An improved synthesis of cis-4-phenyl-2-propionamidotetralin (4-P-PDOT): a selective MT2melatonin receptor antagonist
Simone
Lucarini
,
Annalida
Bedini
,
Gilberto
Spadoni
and
Giovanni
Piersanti
*
Institute of Medicinal Chemistry, University of Urbino “Carlo Bo”, Piazza del Rinascimento 6, 61029 Urbino (PU), Italy. E-mail: giovanni.piersanti@uniurb.it; Fax: +39 0722 303313; Tel: +39 0722 303323
First published on 13th November 2007
Abstract
A novel, efficient and diastereoselective procedure was developed for the gram-scale synthesis of cis-4-phenyl-2-propionamidotetralin (4-P-PDOT), a selective MT2melatonin receptor antagonist. The synthetic strategy involved the conversion of 4-phenyl-2-tetralone to enamide followed by diastereoselective reduction affording cis-4-P-PDOT in good yield. The mechanism of the reduction step was explored by employing deuterated reagents.
Introduction
Most compounds endowed with biological interest are molecules with a high degree of conformational freedom, which produces several conformational isomers. Increasing the rigidity of bioactive molecules is a valuable tool for investigating the stereochemical features of small-molecule binding sites. In the case of simple flexible substances, such as arylethylamines, a method commonly used to reduce the flexibility has been the modification of the ethylamine fragment into a bicyclic system. For instance, the tetralin skeleton has been successfully used as a rigid template for the synthesis of non-indolic melatonin-like agents,1 and several other substances possessing important biological activities.2,3Melatonin (N-acetyl-5-methoxytryptamine, MLT), a neurohormone mainly secreted by the pineal gland during dark periods, is an indole derivative with a flexible ethylamido chain attached at the C3 position. In mammals, melatonin modulates a variety of cellular, neuroendocrine and physiological processes through activation of at least two high-affinity G-protein coupled receptors, named MT1 and MT2.4
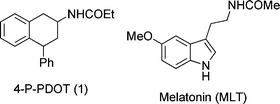
Converging evidence from pharmacophore analysis, 3D-QSAR and GPCR comparative modelling in the field of melatonin receptor ligands has allowed the definition of the structural requirements for MT2 selective antagonism.5–7 In particular, we evidenced that the presence of a bulky substituent in an area corresponding to positions 1 and 2 of the indole nucleus of MLT, and “out-of-plane” from the indole ring, confers selectivity for the MT2receptor and leads to a reduction of intrinsic activity.8 The melatonin receptor ligand 4-phenyl-2-propionamidotetralin (4-P-PDOT, Fig. 1) fulfilled this requirement9 and is one of the most interesting melatonin MT2 selective antagonists.
 | ||
| Fig. 1 | ||
Recently, we examined the influence of different stereochemistries of 4-phenyl-2-propionamidotetralin on the binding affinity and intrinsic activity at human MT1 and MT2 receptors.10 The (±)-cis diastereoisomer of compound1 (Fig. 1) has higher MT2 binding affinity (pKi = 10.8), as compared to its corresponding trans-isomer (pKi = 8.45) and good selectivity for the MT2receptor (MT2/MT1 = 225) rendering cis-4-P-PDOT one of the most used pharmacological tools to identify functional MT2 receptors.
Although this compound has been used in small amounts in in vitro tests to provide some understanding of the roles of MT1 and MT2melatonin receptors,11–13 more precise and specific in vivo animal experiments, where large amounts of compound are required, are necessary to clarify the physiological role of both receptors.
The need for multi-gram amounts of this very expensive substance14 for in vivo studies prompted us to design a more efficient, diastereoselective route to the more active cis-4-P-PDOT.
The method used in the first patent route,15 involved a base-catalyzed condensation between phenylacetone and benzaldehyde, followed by ring closure via an intramolecular Friedel–Crafts alkylation with AlCl3 in CS2 to give 4-phenyl-2-tetralone. The 4-phenyl-2-propionamidotetralin 1 was obtained by reductive amination with benzylamine, deprotection and finally acylation with propionic anhydride according to the Schotten–Baumann procedure. The overall yield for 4-P-PDOT prepared according to this synthetic route was very low (ca. 1%) and moreover there is no mention of the stereochemistry of the product synthesized.
Our initial approach to the synthesis of the cis-diastereoisomer of compound 1 (Scheme 1)10 was based on the protocol reported by Wyrick et al.16 and involved the synthesis of 4-phenyl-2-tetralone 4 by cyclization of trans-1,4-diphenyl-3-buten-2-one 3 in polyphosphoric acid followed by reduction with NaBH4 to give a 85 : 15 cis–trans mixture of tetralols 5. The predominantly cis-tetralol was epimerized via the ester intermediate 6 to the trans-alcohol 7. The latter was converted to the cis-primary amine 9 by tosylation, followed by reaction with sodium azide in aqueous DMF then catalytical (10% Pd/C) hydrogenation. Finally, the primary amine was converted to (±)-cis-4-phenyl-2-propionamidotetralin 1 by acylation with propionic anhydride.
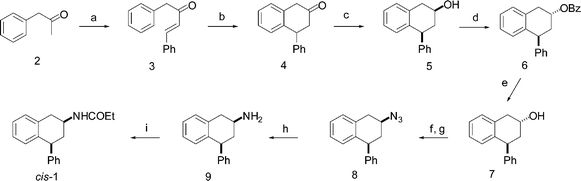 | ||
| Scheme 1 Precedented synthetic approach. Reagents: (a) PhCHO, NaOH, water, 60 °C, 18 h; (b) PPA, xylene, reflux, 2 h; (c) NaBH4, MeOH, reflux, 16 h; (d) DEAD, (C6H5)3P, PhCOOH, THF, 55 °C, 4 h; (e) NaOH, MeOH, reflux, 2 h; (f) TsCl, Py, 5 °C, 5 days; (g) NaN3, DMF–H2O, 50 °C, 4 h; (h) H2 (4 atm.) 10% Pd/C, 2-propanol, rt, 24 h; (i) (C2H5CO)2O, Et3N, toluene, rt, 24 h. | ||
The described synthetic procedure (Scheme 1) involved overall nine tedious steps, several flash chromatography purifications, the use of some hazardous reagents and the overall transformations are not very efficient.
Herein we describe a more efficient synthesis of (±)-cis-4-P-PDOT in only three steps and high overall yield that is suitable for multigram-scale preparations.
Results and discussion
As presented, with the improved synthesis of cis-1 reported in Schemes 2 and 3, we first sought a more direct single step synthesis of the key tetralone 4 through addition of styrene to phenylacetyl chloride in the presence of aluminium chloride as a catalyst, by analogy to what has been reported previously.17 Then, we explored the reactivity of this cyclic ketone with nitrogen nucleophiles. Attempts to convert 4-phenyl-2-tetralone to 4-phenyl-2-aminotetraline by reductive amination procedures gave low yields of product15 or even no product at all16 as the tetralone dimerized immediately in the presence of weak bases such as the amines employed.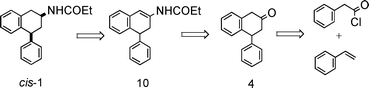 | ||
| Scheme 2 Retrosynthetic analysis. | ||
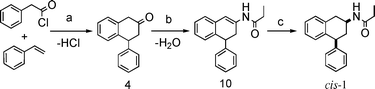 | ||
| Scheme 3 Novel synthetic approach. Reagents: (a) AlCl3, CH2Cl2, 0 °C 30 min; (b) propylamide, PTSA cat., toluene, reflux, 4 h; (c) TES, TFA, −10 °C, 10 min. | ||
We envisioned, then, propionamide, as a less basic and nucleophilic enough alternative to the amine, to couple with the tetralone 4. In fact it is well-known that enamides can be easily prepared by the acid catalyzed condensation of primary amides with carbonyl compounds18 and we took advantage of this trivial fact in designing our synthetic route. Stereoselective reduction of the newly formed enamide 10 (Scheme 2) would complete the synthesis of the target compound.
The easy and direct route to the key enamide 10 was achieved by condensation of primary propylamide with the cyclic ketone 4 in the presence of p-toluenesulfonic acid as catalyst and continuous removal of water using Dean–Stark apparatus.
Given our project’s ongoing synthesis of novel analogs of 4-P-PDOT we required a general reduction procedure for enamides which would be tolerant to functional groups. In addition, we required the regiospecific delivery of a hydride equivalent to the vinyl group in anticipation of the preparation of several 3H-labeled analogs.
With all this in mind we thought to use ionic reduction conditions for a chemo-, regio- and stereoselective reduction of the enamide. We were delighted to find that treatment of the substrate 10 with Et3SiH (TES) in the presence of trifluoroacetic acid (TFA) furnished the desired product in excellent yield and good diastereoselectivity (91% yield; 81 : 19 cis–trans ratio). No reduction took place when acetic acid was used instead of TFA. Attempts to further improve the diastereoselectivity by lowering the temperature to −78 °C resulted in unacceptable low conversion rates.
It is noteworthy, that the present method offers a highly atom economic process to 4-P-PDOT.
The pure cis-isomer can be easily isolated by crystallization (acetone–n-hexane). The physical–chemical properties and NMR-spectra of compound cis-1 synthesized by the novel procedure (Scheme 3) were identical to those previously reported.11
The mechanism of the reduction was briefly explored by carrying out the reaction in the presence of deuterated and non-deuterated reagents. Reduction of 10 using Et3SiD in place of Et3SiH, resulted in high conversion to deuterium labeled 4-P-PDOT d1-cis-1a (Fig. 2) in about the same time (10 min). In contrast, by using CF3COOD only 4-P-PDOT d1-cis-1b was obtained. Comparison of the 1H NMR signals for the CH and -CH2groups of cis-1a and cis-1b with those of 1 showed simplified 1H NMR multiplet patterns due to substitution by 2H. This was confirmed by analysis of the MS spectrum which revealed almost complete mono-deuteriation (d1 95 atom%) and 13C NMR confirmed the location of the deuterium exclusively at the C2 (45.3) and C1 (36.5) for cis-1a and cis-1b respectively.
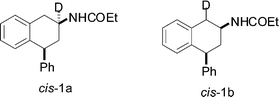 | ||
| Fig. 2 | ||
Based on these results we hypothesized the mechanism shown in Scheme 4. The diastereoselectivity outcome of the reduction can be explained by both the Burgi–Dunitz trajectory for hydride approach to carbonyls and the Cieplak19 mode for the nucleophilic addition to a carbonyl group with non-chelating α-substituents. It is known that the stereochemistry of nucleophilic addition to cyclohexanones is determined by two factors: steric hindrance which favours the equatorial approach (b, Scheme 4), and electron donation from the cyclohexanone’s σC–C bonds into the σ* which favours the axial approach (a, Scheme 4) since the carbon–hydrogen bonds are better electron donors. Given the predominant cis product, we can confidently assume that the electronic factors prevail over the steric hindrance on substrate 10 in our reaction conditions.
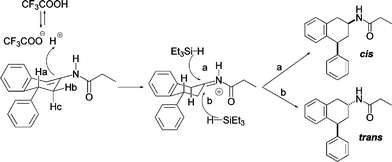 | ||
| Scheme 4 Proposed mechanism. | ||
There are certain advantages to our synthetic approach (Scheme 3) over the previous routes as presented and all of them can be included within the concepts of “atom economy”20 and “step economy”.21 Indeed precise control over the individual reactivity of functional groups (chemoselectivity) without the need to use protecting groups or useless interconversions of functional groups, has been achieved increasing the brevity and efficiency of the synthesis (only three steps and a high overall yield of 38%).
Conclusion
In summary, we have developed a new, practical and convenient protocol for the synthesis of the MT2melatonin receptor antagonist cis-4-P-PDOT, improving yields and reducing the time and cost of the procedure, making the compound more easily accessible for further biological evaluations.Experimental
General
Melting points were determined on a Büchi B-540 capillary melting point apparatus and are uncorrected. 1H NMR and 13C NMR spectra were recorded on a Bruker AVANCE 200 spectrometer, using CDCl3 as solvent unless otherwise noted. Chemical shifts (δ scale) are reported in parts per million (ppm) relative to the central peak of the solvent. Coupling constants (J values) are given in hertz (Hz). ESI-MS spectra were taken on a Waters Micromass ZQ instrument. Only molecular ions (M + 1) are given. Infrared spectra were obtained on a Nicolet Avatar 360 FTIR spectrometer, absorbances are reported in cm−1. Column chromatography purifications were performed under “flash” conditions using Merck 230–400 mesh silica gel. Analytical thin-layer chromatography (TLC) was carried out on Merck silica gel 60 F254 plates. All chemicals were purchased from commercial suppliers and used directly without any further purification, including the deuterated reagents [triethyl(silane-d) and trifluoroacetic acid-d].(±)-4-Phenyl-3,4-dihydronaphthalen-2-one (4)
Freshly distilled styrene (1 g, 9.7 mmol, 1 equiv.) in dry dichloromethane (120 mL) was added dropwise under nitrogen at 0 °C during 15 min to a stirred suspension of anhydrous aluminium trichloride (3.9 g, 29 mmol, 3 equiv.) in dry dichloromethane (130 mL) containing phenylacetyl chloride (1.5 g, 9.7 mmol, 1 equiv.) and the mixture was stirred at 0 °C for a further 15 min. The reaction was quenched by a saturated solution of Seignette salt (100 mL) and the organic solution was washed by a saturated solution of sodium hydrogen carbonate (3 × 50 mL), dried over sodium sulfate and concentrated under reduced pressure. The crude material was filtered through a silica gel plug (eluent cyclohexane–ethyl acetate, 7 : 3) and the resulting yellow oil was purified by Kugelrohr distillation (bp ca. 130 °C–0.1 Torr)17,22 obtaining a light yellow oil (1.2 g, 5.4 mmol, yield 56%).NMR δH (200 MHz, CDCl3) 2.90–3.01 (2 H, m, CH2), 3.65 (2 H, AB, JAB = 20.0 Hz, CH2), 4.48 (1 H, t, J = 6.6 Hz, CH), 7.01 (1 H, d, J = 7.0 Hz, ArH), 7.12–7.36 (8 H, m, ArH); IR (nujol) νmax/cm−1 1720 (CO); ESI-MS (m/z) 223 (M + 1).
(±)-N-(4-Phenyl-3,4-dihydronaphthalen-2-yl) propionamide (10)
To a 50 mL round-bottom flask equipped with a Dean–Stark apparatus were introduced the tetralone 4 (1.5 g, 6.8 mmol, 1 equiv.), the propionamide (1.2 g, 16.9 mmol, 2.5 equiv.) and p-toluenesulfonic acid (0.13 g, 0.7 mmol, 0.1 equiv.) in 30 mL of toluene. The mixture was refluxed for 4 h under a nitrogen atmosphere. After cooling down to room temperature, the propionamide was precipitated and filtered off. The organic solution was washed by a saturated solution of sodium hydrogen carbonate (3 × 50 mL) and water (3 × 50 mL), dried over sodium sulfate and concentrated under reduced pressure. The enamide 10 was purified by chromatography on silica gel (eluent cyclohexane–ethyl acetate, 7 : 3) and re-crystallized (diethyl ether–petroleum ether) obtaining a light yellow solid (1.78 g, 6.4 mmol, yield 95%).
NMR
δH
(200 MHz, CDCl3) 1.18 (3 H, t, J = 7.6 Hz, NHCOCH2CH3), 2.29 (2 H, q, J = 7.6 Hz, NHCOCH2CH3), 2.70 (1 H, dd, J1 = 16.0 Hz, J2 = 9.4 Hz, C3Ha), 2.77 (1 H, dd, J1 = 16.0 Hz, J2 = 7.6 Hz, C3Hb), 4.21 (1 H, t, J = 8.2 Hz, CHPh2), 6.42 (1 H, br s, NH), 6.79 (1 H, d, J = 7.6 Hz, C1H), 7.01 (1 H, td, J1 = 7.4 Hz, J2 = 1.6 Hz, ArH), 7.11–7.32 (8 H m, ArH); δC (50 MHz, CDCl3) 9.5, 30.7, 36.0, 44.3, 111.2, 126.0, 126.4, 126.7, 127.1, 127.4, 128.3, 128.6, 133.4, 134.9, 135.2, 143.6, 172.4; IR (nujol) νmax/cm−1 3337 (NH), 1655 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C); ESI-MS (m/z): 278 (M + 1); mp 123–124 °C.
C); ESI-MS (m/z): 278 (M + 1); mp 123–124 °C.
(±)-cis-N-(4-Phenyl-1,2,3,4-tetrahydronaphthalen-2-yl) propionamide (1)
A solution of enamide 10 (0.07 g, 0.25 mmol, 1 equiv.) in CF3COOH (2 mL) was cooled down to −10 °C and triethylsilane was added dropwise (0.4 mL, 2.5 mmol, 10 equiv.). After 10 minutes the reaction was completed and a saturated solution of sodium hydrogen carbonate was carefully added until the pH was neutral. The mixture was extracted by dichloromethane (3 × 50 mL) and the organic solution was washed by brine (3 × 50 mL). The resulting organic solution was dried over sodium sulfate and concentrated under reduced pressure. The crude material was purified by flash chromatography (silica gel, eluent cyclohexane–ethyl acetate, 7 : 3) to yield a mixture of cis- and trans-diastereoisomers (cis–trans ratio 81 : 19, 0.064 g, 0.23 mmol, yield 91%). The pure cis-1 (0.05 g, 0.18 mmol, yield 71%) can be obtained by a single re-crystallization (acetone–n-hexane).NMR δH (200 MHz, CDCl3) 1.15 (3 H, t, J = 7.6 Hz, NHCOCH2CH3), 1.79 (1 H, apt q, J = 11.6 Hz, C3Ha), 2.22 (2 H, q, J = 7.6 Hz, NHCOCH2CH3), 2.37–2.46 (1 H, m, C3Hb), 2.79 (1 H, dd, J1 = 11.0 Hz, J2 = 15.8 Hz, C1Ha), 3.23 (1 H, ddd, J1 = 1.8 Hz, J2 = 5.3 Hz, J3 = 15.8 Hz, C1Hb), 4.25 (1 H, dd, J1 = 5.5 Hz, J2 = 11.5 Hz, C4H), 4.32–4.46 (1 H, m, C2H), 5.54 (1 H, brs d, J = 7.1 Hz, NH), 6.80 (1 H, d, J = 7.4 Hz, ArH), 7.01–7.37 (8 H, m, ArH); δC (50 MHz, CDCl3) 9.8, 29.8, 36.8, 40.4, 45.6, 46.1, 126.21, 126.24, 126.4, 128.56, 128.64, 129.0, 129.5, 134.9, 138.7, 146.0, 173.1; IR (nujol) νmax/cm−1 3289 (NH), 3024 (ArH), and 1670 (CO); ESI-MS (m/z): 280 (M + 1); mp 170–172 °C.
(±)-cis-N-(2-Deutero-4-phenyl-1,2,3,4-tetrahydronaphthalen-2-yl) propionamide (cis-1a)
The compound cis-1a was synthesized by reduction of 10 by triethyl(silane-d) (TESD) following the procedure described above. The pure cis-1a (yield 70%) was obtained by re-crystallization (acetone).δ H (200 MHz, CDCl3) 1.15 (3 H, t, J = 7.6 Hz, NHCOCH2CH3), 1.79 (1 H, apt t, J = 12.0 Hz, C3Ha), 2.22 (2 H, q, J = 7.6 Hz, NHCOCH2CH3), 2.41 (1 H, ddd, J1 = 12.4 Hz, J2 = 5.8 Hz, J3 = 2.1 Hz, C3Hb), 2.80 (1 H, d, J = 15.6 Hz, C1Ha), 3.22 (1 H, d, J = 15.6 Hz, C1Hb), 4.24 (1 H, dd, J1 = 5.6 Hz, J2 = 11.2 Hz, C4H), 5.62 (1 H, br s, NH), 6.79 (1 H, d, J = 7.5 Hz, ArH), 7.00–7.36 (8 H, m, ArH); δC (50 MHz, CDCl3) 9.9, 29.8, 36.8, 40.3, 45.3 (1 C, t, J = 21.5 Hz), 46.1, 126.24, 126.26, 126.5, 128.6, 128.7, 129.1, 129.5, 135.0, 138.7, 146.0, 173.2; IR (nujol) νmax/cm−1 3291 (NH), 3026 (ArH), and 1670 (CO); ESI-MS (m/z): 281 (M + 1); mp 172–174 °C.
(±)-cis-N-(1-Deutero-4-phenyl-1,2,3,4-tetrahydronaphthalen-2-yl) propionamide (cis-1b)
The compound cis-1b was synthesized by reduction of 10 by triethylsilane using trifluoroacetic acid-d as a solvent following the procedure described above. The pure cis-1b (yield 67%) was obtained by re-crystallization (acetone).δ H (200 MHz, CDCl3) 1.15 (3 H, t, J = 7.6 Hz, NHCOCH2CH3), 1.78 (1 H, apt q, J = 11.7 Hz, C3Ha), 2.21 (2 H, q, J = 7.6 Hz, NHCOCH2CH3), 2.36–2.47 (1 H, m, C3Hb), 2.75–2.86 (0.5 H, m, C1Ha/Da), 3.19–3.27 (0.5 H, m, C1Hb/Db), 4.24 (1 H, dd, J1 = 5.6 Hz, J2 = 11.6 Hz, C4H), 4.31–4.44 (1 H, m, C2H), 5.54 (1 H, br d, J = 7.4 Hz, NH), 6.80 (1 H, d, J = 7.4 Hz, ArH), 7.01–7.34 (8 H, m, ArH); δC (50 MHz, CDCl3) 9.9, 29.9, 36.5 (1 C, t, J = 19.0 Hz), 40.4, 45.5, 46.1, 126.23, 126.27, 126.5, 128.6, 128.7, 129.1, 129.5, 134.9, 138.8, 146.0, 173.2; IR (nujol) νmax/cm−1 3291 (NH), 3023 (ArH), 1671 (CO); ESI-MS (m/z): 281 (M + 1); mp 171–173 °C.
Acknowledgements
This work was supported by the Italian M. I. U. R. (Ministero dell'Istruzione, dell'Università e della Ricerca). We would like to thank Prof. Giuseppe Diamantini for analytical support.References
- S. Copinga, P. G. Tepper, C. J. Grol, A. S. Horn and M. L. Dubocovich, J. Med. Chem., 1993, 36, 2891 CrossRef CAS.
- N. P. Choksi, W. B. Nix, S. D. Wyrick and R. G. Booth, Brain Res., 2000, 852, 151 CrossRef CAS.
- E. C. Bucholtz, R. L. Brown, A. Tropsha, R. G. Booth and S. D. Wyrick, J. Med. Chem., 1999, 42, 3041 CrossRef CAS.
- J. Barrenetxe, P. Delagrange and J. A. Martinez, J. Physiol. Biochem., 2004, 60, 61 Search PubMed; M. L. Dubocovich, D. P. Cardinali, P. Delagrange, D. N. Krause, A. D. Strosberg, D. Sugden, F. D. Yocca, in The IUPHAR Compendium of Receptor Characterization and Classification, ed. D. Girdlestone, IUPHAR Media, London, 2nd edn, 2000, pp. 271 Search PubMed.
- G. Spadoni, C. Balsamini, G. Diamantini, A. Tontini, G. Tarzia, M. Mor, S. Rivara, P. V. Plazzi, R. Nonno, V. Lucini, M. Pannacci, F. Fraschini and B. M. Stankov, J. Med. Chem., 2001, 44, 2900 CrossRef CAS.
- S. Rivara, M. Mor, C. Silva, V. Zuliani, F. Vacondio, G. Spadoni, A. Bedini, G. Tarzia, V. Lucini, M. Pannacci, F. Fraschini and P. V. Plazzi, J. Med. Chem., 2003, 46, 1429 CrossRef CAS.
- S. Rivara, S. Lorenzi, M. Mor, P. V. Plazzi, G. Spadoni, A. Bedini and G. Tarzia, J. Med. Chem., 2005, 48, 4049 CrossRef CAS.
- M. Mor, S. Rivara, A. Lodola, S. Lorenzi, F. Bordi, P. V. Plazzi, G. Spadoni, A. Bedini, A. Duranti, A. Tontini and G. Tarzia, Chem. Biodiversity, 2005, 2, 1438 CrossRef CAS.
- M. L. Dubocovich, M. L. Masana, S. Iacob and D. M. Sauri, Naunyn-Schmiedeberg’s Arch. Pharmacol., 1997, 355, 365–375 CrossRef CAS.
- G. Gatti, G. Piersanti and G. Spadoni, Farmaco, 2003, 58, 469 CrossRef CAS.
- M. L. Dubocovich, K. Yun, W. M. Al-Ghoul, S. Benloucif and M. I. Masana, FASEB J., 1998, 12, 1211 CAS.
- S. Doolen, D. N. Krause, M. L. Dubocovich and S. P. Duckles, Eur. J. Pharmacol., 1998, 345, 67 CrossRef CAS.
- A. E. Hunt, W. M. Al-Ghoul, M. U. Gillette and M. L. Dubocovich, Am. J. Physiol.: Cell Physiol., 2001, 280, C110 CAS.
- Tocris 10 mg/€95 in stock.
-
A. S. Horn and M. L. Dubocovich, Eur. Pat. Appl., Patent no. EP 420
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 064 A2 19910403, 1991, 30 pp Search PubMed.
064 A2 19910403, 1991, 30 pp Search PubMed. - S. D. Wyrick, R. G. Booth, A. M. Myers, C. E. Owens, N. S. Kula, R. J. Baldessarini, A. T. McPhail and R. B. Mailman, J. Med. Chem., 1993, 36, 2542–2551 CrossRef CAS.
- I. Fleming and A. Pearce, J. Chem. Soc., Perkin Trans. 1, 1980, 2485 RSC;
N. Griebenow, T. Flessner, M. Raabe and H. Bischoff, Ger. Offen., Patent no. WO 2005077907, 2005, 49 pp Search PubMed.
- J. L. Renaud, P. Dupau, P. Le Gendre, C. Bruneau and P. H. Dixneuf, Synlett, 1832, 1999 Search PubMed.
- A. S. Cieplak, J. Am. Chem. Soc., 1981, 103, 4540 CrossRef CAS.
- B. M. Trost, Science, 1991, 254, 1471 CrossRef CAS; B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259 CrossRef CAS.
- P. A. Wender, F. C. Bi, G. G. Gamber, F. Gosselin, R. D. Hubbard, M. J. C. Scanio, R. Sun, T. J. Williams and L. Zhang, Pure Appl. Chem., 2002, 74, 25 CrossRef CAS; P. A. Wender, S. T. Handy and D. L. Wright, Chem. Ind., 1997, 765 Search PubMed; P. A. Wender and B. L. Miller, in Organic Synthesis: Theory and Applications, ed. T. Hudlicky, JAI Press, Greenwich, Connecticut, vol. 2, 1993, pp. 27–66 Search PubMed.
- S. A. Fine and R. L. Stern, J. Org. Chem., 1967, 32, 4132 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2008 |