520(4); Co(1)–Co(2) 2.4682(5); O(4)⋯H–O(9) 2.720(3). Key angles [deg]: C(5)–C(7)–C(8) 146.6(3); C(7)–C(8)–C(9) 141.3(2).](/image/article/2008/OB/b713371e/b713371e-f2.gif) | ||
| Fig. 2 An ORTEP view of the 8c with the atom-labeling scheme. Thermal ellipsoids at the 50% probability level. Selected interatomic distances [Å]: C(5)–C(7) 1.456(4); C(7)–C(8) 1.347(4); C(8)–C(9) 1.520(4); Co(1)–Co(2) 2.4682(5); O(4)⋯H–O(9) 2.720(3). Key angles [deg]: C(5)–C(7)–C(8) 146.6(3); C(7)–C(8)–C(9) 141.3(2). | ||
We also noticed a hydrogen bond between pyrimidine O(4) and cyclohexanol O(9), which is specific for compound 8c. Unlike the structure reported for 5-ethynylferrocenyl-2′-deoxycytidine,388c lacks Watson–Crick hydrogen bond motifs. This is due in part to the intramolecular O(4)⋯H–O(9) contact and non-covalent contacts between the sugar and base portions of 8c. Base N(3) and each of the sugar OH groups participate in hydrogen bonds that contribute to the organization of 8c. Packing diagrams are available in the ESI.†
Cytotoxicity
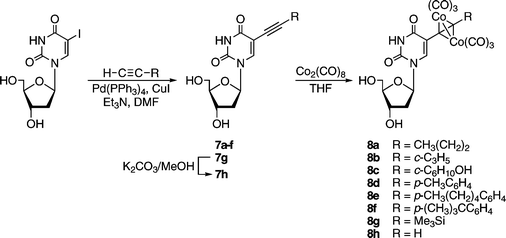
After completion of synthesis and characterization, the compounds were investigated for their antitumor activity in vitro against two different human breast cancer cultures MCF-7 and MDA-MB-231. The results are summarized in Table 1 and illustrated in comparison with the non-coordinated alkynyl nucleosides 7a–h30 in Fig. 3.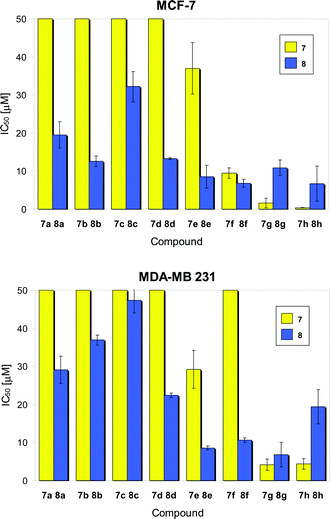 | ||
| Fig. 3 Effect of the substituents of cobalt complexes 8 and their alkynyl precursors 7 for the proliferation of MCF-7 (top) and MDA-MB-231 (bottom) human breast cancer cell lines.39 | ||
All target compounds displayed significant antiproliferative effects with IC50 values in the range of 6.7 µM (8h in MCF-7 cells) to 47.3 µM (8c in MDA-MB-231 cells). Thus, the potency of the more active target compounds is well within the range of established anticancer drugs such as 5-fluorouracil and cisplatin or the lead compound for cytotoxic hexacarbonyldicobalt complexes 6 (IC50 values in the range of 1–10 µM in this assay ).24 With the exception of 8g, MCF-7 cells were more sensitive towards the action of metallo-nucleosides, which is in good agreement with the results obtained with the non-coordinated alkynyl deoxyuridines 7a–h. Interestingly, 7a–d were inactive (IC50 values above 50 µM) whereas their cobalt carbonyl derivatives 8a–d displayed activity in both tumor cell cultures investigated (Fig. 3). The alkynyl deoxyuridine 7f had shown significant selectivity towards MCF-7 cells and was inactive in MDA-MB-231 cells. This selectivity almost disappeared for the corresponding Co2(CO)6 derivative 8f as good antiproliferative effects could be noted in MDA-MB-231 cells. Concerning the alkyne precursors, derivatives with a hydrogen or silyl group at one end of the alkyne moiety were most active (IC50 values lower than 5 µM in all experiments with 7g and 7h). For the respective cobalt carbonyl species 8g and 8h the antiproliferative activity was decreased (IC50 values 6.7 to 19.4 µM).
Obviously, the coordination of alkynes to Co2(CO)6 has a strong influence on the biological activity of the respective alkyne compounds. In general, the coordination process led to a significant increase in the cytotoxic potency for alkyl/aryl substituents at the non-nucleoside side of the alkyne , but in the case of hydrogen (terminal alkyne ) or a silyl group, a decrease of the cytotoxic effect was observed.
Uptake into cells and nuclei
Previous experiments proved that the hexacarbonyl dicobalt structure strongly increases the lipophilicity of alkyne species.24 Thus, derivatization of deoxyuridines as cobalt carbonyl derivatives will alter their cellular uptake, intracellular distribution and interaction with biomolecules such as DNA, which can be considered the main target for novel nucleoside analogues. HPLC experiments using a reversed phase stationary phase were carried out for the selected alkyne /cobalt alkyne pairs 7g/8g and 7h/8h, and confirmed the lipophilicity increase associated with addition of the dicobalt hexacarbonyl unit (as indicated by the higher retention times of the complexes compared to the free ligands, see ESI for more details† ).One of the least active (8c) and one of the most active (8f) target compounds were selected for further studies on the uptake of the complexes into the tumor cells and into the nuclei of the cells. For this purpose, an established analytical method based on atomic absorption spectroscopy was applied. Fig. 4 shows the time dependent uptake of 8c and 8f into MCF-7 and MDA-MB-231 tumor cells. In general, the cellular levels were significantly higher for 8f than for 8c, which is in good agreement with the results from the cytotoxicity studies. Complex 8f reached the highest levels after 6 h exposure while 8c accumulated faster (maxima after 2 h incubation). During 24 h the cellular cobalt contents decreased for both complexes. This trend was less striking in the MCF-7 cell line and probably contributes to the higher overall sensitivity of MCF-7 compared to MDA-MB-231 cells. Altogether these results indicate that cellular uptake plays an important role for the bioactivity of cobalt carbonyl nucleosides . It can be assumed that the hydroxyl group positioned on the cyclohexanol moiety of 8c has a negative effect on cellular drug accumulation (due to a decrease of lipophilicity) and influences excretion or metabolism.
 | ||
| Fig. 4 Cellular uptake of complexes 8c and 8f into MCF-7 (top) and MDA-MB-231 (bottom); results are expressed as nmol compound per mg cellular protein (n = 6). | ||
In order to evaluate if the target compounds reached the cellular location of their biological target DNA, the nuclei were isolated and the drug amount was quantified after exposure for 24 h (Table 2). The uptake into the nuclei increased in the same order as the cytotoxicity and cellular uptake (8c < 8h). Thus, it can be concluded that both the intracellular distribution into the nuclei and the cellular uptake determine the antiproliferative properties of the investigated target compounds.
| Cell line | Nuclear uptake/nmol mg−1 | |
|---|---|---|
| 8c | 8f | |
| MCF-7 | 0.051 (±0.020) | 0.217 (±0.096) |
| MDA-MB-231 | 0.043 (±0.014) | 0.321 (±0.159) |
Conclusions
The present study shows that the concept of Co2(CO)6 derivatization can be used to change the potency of bioactive nucleoside compounds but a critical selection of the alkyne ligand remains an important issue. As summarized in Fig. 3, the Co2(CO)6 moiety may be a useful tool to convert inactive nucleosides into active ones but does not necessarily improve the cytotoxicity of already strong antiproliferative active derivatives. Explanations for this discrepancy may be found by the investigation of the molecular interactions with potential biological targets such as DNA or DNA related enzymes, which is the subject of ongoing studies.In general, the presented results are in line with the former investigation of derivatives of benzoic acids, which showed that small structural modifications of the ligand structure can cause significant changes in the biological activity.24 Moreover, the crystallographic data presented in this report underlined the impact of Co2(CO)6 coordination on the three dimensional structure of the nucleoside ligand. As demonstrated by means of an active and a poorly active complex, the uptake into the cells and intracellular biodistribution into the nuclei—parameters that are strongly influenced by the presence of the cobalt carbonyl complex—play an important role in the bioactivity of cobalt metallo-nucleosides.
Previous studies on 6 showed a high chemical stability of the Co2(CO)6-alkyne moiety,23 as well as an extremely low cellular uptake of the precursor compound Co2(CO)8.24 Therefore, it can be assumed that only intact complexes are accumulated inside the cells where they supposedly interact with biological targets and/or are transformed to active metabolites. The fate of the complexes inside the cells remains to be elucidated in detail. However, as indicated by evaluation of the cobalt content of the nuclei, some amounts of the complexes reach the main location of the most probable primary target DNA.
As an overall result, our studies showed that the alkyne cobalt carbonyl organometallic fragment is a useful tool in medicinal chemistry research concerning the modulation of the properties of known drugs or bioactive compounds.
Experimental section
General
Commercial chemicals were treated as follows: THF distilled from Na–benzophenone. Co2(CO)8 (Acros or Strem), silica gel (J. T. Baker, 60–200 mesh), TLC plates Analtech GF, cat. number 2521 or Merck 60, cat. number 5715 used as received. 5-Alkynyl-2′-deoxyuridines 7 were obtained as described.30,40 Other materials not listed were used as received.IR spectra were recorded on a Bio-Rad FTS-175C spectrometer. NMR spectra were obtained on a Bruker Avance DPX-200 spectrometer (1H of 200 MHz and 13C of 50 MHz). Chemical shift values (δ) are in ppm and coupling constant values (J) are in Hz. Mass spectra were recorded on a Micromass ZQ instrument; m/z indicates the most intense peak of the isotope envelope. Microanalyses were conducted by Atlantic Microlab.
Synthesis of hexacarbonyl dicobalt 5-alkynyl-2′-deoxyuridines (8). General procedure:
A Schlenk flask was charged under a nitrogen atmosphere with Co2(CO)8 (typically 0.36–0.46 mmol, 1.2 equiv.), THF (typically 4–6 mL), and 7 (typically 0.30–0.38 mmol). The mixture was stirred for ca. 1 h at room temperature (22 °C). The solvent was removed by rotary evaporation and the residue was dried by oil pump vacuum for 1 h. The solid was dissolved in a minimum amount of CHCl3 (ca. 1 mL). Silica gel column chromatography (typically 25 × 2.5 cm; CHCl3–CH3OH 100 : 0 → 90 : 10) gave a red-brown fraction. Solvent was removed by rotary evaporation and the residue was dried by an oil pump vacuum to give the dicobalt hexacarbonyl compounds 8a–h as a brown or dark red powder that were stored, under a nitrogen atmosphere, in a freezer at −10 °C.Hexacarbonyl dicobalt 5-pent-1-yn-1-yl-2′-deoxyuridine (8a)
From 7a (0.0997 g, 0.339 mmol) and Co2(CO)8 (0.1390 g, 0.4065 mmol), in THF (6 mL). Dark brown solid of 8a (0.1711 g, 0.2949 mmol, 87%). Anal. (C20H18Co2N2O11) C, H: calcd, 41.40, 3.13; found, 41.40, 3.32%. IR (cm−1, KBr) νCoCO 2091 s, 2050 vs, 2021 vs; 1702 s, 1686 s, 1638 w. MS (ES+, AcOK, MeOH) 619 ((M + K)+, 15%), 591 ((M − CO + K)+, 8%), 563 ((M − 2CO + K)+, 6%), 535 ((M − 3CO + K)+, 8%), 507 ((M − 4CO + K)+, 6%), 333 ((M − Co2(CO)6 + K)+, 100%). NMR (acetone-d6):361H 10.28 (s, 1H, N-3), 8.37 (s, 1H, H-6), 6.39 (dd, J = 7.7, 6.2, 1H, H-1′), 4.58–4.48 (m, 1H, OH-5′), 4.43 (d, J = 3.7, 1H, OH-3′), 4.32 (t, J = 4.8, 1H, H-3′), 4.04 (q, J = 2.2, 1H, H-4′), 3.90–3.75 (m, 2H, H-5′), 3.09 (t, 2H, J = 7.9, H-1″), 2.36–2.25 (m, 2H, H-2′), 1.71 (sextet, 2H, J = 7.7, H-2″), 1.06 (t, 3H, J = 7.3, H-3″); 13C{1H} 201.0 (CoCO), 161.5 (C-4), 150.8 (C-2), 140.2 (C-6), 113.3 (C-5), 104.8 (dU–CC), 89.3 (C-4′), 86.6 (C-1′), 85.6 (dU-CC), 73.0 (C-3′), 63.3 (C-5′), 41.9 (C-2′), 37.4 (C-1″), 26.0 (C-2″), 14.4 (C-3″).
Hexacarbonyl dicobalt 5-(cyclopropylethynyl)-2′-deoxyuridine (8b)
From 7b (0.1012 g, 0.3462 mmol) and Co2(CO)8 (0.1421 g, 0.4155 mmol), in THF (6 mL). Dark brown solid of 8b (0.1851 g, 0.3201 mmol, 93%). Anal. (C20H16Co2N2O11) C, H: calcd, 41.54, 2.79; found, 41.01, 2.90%. IR (cm−1, KBr) νCoCO 2091 s, 2051 vs, 2022 vs; 1702 s, 1686 s, 1637 w. MS (ES+, AcOK, MeOH) 617 ((M + K)+, 30%), 589 ((M − CO + K)+, 11%), 561 ((M − 2CO + K)+, 6%), 533 ((M − 3CO + K)+, 13%), 505 ((M − 4CO + K)+, 10%), 331 ((M − Co2(CO)6 + K)+, 100%). NMR (acetone-d6):361H 10.27 (s, 1H, N-3), 8.35 (s, 1H, H-6), 6.39 (dd, J = 7.6, 6.3, 1H, H-1′), 4.59–4.47 (m, 1H, OH-5′), 4.43 (d, J = 3.6, 1H, OH-3′), 4.31 (t, J = 4.6, 1H, H-3′), 4.08–3.98 (m, 1H, H-4′), 3.90–3.65 (m, 2H, H-5′), 2.62–2.41 (m, 1H, H-1″), 2.38–2.21 (m, 2H, H-2′), 1.24–1.06 and 0.95–0.65 (2 m, 2 × 2H, H-2″ and H-3″); 13C{1H} 200.8 (CoCO), 161.3 (C-4), 150.8 (C-2), 139.6 (C-6), 113.2 (C-5), 109.2 (dU–CC), 89.2 (C-4′), 86.4 (C-1′), 85.0 (dU–CC), 73.0 (C-3′), 63.2 (C-5′), 41.8 (C-2′), 16.3 (C-1″), 13.1 and 12.9 (C-2″ and C-3″).
Hexacarbonyl dicobalt 5-[(1-hydroxycyclohexyl)ethynyl]-2′-deoxyuridine (8c)
From 7c (0.1332 g, 0.3802 mmol) and Co2(CO)8 (0.1560 g, 0.4562 mmol), in THF (6 mL). Dark brown solid of 8c (0.1850 g, 0.2912 mmol, 77%). Anal. (C23H22Co2N2O12) C, H: calcd, 43.42, 3.49; found, 43.41, 3.77%. IR (cm−1, KBr) νCoCO 2093 s, 2054 vs, 2025 vs; 1702 s, 1670 s, 1637 w. MS (ES+, AcOK, MeOH) 1311 ((2 M + K)+, 12%), 675 ((M + K)+, 53%), 647 ((M + K − CO)+, 35%), 619 ((M − 2CO + K)+, 8%), 591 ((M − 3CO + K)+, 21%), 389 ((M − Co2(CO)6 + K)+, 100%). NMR (acetone-d6):361H 10.60 (s, 1H, N-3), 8.47 (s, 1H, H-6), 6.38 (dd, J = 7.9, 5.9, 1H, H-1′), 5.59 (s, 1H, HO–C6H10), 4.53 (br s, 1H, OH-5′), 4.45 (br s, 1H, OH-3′), 4.34 (br s, 1H, H-3′), 4.11–4.01 (m, 1H, H-4′), 3.92–3.70 (m, 2H, H-5′), 2.45–2.15 (m, 2H, H-2′), 2.00–1.45 (m, 9H, c-C6H10), 1.35–1.10 (m, 1H, c-C6H10); 13C 200.8 (CoCO), 162.9 (d, J = 9.6, C-4), 150.3 (d, J = 8.0, C-2), 142.2 (d, J = 183.5, C-6), 113.1 (s, C-5), 112.6 (dU–CC), 89.4 (d, J = 149.4, C-4′), 86.7 (d, J = 170.4, C-1′), 84.4 (d, J = 3.5, dU–CC), 73.2 (s, C-1″), 73.0 (d, J = 149.1, C-3′), 63.1 (d, J = 139.9, C-5′), 42.0 (t, J = 133.2, C-2′), 40.6 and 40.4 (2t, J = 132.1, C-2″), 26.5 (t, J = 125.0, C-4″), 23.9 and 22.8 (2t, J = 129.1, C-3″).
Hexacarbonyl dicobalt 5-[(4-methylphenyl)ethynyl]-2′-deoxyuridine (8d)
From 7d (0.1016 g, 0.2968 mmol) and Co2(CO)8 (0.1218 g, 0.3561 mmol), in THF (5 mL). Dark brown solid of 8d (0.1716 g, 0.2731 mmol, 92%). Anal. (C24H18Co2N2O11) C, H: calcd, 45.88, 2.89; found, 45.66, 3.36%. IR (cm−1, KBr) νCoCO 2092 s, 2055 vs, 2023 vs; 1690 s, 1603 w. MS37 (ES+, KCl, MeOH) 1295 ((2 M + K)+, 36%), 667 ((M + K)+, 92%), 639 ((M + K − CO)+, 100%), 629 ((M + H)+, 12%). NMR (acetone-d6):361H 10.32 (s, 1H, N-3), 8.43 (s, 1H, H-6), 7.53 (d, J = 8.0, 2H, o-C6H4CC), 7.19 (d, J = 8.0, 2H, m-C6H4CC), 6.43 (t, J = 6.9, 1H, H-1′), 4.52 (br s, 1H, OH-5′), 4.43 (d, J = 2.7, 1H, OH-3′), 4.24 (t, J = 4.5, 1H, H-3′), 4.06–4.02 (m, 1H, H-4′), 3.83–3.68 (m, 2H, H-5′), 2.32 (s, 3H, CH3), 2.40–2.25 (m, 2H, H-2′); 13C{1H} 200.5 (CoCO), 160.8 (C-4), 150.8 (C-2), 139.8 (C-6), 138.8 (p-C6H4CC), 136.3 (i-C6H4CC), 130.6 (o-C6H4CC), 130.2 (m-C6H4CC), 113.6 (C-5), 96.7 (dU-CC), 89.2 (C-4′), 86.4 (C-1′), 86.2 (dU–CC), 73.1 (C-3′), 63.3 (C-5′), 41.9 (C-2′), 21.4 (CH3).
Hexacarbonyl dicobalt 5-[(4-pentylphenyl)ethynyl]-2′-deoxyuridine (8e)
From 7e (0.1200 g, 0.3012 mmol) and Co2(CO)8 (0.1236 g, 0.3614 mmol), in THF (5 mL). Dark brown solid of 8e (0.1643 g, 0.2402 mmol, 80%). Anal. (C28H26Co2N2O11) C, H: calcd, 49.14, 3.83; found, 48.76, 3.93%. IR (cm−1, KBr) νCoCO 2092 s, 2056 vs, 2024 vs; 1700 s, 1686 s, 1606 w. MS (ES+, KCl, MeOH) 721 ((M + K)+, 65%), 693 ((M − CO + K)+, 100%), 666 ((M − 2CO + K)+, 32%), 638 ((M − 3CO + K)+, 37%), 610 ((M − 4CO + K)+, 64%), 582 ((M − 5CO + K)+, 59%), 554 ((M − 6CO + K)+, 42%), 436 ((M − Co2(CO)6 + K)+, 25%). NMR (acetone-d6):361H 10.34 (s, 1H, N-3), 8.43 (s, 1H, H-6), 7.56 (d, J = 8.2, 2H, o-C6H4CC), 7.22 (d, J = 8.2, 2H, m-C6H4CC), 6.43 (dd, J = 7.9, 6.2, 1H, H-1′), 4.58–4.48 (m, 1H, OH-5′), 4.46 (d, J = 3.6, 1H, OH-3′), 4.26 (t, J = 4.6, 1H, H-3′), 4.11–4.00 (m, 1H, H-4′), 3.90–3.67 (m, 2H, H-5′), 2.61 (t, J = 7.3, 2H, H-1″), 2.42–2.18 (m, 2H, H-2′), 1.74–1.54 (m, 2H, H-2″), 1.43–1.25 (m, 4H, H-3″, H-4″), 0.89 (t, J = 6.6, 3H, H-5″); 13C{1H} 200.6 (CoCO), 160.8 (C-4), 150.8 (C-2), 143.8 (C-6), 139.8 (p-C6H4CC), 136.5 (i-C6H4CC), 130.6 (o-C6H4CC), 129.5 (m-C6H4CC), 113.6 (C-5), 96.7 (dU–CC), 89.2 (C-4′), 86.4 (C-1′), 86.2 (dU–CC), 73.1 (C-3′), 63.2 (C-5′), 41.8 (C-2′), 36.4 (C-1″), 32.3 (C-3″), 31.8 (C-2″), 23.2 (C-4″), 14.3 (C-5″).
Hexacarbonyl dicobalt 5-[(4-tert-butylphenyl)ethynyl]-2′-deoxyuridine (8f)
From 7f (0.0622 g, 0.162 mmol) and Co2(CO)8 (0.0665 g, 0.194 mmol), in THF (4 mL). Dark brown solid of 8f (0.0935 g, 0.140 mmol, 86%). Anal. (C27H24Co2N2O11) C, H: calcd, 48.38, 3.61; found, 48.08, 3.63%. IR (cm−1, KBr) νCoCO 2091 s, 2055 vs, 2025 vs; 1702 s, 1686 s, 1637 w. MS (ES+, AcOK, MeOH) 709 ((M + K)+, 100%), 681 ((M + K − CO)+, 71%), 653 ((M − 2CO + K)+, 29%), 625 ((M − 3CO + K)+, 22%), 597 ((M − 4CO + K)+, 6%), 423 ((M − Co2(CO)6 + K)+, 48%). NMR (acetone-d6):361H 10.33 (s, 1H, N-3), 8.44 (s, 1H, H-6), 7.60 (d, J = 8.2, 2H, o-C6H4CC), 7.44 (d, J = 8.2, 2H, m-C6H4CC), 6.44 (dd, J = 7.8, 6.2, 1H, H-1′), 4.58–4.47 (m, 1H, OH-5′), 4.43 (d, J = 3.7, 1H, OH-3′), 4.21 (t, J = 4.6, 1H, H-3′), 4.10–4.00 (m, 1H, H-4′), 3.90–3.65 (m, 2H, H-5′), 2.43–2.16 (m, 2H, H-2′), 1.35 (s, 9H, C(CH3)3); 13C 200.5 (s, CoCO), 160.9 (d, J = 9.3, C-4), 151.8 (br s, p-C6H4CC), 151.0 (d, J = 7.4, C-2), 139.8 (d, J = 181.0, C-6), 136.3 (t, J = 7.8, i-C6H4CC), 130.4 (dd, J = 159.8, 6.0, o-C6H4CC), 126.5 (dd, J = 157.2, 6.6, m-C6H4CC), 113.6 (s, C-5), 96.5 (s, dU–CC), 89.2 (d, J = 147.3, C-4′), 86.4 (d, J = 169.6, C-1′), 86.2 (d, J = 4.8, dU–CC), 73.1 (d, J = 151.0, C-3′), 63.3 (t, J = 141.0, C-5′), 41.9 (t, J = 133.8, C-2′), 35.3 (s, C(CH3)3), 31.5 (qt, J = 126.0, 4.4, C(CH3)3).
Hexacarbonyl dicobalt 5-[(trimethylsilyl)ethynyl]-2′-deoxyuridine (8g)
From 7g (0.1000 g, 0.3083 mmol) and Co2(CO)8 (0.1266 g, 0.3700 mmol), in THF (5 mL). Dark brown solid of 8g·1/3(CHCl3) (0.1680 g, 0.2584 mmol, 84%). Anal. (C20H20Co2N2O11Si)·1/3(CHCl3): C, H: calcd, 37.56, 3.15; found, 37.66, 3.63%. IR (cm−1, KBr) νCoCO 2090 s, 2052 vs, 2021 vs; 1700 s, 1685 s, 1638 w. MS (ES+, KCl, MeOH) 648 ((M + K)+, 76%), 620 ((M − CO + K)+, 61%), 592 ((M − 2CO + K)+, 37%), 564 ((M − 3CO + K)+, 11%), 536 ((M − 4CO + K)+, 65%), 508 ((M − 5CO + K)+, 6%), 480 ((M − 6CO + K)+, 51%), 409 ((M − 6CO − Si(CH3)3 + K)+, 57%), 363 ((M − Co2(CO)6 + K)+, 100%). NMR (acetone-d6):361H 10.38 (s, 1H, N-3), 8.29 (s, 1H, H-6), 6.38 (dd, J = 7.9, 5.9, 1H, H-1′), 4.65–4.47 (m, 1H, OH-5′), 4.44 (d, J = 3.6, 1H, OH-3′), 4.26 (t, J = 4.7, 1H, H-3′), 4.05 (q, J = 2.2, 1H, H-4′), 3.95–3.75 (m, 2H, H-5′), 2.45–2.15 (m, 2H, H-2′), 0.37 (s, 9H, Si(CH3)3); 13C{1H} 201.1 (CoCO), 160.8 (C-4), 150.6 (C-2), 140.7 (C-6), 113.7 (C-5), 98.1 (dU–CC), 89.2 (C-4′), 86.5 (C-1′), 85.7 (dU–CC), 73.0 (C-3′), 63.1 (C-5′), 41.7 (C-2′), 1.0 (Si(CH3)3).
Hexacarbonyl dicobalt 5-ethynyl-2′-deoxyuridine (8h)
From 7h (0.0802 g, 0.318 mmol) and Co2(CO)8 (0.1305 g, 0.3816 mmol), in THF (6 mL). Dark brown solid of 8h (0.1490 g, 0.2769 mmol, 87%). Anal. (C17H12Co2N2O11) C, H: calcd, 37.94, 2.25; found, 38.04, 2.46%. IR (cm−1, KBr) νCoCO 2096 s, 2055 vs, 2027 vs; 1702 s, 1686 s, 1638 w. MS (ES+, AcOK, MeOH) 577 ((M + K)+, 58%), 549 ((M − CO + K)+, 34%), 521 ((M − 2CO + K)+, 6%), 493 ((M − 3CO + K)+, 20%), 465 ((M − 4CO + K)+, 20%), 291 ((M − Co2(CO)6 + K)+, 100%). NMR (acetone-d6):361H 10.25 (s, 1H, N-3), 8.57 (s, 1H, H-6), 6.71 (s, 1H, CCH), 6.35 (t, J = 6.6, 1H, H-1′), 4.53 (br s, 1H, OH-5′), 4.44 (br s, 1H, OH-3′), 4.36 (t, J = 4.2, 1H, H-3′), 4.10–3.93 (m, 1H, H-4′), 3.93–3.72 (m, 2H, H-5′), 2.39–2.20 (m, 2H, H-2′); 13C 200.9 (s, CoCO), 161.5 (d, J = 9.3, C-4), 150.8 (d, J = 8.0, C-2), 141.0 (d, J = 179.5, C-6), 111.9 (s, C-5), 89.2 (d, J = 148.9, C-4′), 86.6 (d, J = 170.5, C-1′), 83.1 (s, dU–CC), 76.6 (d, J = 224.4, dU–CC), 72.5 (d, J = 150.3, C-3′), 62.9 (t, J = 140.5, C-5′), 41.9 (t, J = 133.7, C-2′).
Crystallography
Light brown plates of 8c were grown by evaporation of a CHCl3–MeOH (1 : 1 v/v) solution during 14 days, placed over molecular sieves in a closed jar under a nitrogen atmosphere in a glove box. Data were collected as outlined in the ESI (Table S3).†Biological evaluation
Experiments concerning cytotoxicity and uptake into the cells and nuclei were performed according to recently described procedures.24Acknowledgements
We thank the National Institutes of Health (NIH, grant CA111329), the Deutsche Forschungsgemeinschaft (DFG, grant FOR630), and the Oakland University Research Excellence Program in Biotechnology for support of this research. The Thompson Award for C.D.S. is acknowledged, and Prof. S. Szafert and Prof. K. Wheeler for helpful discussions.References
- Concepts and Models in Bioinorganic Chemistry, ed. H. B. Kraatz and N. Metzler-Nolte, Wiley-VCH, Weinheim, 2006, pp. 1–443 Search PubMed.
- R. H. Fish and G. Jaouen, Organometallics, 2003, 22, 2166–2177 CrossRef CAS.
- N. Metzler-Nolte, Angew. Chem., Int. Ed., 2001, 40, 1040–1043 CrossRef CAS.
- (a) R. Dagani, Chem. Eng. News, 2002, 80, 23–29; (b) Z. Guo and P. J. Sadler, Angew. Chem., Int. Ed., 1999, 38, 1512–1531 CrossRef CAS.
- U. Schatzschneider and N. Metzler-Nolte, Angew. Chem., Int. Ed., 2006, 45, 1504–1507 CrossRef CAS.
- A. R. Timerbaev, C. G. Hartinger, S. S. Aleksenko and B. K. Keppler, Chem. Rev., 2006, 106, 2224–2248 CrossRef CAS.
- N. Metzler-Nolte, Nachr. Chem., 2006, 54, 966–970 Search PubMed.
- I. Ott and R. Gust, Arch. Pharm. Chem. Life Sci., 2007, 340, 117–126 Search PubMed.
- S. Tzanopoulou, I. C. Pirmettis, G. Patsis, M. Paravatou-Petsotas, E. Livaniou, M. Papadopoulos and M. Pelecanou, J. Med. Chem., 2006, 49, 5408–5410 CrossRef CAS.
- For other Re and Tc tricarbonyl complexes designed as myocardial imaging agents see: L. Maria, S. Cunha, M. Videira, L. Gano, A. Paulo, I. C. Santos and I. Santos, Dalton Trans., 2007, 3010–3019 Search PubMed.
- (a) M. Salmain, N. Fischer-Durand, L. Cavalier, B. Rudolf, J. Zakrzewski and G. Jaouen, Bioconjugate Chem., 2002, 13, 693–698 CrossRef CAS; (b) M. Salmain, A. Vessières, A. Varenne, P. Brossier and G. Jaouen, J. Organomet. Chem., 1999, 589, 92–97 CrossRef CAS.
- (a) H. Bregman and E. Meggers, Org. Lett., 2006, 8, 5465–5468 CrossRef CAS; (b) J. É. Debreczeni, A. N. Bullock, G. E. Atilla, D. S. Williams, H. Bregman, S. Knapp and E. Meggers, Angew. Chem., Int. Ed., 2006, 45, 1580–1585 CrossRef CAS; (c) E. Meggers, G. E. Atilla-Gokcumen, H. Bregman, J. Maksimoska, S. P. Mulcahy, N. Pagano and D. S. Williams, Synlett, 2007, 1177–1189 CrossRef CAS.
- T. R. Johnson, B. E. Mann, J. E. Clark, R. Foresti, C. J. Green and R. Motterlini, Angew. Chem., Int. Ed., 2003, 42, 3722–3729 CrossRef CAS.
- D. Colangelo, A. Ghiglia, A. Ghezzi, M. Ravera, E. Rosenberg, F. Spada and D. Osella, J. Inorg. Biochem., 2005, 99, 505–512 CrossRef CAS.
- D. Schlawe, A. Majdalani, J. Velcicky, E. Heßler, T. Wieder, A. Prokop and H.-G. Schmalz, Angew. Chem., Int. Ed., 2004, 43, 1731–1734 CrossRef CAS.
- Selected representative recent reference of metal carbonyls attached to nucleosides: L. Wei, J. Babich, W. C. Eckelman and J. Zubieta, Inorg. Chem., 2005, 44, 2198–2209 Search PubMed.
- For electronic structure of the alkyne-bridged dicobalt hexacarbonyl complex see: J. A. Platts, G. J. S. Evans, M. P. Coogan and J. Overgaard, Inorg. Chem., 2007, 46, 6291–6298 Search PubMed.
- Peptide labeling using a dicobalt hexacarbonyl alkynyl complex has been reported: M. A. Neukamm, A. Pinto and N. Metzler-Nolte, Chem. Commun., 2008 10.1039/b712886j.
- (a) S. Top, H. El Hafa, A. Vessières, M. Huché, J. Vaissermann and G. Jaouen, Chem.–Eur. J., 2002, 8, 5241–5249 CrossRef CAS; (b) D. Osella, F. Galeotti, G. Cavigiolio, C. Nervi, K. I. Hardcastle, A. Vessières and G. Jaouen, Helv. Chim. Acta, 2002, 85, 2918–2925 CrossRef CAS.
- (a) I. Ott, B. Kircher and R. Gust, J. Inorg. Biochem., 2004, 98, 485–489 CrossRef CAS; (b) T. Roth, C. Eckert, H.-H. Fiebig and M. Jung, Anticancer Res., 2002, 22, 2281–2284 CAS; (c) M. Jung, D. E. Kerr and P. D. Senter, Arch. Pharm. Pharm. Med. Chem., 1997, 330, 173–176 CrossRef CAS.
- Hexacarbonyl dicobalt propargylfructopyranose: I. Ott, T. Koch, H. Shorafa, Z. Bai, D. Poeckel, D. Steinhilber and R. Gust, Org. Biomol. Chem., 2005, 3, 2282–2286 Search PubMed.
- K. Schmidt, M. Jung, R. Keilitz, B. Schnurr and R. Gust, Inorg. Chim. Acta, 2000, 306, 6–16 CrossRef CAS.
- For interaction with proteins and stability see: I. Ott and R. Gust, Biometals, 2005, 18, 171–177 Search PubMed.
- I. Ott, K. Schmidt, B. Kircher, P. Schumacher, T. Wiglenda and R. Gust, J. Med. Chem., 2005, 48, 622–629 CrossRef CAS.
- (a) Deoxynucleoside Analogs in Cancer Therapy, ed. J. P. Godefridus, Humana Press, Totowa, 2006 Search PubMed; (b) Nucleosides and Nucleotides as Antitumor and Antiviral Agents, ed. C. K. Chu and D. C. Baker, Plenum Press, New York, 1993 Search PubMed.
- (a) C. M. Galmarini, J. R. Mackey and C. Dumontet, Lancet Oncol., 2002, 3, 415–424 Search PubMed; (b) C. M. Galmarini, Electr. J. Oncol., 2002, 1, 22–32 Search PubMed.
- S. Hatse, E. De Clercq and J. Balzarini, Biochem. Pharmacol., 1999, 58, 539–555 CrossRef CAS.
- I. E. Smith, Lancet, 2002, 360, 790–792 CrossRef.
- A representative collection of articles related to antiviral investigations can be found in the special issue: Antiviral Res., 2006, 71, 75–414.
- S. Meneni, I. Ott, C. D. Sergeant, A. Sniady, R. Gust and R. Dembinski, Bioorg. Med. Chem., 2007, 15, 3082–3088 CrossRef CAS , and references therein.
- Selected recent representative examples: (a) R. E. Hudson and A. Ghorbani-Choghamarani, Org. Biomol. Chem., 2007, 5, 1845–1848 RSC; (b) W. A. Cristofoli, L. I. Wiebe, E. De Clercq, G. Andrei, R. Snoeck, J. Balzarini and E. E. Knaus, J. Med. Chem., 2007, 50, 2851–2857 CrossRef CAS.
- N. Esho, B. Davies, J. Lee and R. Dembinski, Chem. Commun., 2002, 332–333 RSC.
- M. S. Rao, N. Esho, C. Sergeant and R. Dembinski, J. Org. Chem., 2003, 68, 6788–6790 CrossRef CAS.
- The nucleoside 7h was prepared by desilylation of 7g.
- N. M. Deschamps, J. H. Kaldis, P. E. Lock, J. F. Britten and M. J. McGlinchey, J. Org. Chem., 2001, 66, 8585–8591 CrossRef CAS.
-
(a) We were not able to achieve an NMR spectral resolution comparable to that of the 5-alkynyl-2′-deoxyuridines.30 This can likely be attributed to the presence of cobalt. Thus, more advanced techniques
that would help to facilitate complete assignments of signals were not pursued. Doublet/broad singlet and multiplet peaks (acetone-d6) were assigned to 3′- and 5′-OH, respectively. For 7, the 3′- and 5′-OH (also assigned based upon multiplicity; doublet and triplet, DMSO-d6) exhibit reversed relative positions of chemical shifts;
(b) Pairs of o- and m- of the phenyl rings 1H and 13C signals, C-1′ and C-4′, and CC signals were tentatively assigned following trends for 7,30 except the CC for 8h, where we were able to observe 1JC–H.
- MS ES+ (positive) data are given. We observed that negative spectra also show consecutive fragmentations with a loss of CO. A representative ES− (negative) spectrum for 8d is available in the supplementary materials of our earlier work32.
- A. R. Pike, L. C. Ryder, B. R. Horrocks, W. Clegg, B. A. Connolly and A. Houlton, Chem.–Eur. J., 2005, 11, 344–353 CrossRef.
- Corrected value for 7d/MCF-7 from the one given in ref. 30.
- The equivalents reported in the general procedure in ref. 30 apply to volatile alkynes. To produce 7c–f, 1.2 equiv. of a less volatile alkyne may be used. For an alternate procedure utilizing volatile alkynes see: S. Ghilagaber, W. N. Hunter and R. Marquez, Tetrahedron Lett., 2007, 48, 483–486 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Tables of comparison of 1H and 13C NMR signals for compounds 8a–h. 1H and 13C NMR spectra for compounds 8a–c, e–h. Crystal and refinement data, and packing diagrams for compound 8c. See DOI: 10.1039/b713371e |
| ‡ CCDC reference number 646479. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b713371e |
| This journal is © The Royal Society of Chemistry 2008 |