Synthesis of functionalised aromatic oligamide rods†‡
Jeffrey
Plante
ab,
Fred
Campbell
ab,
Barbora
Malkova
b,
Colin
Kilner
a,
Stuart L.
Warriner
ab and
Andrew J.
Wilson
*ab
aSchool of Chemistry, University of Leeds, Woodhouse Lane, Leeds, LS29JT, United Kingdom. E-mail: A.J.Wilson@leeds.ac.uk; Fax: +44 (0)113 3431409; Tel: +44 (0)113 3436565
bAstbury Centre for Structural Molecular Biology, University of Leeds, Woodhouse Lane, Leeds, LS29JT, United Kingdom
First published on 7th November 2007
Abstract
A current goal in synthetic chemistry is the design and synthesis of molecules that adopt well defined conformations—so called foldamers. In this manuscript we describe a modular approach for construction of rod shaped para-oligobenzamide molecules. Our approach permits regiospecific incorporation of side chains through a phenolic ether linkage on the scaffold; a feature that partly restricts the conformation of the rod through intramolecular hydrogen-bonding.
Introduction
A current goal of biomolecular chemistry is to replicate the functions performed by nature's biomacromolecules with synthetic mimics. Nature uses polymers comprising well defined monodisperse sequences of monomers that adopt highly defined secondary and tertiary three dimensional architectures. Therefore, significant effort has been devoted to identifying model systems that give rise to well defined secondary structures termed ‘foldamers’.1,2 More recently functional properties3 have been illustrated as evidenced by reports on foldamers that inhibit protein–protein interactions,4 act as bacteriocidal agents5 and recognise small molecules.6 Similarly, the knowledge derived from fundamental studies of secondary structural motifs has recently allowed some simple tertiary structures to be prepared.7–9 For the most well studied class of foldamer, i.e. those derived from β-peptides,10 a key feature that has permitted these studies is the availability of iterative syntheses that give rise to well defined sequences incorporating many different monomers.We have become interested in the study of aromatic oliogoamides11–13 because their syntheses are in principle amenable to the methods used for conventional α-amino acid derived peptides. Most aromatic oligoamides adopt helical conformations pre-organised by intramolecular hydrogen-bonds, with extended or rod-like conformations less common.14–18 Rod-like oligomers may find use in protein,19peptide20 and oligonucleotide recognition.21 We recently described a route to oligoamide macrocycle synthesis with regiospecific incorporation of functional groups.22 The approach does not require hydrogen-bonding but instead exploits the helical conformation23 adopted by linear N-alkylated aromatic oligoamides which preferentially adopt the cis conformer at the amide linkage.24 Given that phenyl-benzamides prefer the trans conformation,25 we hypothesized that it should be possible to construct short rod-like oliogoamides using a similar strategy. Indeed, a similar approach was used to assemble nanometre sized rods from 4′-amino-[1,1′-biphenyl]-4-carboxylic acid derivatives.26 Herein we describe a robust modular synthesis of aromatic oligoamides and their structural characterisation. These O-alkylated oligomers were recently proposed to be potential α-helix mimetics.27
Results and discussion
Synthesis
Our design of rod-like oligomers necessitates placing the carboxylate and amine funtionalities that form the linking amide in a para relationship. We also required a robust method to incorporate side-chains into each monomer unit and were attracted by O-alkylation. This has the advantage of pre-organising the scaffold further through 5-membered intramolecular hydrogen-bonding between the oxygen of the alkoxy substituent and the amide NH. Iterative synthesis via sequential coupling of masked 4-nitro-3-alkoxybenzoic acid monomers (Scheme 1) as described for synthesis of β-sheet binding pyrazole oligoamides20 was attractive to us. Such an approach is fully modular, so once a monomer has been synthesised it can be rapidly incorporated into any desired sequence. In previously described studies,27N to C terminal assembly was employed, but amide bond formation with alkylated monomers failed and a less modular amide formation then alkylation was necessary.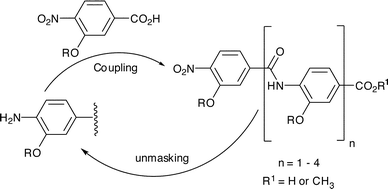 | ||
| Scheme 1 Iterative synthetic strategy for construction of aromatic oligoamide rods. | ||
For these solution phase studies we chose 4-nitro-3-hydroxy-benzoic acid as the starting material due to the ease of monomer synthesis. Scheme 2 outlines our approach which furnishes both 4-nitro-3-alkoxybenzoic acids 4a–f for chain elongation and unmasked amines 5a–b that can be used as the starting monomer. Alkylation of the phenolic oxygen is possible either by reaction with commercially available alkyl halides or Mitsunobu reaction with alcohols. The latter approach gives poorer yields and more difficult purification; however, the compatibility of both reactions coupled with the wide variety of commercially available alkyl halides and alcohols, augers well for future studies.
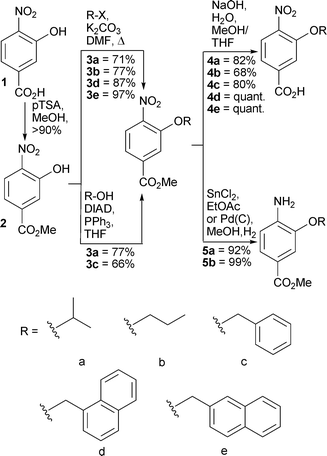 | ||
| Scheme 2 Synthesis of monomers for iterative synthesis of aromatic oligoamide rods. | ||
The synthesis of trimers is shown in Scheme 3. The key steps involve (i) amide bond formation, for which we selected dichlorotriphenyl phosphorane as activating agent and (ii) unmasking of an amino group, for which we selected tin (II) chloride. For amide bond formation, only coupling reagents that generate acid chlorides gave decent yields and then only when heated. For nitro group reduction, although hydrogenation over palladium on carbon proceeded smoothly, this is not compatible with benzylic O-alkyl substituents. We found reduction with tin (II) chloride to be a viable alternative. Yields were consistently above 90% for each of these steps. The final compounds 10 were fully deprotected to expose the terminal amine and carboxylate functionalities (hydrolysis followed hydrogenation—see below).
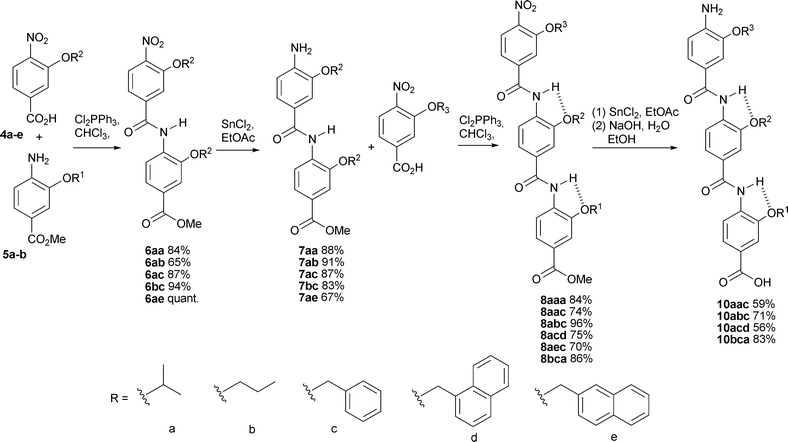 | ||
| Scheme 3 Synthesis of fully unmasked trimers 10via an iterative chain elongation approach. Identity of side chains in oligomers is given from the C terminus e.g. for 10aac R1 = a, R2 = a, and R3 = c. | ||
The synthesis of tetramers and pentamers was found to be more challenging. Initially we conceived a 2 + 3 fragment based strategy; however, our attempts to cleave the methyl ester of trimer 8aaa resulted in hydrolysis of the amide grouppara to the terminal nitro group. This is perhaps unsurprising given the electron withdrawing nature of the nitro group. Thus, although we could have also explored a 3 + 2 strategy, this result suggested that clean hydrolysis of nitro masked dimers 6 was unlikely and we opted instead for a simple 3 + 1 then 4 + 1 chain elongation approach (Scheme 4). Only small reductions in yield were observed for the coupling steps, however, the tetramers and pentamers underwent sluggish nitro group reduction. The most likely reason for this is the poorer solubility of these compounds in the reaction media. Indeed, we were unable to reduce tetramer 12acde whilst 12aecb gave a poor conversion resulting in only 22% of the reduced product although it should be noted that unreacted starting material was also recovered.
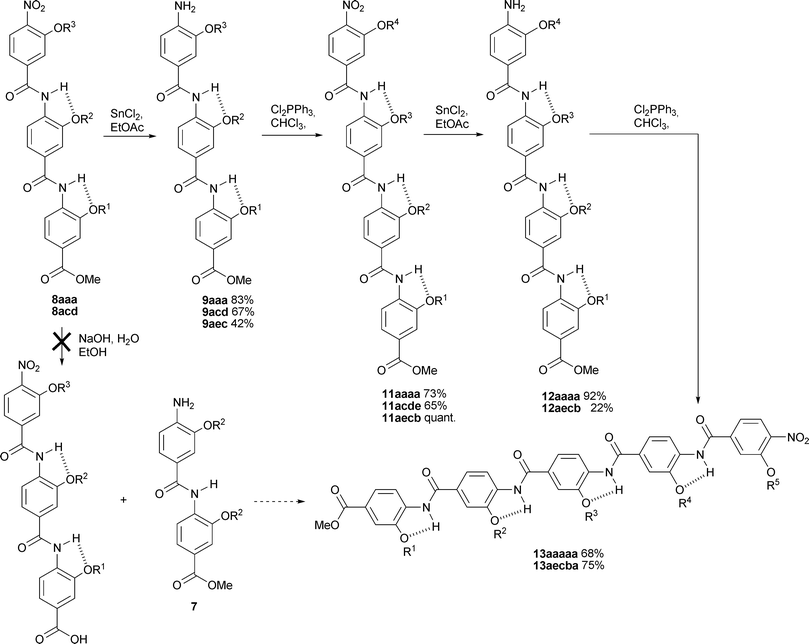 | ||
| Scheme 4 Synthesis of oligoamide tetramers and pentamers (letters correspond to R groups as for Schemes 2 and 3). | ||
Structural studies
Single crystals were obtained by the slow evaporation of a solution of trimer 8abc in ethyl acetate. The X-ray structure confirms the oligomer adopts an extended structure and the presence of an intramolecular hydrogen-bond between the amide NH and the alkoxy oxygen on the neighbouring phenyl ring (Fig. 1). NMR studies indicate that this behaviour is mirrored in solution. For example 8aaa, in both DMSO-d6 and chlorinated solvents, exhibits strong hydrogen bonds as evidenced by the downfield shift of the NH protons (Hf and Hl) (removed in the 1H NMR spectra) (Fig. 2a and b). In both solvents, the NHs undergo no change upon dilution indicating these interactions are intramolecular (Fig. S1 and S2, ESI‡). This is further confirmed upon heating up to 100 °C (Fig S3 and S4, ESI‡); in DMSO-d6, Hf and Hl experience temperature induced shifts of 3.5 ppb K−1 and 4.3 ppb K−1 whilst in C2D2Cl4 they exhibit shifts of 0.8 ppb K−1 and 0.6 ppb K−1. The extended rod-like structure is confirmed in solution by correlations in the 1H–1H NOESY spectrum between NH and ArCH on adjacent aromatic units (i.e. NHl to Hk and Hj and NHf to He and Hd) and the absence of cross-peaks between non-adjacent aromatic resonances (Fig. 2c). Furthermore, this observation confirms free rotation about the ArCO bonds, whilst the absence of cross-peaks between H0 and NH1 or Hi and NHf suggests restricted rotation about the ArNH bonds.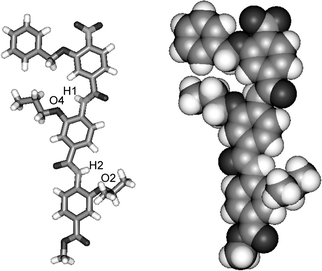 | ||
| Fig. 1 X-Ray crystal structure of compound 8abc shown in stick (left) and CPK (right) format. Key distances H1–O4 = 2.155 Å and H2–O2 = 2.132 Å. | ||
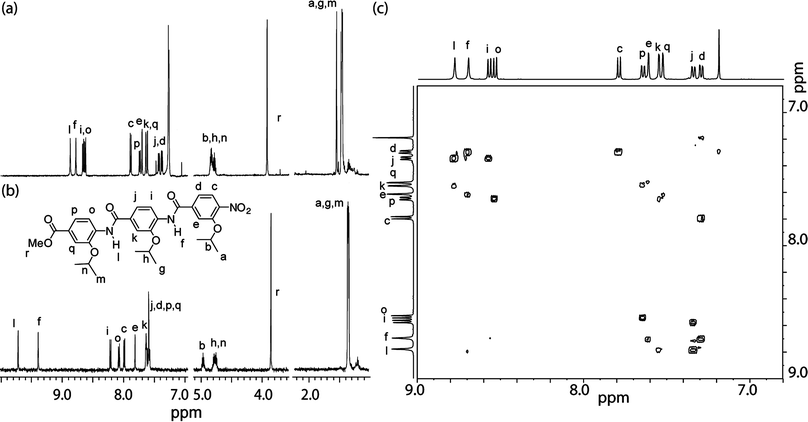 | ||
| Fig. 2 1H NMR spectrum (500 MHz) of trimer 8aaa (a) 3 mM in CDCl3 and (b) 2 mM in DMSO-d6 (c) 1H–1H NOESY spectrum of trimer 8aaa in CDCl3. | ||
The tetramers and pentamers also exhibit downfield shifted NH protons indicative of H-bonding, however signal overlap prevented a thorough conformational analysis as for the trimers. We therefore utilized molecular modelling by performing a full Monte Carlo search in Macromodel28 using the MMFFs force field. This revealed the lowest energy conformation of pentamer 13aaaaa in the gas phase was the extended structure (Fig. 3) and similar results were obtained when the dielectric was set to water. This clearly demonstrated the extended conformation is preferred in both solution phase and gas phase. Furthermore, we tentatively suggest that alternate alkoxy substituents prefer to reside on opposite faces of the rod—all but two of the 20 lowest energy conformers found in the search followed this trend.
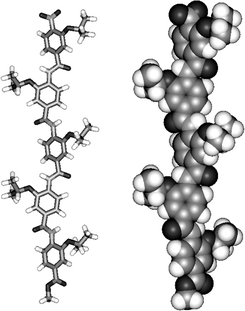 | ||
| Fig. 3 Lowest energy conformer of pentamer 13aaaaa identified by a Monte Carlo search in Macromodel using the MMFFs force field. | ||
Taken together the X-ray, NMR and modelling studies clearly confirm an extended conformation is preferred in the solid state and solution, and that intermolecular hydrogen-bonding fixes rotation about the ArNH bond. However, like the nanometre sized rods described recently by Nowick and co-workers,26 free rotation about the ArCO bond is possible. Our rationale is that the alkoxy substituents minimise steric hindrance by residing on opposing faces, although this conformation would also minimise dipole effects. The NOESY results suggest this preference is not overwhelming as both ArCO conformations are present in solution. Interestingly, in the solid state structure of 8abc (Fig. 1) two of the alkoxy substituents reside on the same face suggesting that crystal packing effects and interactions between adjacent side chains can significantly influence the preferred conformation of these oligomers. This is significant for the following reason: where the molecular recognition properties of these foldamers depend on the alkoxy substituents being presented on one face and available for recognition (e.g. for protein19 or crystal surface29 recognition) an entropic price would need to be paid for fixing the ArCO bond rotation and/or breaking side-chain side-chain interactions. This can be advantageous in that it allows for induced-fit binding. These compounds therefore complement the fully rigidified crescent oligomers and rods described by the groups of Gong,13 Hamilton16 and Li.17,18
Conclusion
We have described the fully modular syntheses of a series of rod like aromatic oligoamides. These molecules can be made from easily accessible monomers and adopt well defined conformations, whilst the building blocks and synthetic methods are compatible with the components of other aromatic oligoamides. Our own future studies will focus on making mixed structures containing building blocks from this pool, elaborating solid phase approaches for their synthesis and studying their molecular recognition properties.Experimental
All chemicals and solvents were purchased from Aldrich and used without further purification. Melting points were determined using a Griffin D5 variable temperature apparatus and are uncorrected. 1H Nuclear magnetic resonance spectra were recorded using a Bruker DRX 500 MHz or DPX 300 MHz machine. 1H spectra are referenced to tetramethylsilane (TMS) and chemical shifts are given in parts per million downfield from TMS. Coupling constants are reported to the nearest 0.1 Hz. Microanalyses were obtained on a Carlo Erba Elemental Analyser MOD 1106 instrument. Infrared spectra were recorded on a Perkin-Elmer FTIR spectrometer and samples analysed in the solid phase. Mass spectra were obtained on a Bruker microTOF using electro-spray ionisation. Representative characterisation is given here for all intermediates leading to a complete trimer 10abc and a complete pentamer 13aecba. The remaining data is available in the ESI.‡Crystal structure determination for 8abc
Single crystals were grown by the slow evaporation of a solution of 8abc in ethyl acetate. X-Ray diffraction data were collected at the University of Leeds. Crystal data. C35H35N3O9, M = 641.66, crystal size 0.3 × 0.1 × 0.1 mm, triclinic, a = 8.0680(2), b = 10.8740(2), c = 19.1980(5) Å, α = 105.2700(10), β = 8.0820(10), γ = 8.0750(10)°, U = 1580.64(6) Å3, T = 150(2) K, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 2, μ = 0.098 mm−1, λ = 0.71073 Å [Mo–Kα], 13724 reflections measured, 7244 unique (Rint = 0.035), observed (I > 2σ(I)). The final R1 was 0.0490 (observed reflections 0.0866) and wR(F2) was 0.1250 (all data 0.1638) for 436 parameters.§
, Z = 2, μ = 0.098 mm−1, λ = 0.71073 Å [Mo–Kα], 13724 reflections measured, 7244 unique (Rint = 0.035), observed (I > 2σ(I)). The final R1 was 0.0490 (observed reflections 0.0866) and wR(F2) was 0.1250 (all data 0.1638) for 436 parameters.§
Molecular modelling
Structures were minimized with Macromodel (MacroModel version 9.0, Schrodinger LLC, New York, NY, 2006) using the Monte Carlo method (5000 structures using the automatic setup) and minimized using the MMFF force field. The results were visualized with Maestro (version 7) and the lowest 20 energy structures were compared, and found to be similar.General procedure A for the alkylation of phenol
Under an inert atmosphere, methyl-3-hydroxy-4-nitrobenzoate (1 equiv.) was dissolved in DMF (5 ml per g of phenol) and K2CO3 (5 equiv.) was added. The alkyl halide (1.4 equiv.) was then added and the solution stirred at 50 °C overnight. The resultant solution was poured into EtOAc (100 mL per g of phenol) and washed twice with H2O (100 mL per g of phenol) and once with brine (100 mL per g of phenol). The organics were dried (Na2SO4) and removed under reduced pressure. The crude oil was subjected to column chromatography (SiO2, CH2Cl2) to yield a pale yellow solid.General procedure B for the saponification of esters
Oligoamide (1 equiv.) was dissolved in THF (25 mL per g of ester) and MeOH (25 mL per g of ester). NaOH (10% aqueous 5 mL per g of ester) was then added and the solution was sealed and stirred overnight. When the reaction was complete as observed by TLC, the organic solvent was removed under reduced pressure. The resulting viscous solution was diluted with water (40 mL per g of ester) and acidified to a pH of 1 using 10% aqueous HCl. The resulting suspension was then filtered and the solid dried overnight yielding a white solid. Alternatively the acid is dissolved in EtOAc and separated from the spurious water and dried (Na2SO4). Removal of the solvents under reduced pressure yielded the acid as a white solid.General procedure C for the coupling reaction
Under an inert atmosphere, to a stirred solution of amine (1.0 equiv.) and acid (1.4 equiv.) in freshly distilled CHCl3 (50 mL per g of amine), dichlorotriphenylphosphorane (4.5 equiv.) was added and the solution allowed to stir at reflux (80 °C) overnight. The solvents were removed under a reduced pressure and the resultant viscous oil subjected to column chromatography (SiO2, CH2Cl2–Et2O gradient) to yield the product.General procedure D for reduction of a nitro group
To a solution of a nitro compound (1 equiv.) in EtOAc (5–10 mL) was added SnCl2·2H2O (5 equiv.) and the reaction mixture was heated to 50 °C under a drying tube. When TLC indicated complete reduction (typically 24 hours), the reaction mixture was cooled, then poured into NaOH solution (10% 100 mL) and the flask rinsed into ethyl acetate (×2). The combined organics were extracted twice with NaOH solution (10% 100 mL) before being washed once with brine (50 mL). The organics were then dried (Na2SO4) and the solvent removed under reduced pressure yielding oils that solidify upon standing. Alternatively, the oil was subjected to column chromatography (SiO2, CH2Cl2–Et2O gradient) to give a cream white to yellow crystalline product.Methyl-3-hydroxy-4-nitrobenzoate 2
A stirred solution of 3-hydroxy-4-nitro benzoic acid 1 (2.50 g, 13.66 mmol) and p-toluene sulfonic acid (0.50 g, 2.63 mmol) in anhydrous methanol (50 mL) under an atmosphere of argon was heated at reflux (65 °C) for 60 h. The reaction mixture was concentrated to leave a bright yellow crystalline solid, which was partitioned between EtOAc and water (4 × 50 mL). The organic fraction was dried (MgSO4), concentrated and dried under vacuum, isolating pure target material (2.38 g, >90%) as a bright yellow powdered solid; mp 90–91 °C (90–91 °C Aldrich); Rf 0.63 (10% EtOAc in CH2Cl2); δH (300 MHz, CDCl3) 4.00 (s, 3H, CO2Me), 7.65 (d, 1H, J = 8.8, ArCH), 7.87 (d, 1H, J = 1.7, ArCH), 8.21 (d, 1H, J = 8.8, ArCH).Methyl-3-isopropyloxy-4-nitrobenzoate 3a
To a stirred solution of methyl-3-hydroxy-4-nitrobenzoate 2 (1.00 g, 5.34 mmol), isopropanol (0.60 mL, 7.85 mmol, excess) and triphenylphosphine (1.33 g, 5.07 mmol) in anhydrous THF (15 mL), under an atmosphere of argon, at 0 °C, was added diisopropyl azodicarboxylate (DIAD) (1.00 mL, 4.82 mmol). The reaction mixture was allowed to warm to room temperature and stirred for a further 72 h. The reaction mixture was concentrated then dissolved in CH2Cl2 and washed with water (2 × 30 mL), saturated NaHCO3 (3 × 30 mL) and brine (30 mL). The organic fraction was concentrated and purified by column chromatography (1 : 1; CH2Cl2–hexane) to yield the product (0.84 g, 77%) as a bright yellow crystalline solid; mp 39–41 °C (found: C, 55.30; H, 5.45; N, 5.75%. C11H13NO5 requires: C, 55.23; H, 5.48; N, 5.86%); Rf 0.55 (CH2Cl2); δH (500 MHz, CDCl3) 1.41 (6H, d, J = 6.0, 2-propyl CH3), 3.96 (3H, s, CO2Me), 4.77 (1H, hep, J = 6.0, CH), 7.65 (1H, d, J = 7.3, ArCH), 7.74 (1H, s, ArCH), 7.76 (1H, d, J = 8.5, Ar 6-CH); δC (75 MHz, CDCl3) 21.8, 52.8, 73.0, 116.9, 121.1, 125.1, 134.4, 143.7, 150.8, 165.4; νmax/cm−1 (solid state) = 3119, 3060, 2989, 1958 (aliph CH), 1725 (CO), 1528; ESI-MSm/z 262 [M + Na]+; ESI-HRMS found m/z 262.0694 [M + Na]+, C11H13NNaO5 requires 262.0686.Methyl-3-isopropyloxy-4-nitrobenzoate 3a
(Alternative preparation using procedure A) methyl-3-hydroxy-4-nitrobenzoate 2 (600 mg, 3.0 mmol), K2CO3 (510 mg, 3.7 mmol), anhydrous DMF (15 mL), 2-bromopropane (450 μL, 5.0 mmol). Purification by column chromatography (20% EtOAc–CH2Cl2) gave a bright yellow crystalline solid: yield: 520 mg, 71% (data as above).3-Isopropyloxy-4-nitro benzoic acid 4a
(Using minor modifications to procedure B) a stirred solution of methyl-3-isopropoxy-4-nitrobenzoate (250 mg, 1.05 mmol) and 1 M NaOH solution (1.5 mL, 1.10 mmol) in methanol (10 mL) was heated at reflux temperature (65 °C) for 12 h until TLC indicated no remaining starting material. On cooling, the reaction mixture was acidified to pH ∼1 (1 N HCl, ∼2 mL) precipitating a solid which was collected by filtration and dried thoroughly under vacuum to give the target acid (194 mg, 82.1%) as a cream solid: mp 173–175 °C (found: C, 53.40; H, 4.90; N, 6.05%. C10H11NO5 requires: C, 53.33; H, 4.92; N, 6.22%); Rf 0.31 (15% MeOH in CH2Cl2); δH (75 MHz, CDCl3) 1.47 (d, 6H, J = 6.1, CH3), 4.82 (q, 1H, J = 6.1, CH), 7.77 (d, 1H, J = 8.1, ArCH), 7.80 (d, 1H, J = 7.6, ArCH), 7.82 (d, 1H, J = 1.3 Hz, ArCH); δC (75 MHz, CDCl3) 22.18, 73.52, 117.70, 122.18, 125.59, 133.70, 144.77, 151.19, 170.69; νmax/cm−1 (solid state) = 2900 (broad, COOH), 1688 (CO), 1524, 1432; ESI-HRMS found m/z 224.0553 [M − H]−, C10H10NO5 requires 224.0553.Methyl-4-amino-3-isopropyloxy benzoate 5a
To a stirred solution of methyl-3-isopropoxy-4-nitrobenzoate 3a (340 mg, 1.42 mmol) in anhydrous methanol (10 mL), under an atmosphere of nitrogen, was added 10% palladium on charcoal (30 mg). The nitrogen atmosphere was evacuated under vacuum and hydrogen gas (1 L, excess) introduced via a balloon. The reaction mixture was stirred for 200 minutes until TLC indicated the reaction was complete. The mixture was passed through a celite pad and concentrated before drying under vacuum to yield the product (272 mg, 92%) as a runny pale brown oil; Rf 0.42 (5% EtOAc in CH2Cl2); δH (500 MHz, CDCl3) 1.29 (d, 6H, J = 6.1, CH3), 3.78 (s, 3H, CO2Me), 4.10 (broad singlet, 2H, NH2), 4.56 (septet, 1H, J = 6.1, CH), 6.59 (d, 1H, J = 8.1, ArCH), 7.39 (d, 1H, J = 1.5, ArCH), 7.45 (dd, 1H, J = 1.6 and 8.2, ArCH); δC (75 MHz, CDCl3) 25.57, 52.05, 71.20, 113.80, 114.44, 119.81, 124.29, 142.62, 144.54, 167.75; νmax/cm−1 (solid state) 3500, 3373 (NH2), 2972, 2950 (CH), 1692, 1613 (CO); ESI-MSm/z 210 [M + H]+; ESI-HRMS found m/z 210.1127 [M + H]+, C11H15NO3 requires 210.1125.Methyl-3-isopropoxy-4-(3-propoxy-4-nitro-benzoylamido)-benzoate 6ab
(Procedure C) 5a (166 mg, 0.8 mmol), 4b (178 mg, 0.8 mmol), chloroform (20 mL), Cl2PPh3 (640 mg, 1.9 mmol). Purification by column chromatography (10% EtOAc in CH2Cl2) afforded the target material (215.7 mg, 65%), as a pale yellow solid; mp 132–133 °C (found C 60.00, H 5.80, N 6.60%. C21H24N2O7 requires: C 60.50, H 5.85, N 6.73%); δH (500 MHz, CDCl3) 1.08 (3H, t, J = 7.4, CH3), 1.44 (6H, d, J = 6.0, CH3), 1.90 (2H, tq, J = 7.1 and 6.9, CH2), 3.92 (3H, s, CO2Me), 4.17 (2H, t, J = 6.4, CH2), 4.78 (1H, hep, J = 6.1, CH), 7.37 (1H, d, J = 8.3, ArCH), 7.61 (1H, s, ArCH), 7.67 (1H, s, ArCH), 7.74 (1H, d, J = 8.4, ArCH), 7.92 (1H, d, J = 8.3, ArCH), 8.59 (1H, d, J = 8.5, ArCH), 8.79 (1H, s, NH); δC (75 MHz, CDCl3) 10.8, 22.6, 22.7, 52.6, 71.8, 72.3, 113.4 (Ar C, CO), 114.5, 117.6, 119.3, 123.6, 126.2, 126.3, 132.6, 140.2, 142.2, 146.3, 153.1, 163.5, 167.0; νmax/cm−1 (solid state) 3425 (NH), 3092, 2977, 2951, 2884, 2621, 1932, 1683 (CO), 1598 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 1516 (NO2); ESI-MSm/z = 417 [M + H]+.
C), 1516 (NO2); ESI-MSm/z = 417 [M + H]+.
Methyl-3-isopropoxy-4-(3-(2-naphthyloxy)-4-nitro-benzoylamido)-benzoate 6ae
(Procedure C) 5a (914.3 mg, 4.4 mmol), 4e (2.0 g, 6.43 mmol), Cl2PPh3 (6.4 g, 19.8 mMol), and chloroform (150 mL) afforded the product (2.2 g, 100%) as a yellow solid; mp 142–143 °C; δH (300 MHz, CDCl3) 1.40 (6H, d, J = 6, iPrCH3), 3.91 (3H, s, CO2Me), 4.75 (1H, hep, J = 6, iPrCH), 5.47 (2H, s, benzylic CH2), 7.37 (1H, d, J = 8.3, ArCH), 7.45–7.51 (2H, m, ArCH), 7.55 (1H, d, J = 8.4, ArCH), 7.59 (1H, s, ArCH), 7.72 (1H, d, J = 8.4, ArCH), 7.79–7.96 (6H, m, ArCH), 8.56 (1H, d, J = 8.5, ArCH), 8.76 (1H, s, NH); δC (75 MHz, CDCl3) 22.6, 52.6, 72.0, 72.3, 113.5, 115.3, 118.1, 119.3, 123.6, 125.0, 126.3, 126.5, 126.7, 126.8, 126.9, 128.2, 128.5, 129.2, 132.5, 132.8, 133.6, 133.7, 140.3, 142.5, 146.3, 152.6, 163.3, 167.0; νmax/cm−1 (solid state) 3423 (NH), 2982, 1713, 1691, 1597, 1522, 1481, 1348, 1273, 1114, 1003, 760; ESI-HRMS found m/z 515.1809 [M + H]+, C29H27N2O7 requires 515.1813.Methyl-3-isopropoxy-4-(3-propoxy-4-amino-benzoylamido)-benzoate 7ab
To a stirred solution of 6ab (175 mg, 0.40 mmol) in anhydrous methanol (7 mL), under an atmosphere of nitrogen, was added palladium on charcoal catalyst (17.5 mg, 10% catalyst). The nitrogen atmosphere was evacuated under vacuum and hydrogen gas (1 L, excess) introduced via a balloon. The reaction mixture was stirred until TLC indicated the reaction was complete and then passed through a celite pad, concentrated and dried under high vacuum to yield the product (141 mg, 91%); mp 103–104 °C; δH (500 MHz, CDCl3) 1.07 (3H, t, J = 7.4, CH3), 1.43 (6H, d, J = 6.0, CH3), 1.88 (2H, tq, J = 6.9 and 6.8, CH2), 3.91 (3H, s, CO2Me), 4.06 (2H, t, J 6.4, CH2), 4.22 (2H, s, NH2), 4.75 (1H, hep, J = 6.1, CH), 6.73 (1H, d, J = 8.5, ArCH), 7.26 (1H, d, J = 8.3, ArCH), 7.44 (1H, s, ArCH), 7.58 (1H, s, ArCH), 7.71 (1H, d, J = 8.6, ArCH), 8.51 (1H, s, NH), 8.62 (1H, d, J = 7.8, ArCH); δC (75 MHz, CDCl3) 22.3, 22.6, 30.9, 52.0, 70.0, 71.8, 91.1, 110.7, 113.2, 113.3, 118.5, 119.9, 123.5, 124.2, 124.5, 133.6, 140.5, 145.6, 146.2, 165.1, 166.9; νmax/cm−1 (solid state) 3492, 3429, 3349, 2970, 2934, 2873, 1704 (CO), 1614; ESI-HRMS found m/z 387.1914 [M + H]+, C21H28N2O5 requires 387.1842.Methyl-3-isopropoxy-4-(3-(2-naphthyloxy)-4-amino-benzoylamido)-benzoate 7ae
(Procedure C) 6ae (2.2 g, 4.4 mmol), SnCl2·2H2O (5.2 g, 23.1 mmol), ethyl acetate (150 mL) afforded the product (1.4 g, 67%) as a yellow oil that solidified upon standing; mp 70–72 °C; δH (300 MHz, CDCl3) 1.40 (6H, d, J = 6, iPrCH3), 3.89 (3H, s, CO2Me), 4.33 (2H, br, NH2), 4.71 (1H, hep, J = 6, iPrCH), 5.29 (2H, s, benzylic CH2), 6.75 (1H, d, J = 8.1, ArCH), 7.29 (1H, d, J = 8.2, ArCH), 7.47–7.52 (2H, m, ArCH), 7.56 (1H, d, J = 8.2, ArCH), 7.58 (1H, s, ArCH), 7.61 (1H, s, ArCH), 7.71 (1H, d, J = 8.4, ArCH), 7.82–7.87 (4H, m, ArCH), 8.63 (1H, d, J = 8.5, ArCH), 8.74 (1H, s, NH); δC (75 MHz, CDCl3) 22.6, 52.4, 71.1, 72.2, 111.8, 113.6, 113.9, 118.9, 120.7, 123.8, 124.5, 124.9, 125.9, 126.7, 126.8, 127.1, 128.2, 128.4, 128.9, 133.6, 133.7, 134.0, 134.4, 141.3, 146.1, 146.4, 165.4, 167.3; νmax/cm−1 (solid state) 3482, 3429, 3364, 2978, 1714, 1668, 1596, 1515, 1481, 1347, 1256, 1203, 1124, 1003, 956, 872, 963, 604; ESI-HRMS found m/z 485.2041 [M]+, C29H28N2O5 requires 485.2071.Methyl-3-isopropoxy-4-(3-propoxy-4-(3-benzyloxy-4-nitro-benzoylamido)-benzoylamido)-benzoate 8abc
(Procedure C) 7ab (125.0 mg, 0.3 mmol), 4c (83.2 mg, 0.3 mmol), chloroform (20 mL), Cl2PPh3 (260.0 mg, 0.8 mmol) afforded the product (193.2 mg, 96%) as a yellow solid; mp 212–214 °C (found: C, 64.55; H, 5.1; N, 6.0%. C35H35N3O9·0.5H2O requires: C, 64.61; H, 5.58; N, 6.46%); δH (500 MHz, CDCl3) 1.11 (3H, t, J = 7.4, CH3), 1.46 (6H, d, J = 6.0, CH3), 1.94 (2H, tq, J = 7.1 and 6.9, CH2), 3.92 (3H, s, CO2Me), 4.18 (2H, t, J = 6.5, CH2), 4.78 (1H, hep, J = 6.0, CH), 5.34 (2H, s, benzylic CH2), 7.43–7.34 (5H, m, ArCH), 7.49 (2H, d, J = 7.3, ArCH), 7.62 (2H, d, J = 10.5, ArCH), 7.73 (1H, d, J = 8.5, ArCH), 7.80 (1H, s, ArCH), 7.97 (1H, d, J = 8.3, ArCH), 8.63 (2H, d, J = 9.1, ArCH), 8.75 (1H, s, NH), 8.87 (1H, s, NH); δC (75 MHz, CDCl3) 10.57, 22.2, 22.5, 52.1, 70.5, 71.4, 71.8, 110.7, 113.1, 114.7, 117.7, 118.6, 118.9, 119.0, 123.3, 125.1, 126.1, 127.2, 128.5, 128.8, 130.6, 130.7, 132.9, 134.9, 139.7, 142.1, 145.8, 147.8, 152.2, 163.0, 164.3, 166.8; νmax/cm−1 (solid state) 3427 (NH2), 2968, 2934, 2879, 1721 (CO), 1596, 1516 (NO2); ESI-HRMS found m/z 642.2445 [M + H]+, C35H36N3O9 requires 642.2446.Methyl-3-isopropoxy-4-(3-(2-napthyloxy)-4-(3-benzyloxy-4-nitro-benzoylamido)-benzoylamido)-benzoate 8aec
(Procedure D) 7ae (1.1 g, 2.1 mmol), 4c (753.7 mg, 2.7 mMol), Cl2PPh3 (3.2 g, 9.9 mmol), and chloroform (100 mL) afforded the product (1.1 g, 70%) as a yellow solid; mp 203–204 °C; δH (300 MHz, CDCl3) 1.45 (6H, d J = 6 Hz, iPrCH3), 3.92 (3H, s, CO2Me), 4.77 (1H, hep, J = 6, CH), 5.13 (2H, s, benzylic CH2), 5.43 (2H, s, benzylic CH2), 7.28–7.53 (8H, m, ArCH), 7.60 (1H, s, ArCH), 7.65 (1H, s, ArCH), 7.71–7.76 (3H, m, ArCH), 7.80–7.88 (5H, m, ArCH), 7.90 (1H, s, ArCH), 8.62 (1H, d J 8.5 Hz, ArCH), 8.67 (1H, d J 8.4 Hz, ArCH), 8.76 (1H, s, NH), 8.87 (1H, br, NH); δC (75 MHz, CDCl3) 22.2, 42.5, 52.1, 71.3, 71.9, 72.1, 111.9, 113.2, 114.4, 116.9, 118.1, 118.7, 119.3, 119.5, 122.3, 123.3, 125.1, 125.2, 125.9, 126.8, 126.9, 127.2, 127.9, 128.5, 128.8, 129.0, 130.8, 131.1, 132.9, 133.0, 134.8, 139.3, 145.8, 147.9, 163.0, 164.0, 166.9; νmax/cm−1 (solid state) 3426, 2978, 1717, 1599, 1492, 1235, 1125, 1013, 957, 849, 746; ESI-HRMS found m/z 740.2565 [M + H]+, C43H38N3O9 requires 740.2603.Methyl-3-isopropoxy-4-(3-propoxy-4-(3-benzyloxy-4-amino-benzoylamido)-benzoylamido)-benzoate 9abc
(Procedure D) 8abc (110.0 mg, 0.2 mmol), SnCl2·2H2O (200.0 mg, 1 mmol) ethyl acetate (40 mL) afforded the product (87.4 mg, 84%) as a pale yellow solid; mp 185 °C (found C 66.2, H 6.10, N 6.40%. C35H37N3O7·H2O requires C 66.67, H 6.24, N 6.67%); δH (500 MHz, CDCl3) 1.12 (3H, t, J = 7.2, CH3), 1.45 (6H, d, J 6.0, CH3), 1.94 (2H, tq, J = 7.0 and 6.9, CH2), 3.91 (3H, s, CO2Me), 4.16 (2H, t, J = 6.4, CH2), 4.77 (1H, hep, J = 5.9, CH), 4.23 (2H, s, NH2), 5.18 (2H, s, benzylic CH2), 6.41 (1H, d, J = 8.8, ArCH), 7.63–7.31 (10H, m, Ar CH), 7.73 (1H, d, J = 8.6, ArCH), 8.62 (1H, d, J = 8.6, ArCH), 8.68 (1H, d, J = 8.7, ArCH), 8.72 (1H, s, NH), 8.87 (1H, s, NH); δC (75 MHz, CDCl3) 9.6, 21.2, 21.5, 51.0, 69.4, 69.6, 70.9, 109.5, 110.3, 112.2, 112.5, 117.6, 118.0, 119.3, 122.4, 123.0, 123.9, 126.8, 127.3, 127.7, 128.3, 131.0, 132.2, 135.5, 139.8, 144.8, 145.0, 146.6, 163.6, 164.1, 165.82; νmax/cm−1 (solid state) 3439, 3368 (NH), 2966, 2873, 1693 (CO), 1601, 1516, 1253; ESI-MSm/z 612 [M + H]+, 650 [M + K]+.Methyl-3-isopropoxy-4-(3-(2-napthyloxy)-4-(3-benzyloxy-4-amino-benzoylamido)-benzoylamido)-benzoate 9aec
(Procedure C) 8aec (1.3 g, 1.8 mmol), SnCl2 (2.5 g, 11.1 mmol) and ethyl acetate (150 mL) afforded the product (530.1 mg, 42%) as a yellow solid; mp 92–93 °C; δH (300 MHz, CDCl3) 1.48 (6H, d, J = 6, iPrCH3), 3.95 (3H, s, CO2Me), 4.27 (2H, br, NH2), 4.77 (1H, hep, J = 6, CH), 4.98 (2H, s, benzylic CH2), 5.42 (2H, s, benzylic CH2), 6.65 (1H, d, J = 8.1, ArCH), 7.28 (1H, d, J = 8.2, ArCH), 7.33–7.41 (6H, m, ArCH), 7.47 (1H, s, ArCH), 7.49–7.60 (3H, m, ArCH), 7.62 (1H, d, J = 8.2, ArCH), 7.64 (1H, s, ArCH), 7.76 (1H, d, J = 8.4, ArCH), 7.78–7.82 (2H, m, ArCH), 7.88–7.91 (2H, m, ArCH), 7.96 (1H, s, ArCH), 8.67 (1H, d, J = 8.5, ArCH), 8.77 (1H, d, J = 8.4, ArCH), 8.80 (1H, s, NH), 8.91 (1H, br, NH); δC (75 MHz, CDCl3) 22.6, 52.5, 70.8, 71.8, 72.3, 111.2, 111.6, 113.6, 113.9, 119.0, 119.1, 119.9, 121.2, 123.7, 123.9, 125.3, 125.5, 126.9, 127.0, 127.2, 127.3, 127.9, 128.3, 128.4, 128.6, 128.9, 129.0, 129.2, 129.6, 132.7, 133.5, 133.6, 133.7, 133.8, 136.8, 141.3, 143.2, 143.3, 147.9, 164.8, 165.4, 167.2; νmax/cm−1 (solid state) 3432, 3344, 2974, 1707, 1682, 1594, 1514, 1487, 1347, 1262, 1144, 1124, 1020, 871, 749, 593; ESI-HRMS found m/z 710.2869 [M + H]+, C43H40N3O7 requires 710.2861.3-Isopropoxy-4-(3-propoxy-4-(3-benzyloxy-4-amino-benzoylamido)-benzoylamido)-benzoic acid 10abc
(Procedure B) 9abc (70 mg, 0.1 mmol) afforded product (58 mg, 85%) isolated by precipitation; mp >246 °C (dec); δH NMR (500 MHz, DMSO-d6) 1.04 (3H, t, J = 7.2, CH2CH3), 1.36 (6H, d, J = 6.0, CH(CH3)2), 1.86 (2H, m, CH2CH2CH3), 4.15 (2H, t, J = 7.2, OCH2), 4.72 (1H, m, OCH), 5.20 (2H, s, OCH2Ar), 5.56 (2H, brs, NH2), 6.74 (1H, d, J = 7.7, ArCH), 7.38 (4H, m, ArCH), 7.48 (3H, m, ArCH), 7.52 (4H, m, ArCH), 8.17 (1H, m, ArCH), 8.24 (1H, m, ArCH), 9.03 (1H, s, NH), 9.32 (1H, m, ArCH); νmax/cm−1 (solid state) 3429, 3364 (NH), 2971, 1681 (CO), 1594, 1488, 1259; ESI-HRMS found m/z 596.2397 [M − H]−, C34H34N3O7 requires 596.2475.Tetramer 11aecb
(Procedure C), 9aec (118.4 mg, 0.17 mmol), 4b (104.5 mg, 0.46 mmol), Cl2PPh3 (641.3 mg, 1.98 mmol), and chloroform (50 mL) afforded the product (147.9 mg, 100%) as a yellow solid; mp 252–253 °C; δH (300 MHz, CDCl3) 1.05 (3H, t, J = 7.2, PrCH3), 1.44 (6H, d, J = 6, iPrCH3), 1.85 (2H, sex, J = 6.3, –OCH2CH2CH3), 3.91 (3H, s, CO2Me), 4.01 (2H, t, J = 6.3, PrCH2), 4.76 (1H, hep, J = 6, iPrCH), 5.05 (2H, s, benzylic CH2), 5.43 (2H, s, benzylic CH2), 7.23 (1H, d, J = 8.3, ArCH), 7.32–7.38 (2H, m, ArCH), 7.39–7.52 (8H, m, ArCH), 7.58–7.61 (3H, m, ArCH), 7.71 (1H, d, J = 8.4, ArCH), 7.76–7.81 (3H, m, ArCH), 7.82–7.93 (3H, m, ArCH), 8.55 (1H, d, J = 8.5, ArCH), 8.61 (1H, d, J = 8.5, ArCH), 8.68 (1H, s, NH), 8.72 (1H, d, J = 8.4, ArCH), 8.84 (1H, br, NH), 8.86 (1H, br, NH); δC (75 MHz, CDCl3) 10.4, 22.2, 52.1, 71.3, 71.5, 71.7, 71.9, 110.8, 111.4, 113.7, 117.3, 117.6, 118.6, 119.0, 119.2, 119.5, 120.1, 123.3, 124.1, 125.7, 126.6, 126.7, 127.1, 127.8, 127.9, 128.9, 130.0, 130.2, 131.1, 131.7, 133.0, 133.1, 133.3, 135.5, 139.3, 141.8, 145.8, 147.6, 147.7, 152.6, 163.0, 164.2, 164.3, 166.8; νmax/cm−1 (solid state) 3433, 3068, 2972, 1708, 1668, 1596, 1519, 1348, 1268, 1125, 1011, 958, 872, 745; ESI-HRMS found 917.3410 m/z [M + H]+, C53H49N4O11 requires 917.3392.Tetramer 12aecb
(Procedure D) 11aecb (151.2 mg, 0.17 mmol), SnCl2·H20 (284.6 mg, 1.26 mmol) ethyl acetate (200 mL) and THF (50 mL) afforded the product (33.4 mg, 22%) as a yellow solid; mp 208–210 °C; δH (300 MHz, CDCl3) 1.04 (3H, t, J = 7.3, PrCH3), 1.44 (6H, d, J = 6, iPrCH3), 1.83 (2H, sex, J = 6.5, OCH2CH2CH3), 3.91 (3H, s, CO2Me), 3.92 (2H, t, J = 6.3, PrCH2), 4.22 (2H, br, NH2), 4.76 (1H, hep, J = 6, iPrCH), 5.06 (2H, s, benzylic CH2), 5.43 (2H, s, benzylic CH2), 6.63 (1H, d, J = 8.1, ArCH), 7.18 (1H, d, J = 8.3, ArCH), 7.29 (1H, s, ArCH), 7.37–7.51 (9H, m, ArCH), 7.57–7.60 (3H, m, ArCH), 7.72 (1H, d, J = 8.4, ArCH), 7.76–7.92 (5H, m, ArCH), 8.61 (1H, d, J = 8.5, ArCH), 8.62 (1H, d, J = 8.5, ArCH), 8.66 (1H, s, NH), 8.73 (1H, d J = 8.4, ArCH), 8.84 (1H, br, NH), 8.87 (1H, br, NH); δC (75 MHz, CDCl3) 10.6, 22.2, 30.3, 52.1, 69.8, 71.2, 71.6, 71.9, 110.3, 110.6, 111.4, 113.2, 113.3, 118.7, 119.0, 119.5, 120.1, 120.3, 123.3, 123.8, 125.0, 126.6, 126.7, 127.0, 127.9, 128.0, 128.6, 128.8, 128.9, 129.8, 131.9, 132.4, 133.1, 133.3, 135.9, 140.6, 145.8, 146.1, 147.3, 147.7, 164.4, 164.6, 165.1, 166.8; νmax/cm−1 (solid state) 3490, 3429, 3356, 2926, 1708, 1665, 1596, 1519, 1348, 1269, 1128, 996, 871, 750; ESI-HRMS found 887.3644 m/z [M + H]+, C53H51N4O9 requires 887.3651.Pentamer 13aecba
(Procedure C) 12aecb (17.6 mg, 0.02 mmol), 4a (14.3 mg, 0.06 mMol), Cl2PPh3 (143.2 mg, 0.44 mmol), and chloroform (30 mL) afforded the product (16.2 mg, 75%) as a yellow solid; mp 255–257 °C; δH (300 MHz, CDCl3) 1.09 (3H, t, J = 7.2 Hz, PrCH3), 1.43 (12H, apparent d, J = 6, iPrCH3), 1.89 (2H, sex, J = 6.3, –OCH2CH2CH3), 3.91 (3H, s, CO2Me), 4.05 (2H, t, J = 6.3, PrCH2), 4.71–4.89 (2H, m overlapping hep, iPrCH), 5.08 (2H, s, benzylic CH2), 5.44 (2H, s, benzylic CH2), 7.30–7.52 (13H, m, ArCH), 7.56–7.61 (3H, m, ArCH), 7.69 (1H, s, ArCH), 7.73 (1H, d J 8.4 Hz, ArCH), 7.79–7.93 (6H, m, ArCH), 8.55 (1H, d J 8.5 Hz, ArCH), 8.61 (1H, d J 8.5 Hz, ArCH), 8.63 (1H, d J 8.4 Hz, ArCH), 8.73 (1H, s, NH), 8.74 (1H, d, J = 8.4, ArCH), 8.78 (1H, br, NH), 8.86 (1H, br, NH), 8.88 (1H, br, NH); νmax/cm−1 (solid state) 3431, 2927, 1671, 1597, 1514, 1424, 1349, 1267, 1123, 1017, 851, 745; ESI-HRMS found 1094.4221 m/z [M + H]+, C63H60N5O13 requires 1094.4182.Acknowledgements
The authors would like to acknowledge the EPSRC (EP/D077842/1) for funding of this research and The Wellcome Trust (080709/Z/06/Z) for a PhD studentship (BM). We would also like to thank Franzi Schramm for early development of monomer synthesis.References
- S. H. Gellman, Acc. Chem. Res., 1998, 31, 178.
- D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893 CrossRef CAS.
- C. M. Goodman, S. Choi, S. Shandler and W. F. DeGrado, Nat. Chem. Biol., 2007, 3, 252 CrossRef CAS.
- J. A. Kritzer, O. M. Stephens, D. A. Guarracino, S. K. Reznik and A. Schepartz, Bioorg. Med. Chem., 2005, 13, 11 CrossRef CAS.
- E. A. Porter, X. F. Wang, H. S. Lee, B. Weisblum and S. H. Gellman, Nature, 2000, 404, 565 CrossRef CAS.
- J.-L. Hou, X.-B. Shao, G.-J. Chen, Y.-X. Zhou, X.-K. Jiang and Z.-T. Li, J. Am. Chem. Soc., 2004, 126, 12386 CrossRef CAS.
- D. S. Daniels, E. J. Petersson, J. X. Qiu and A. Schepartz, J. Am. Chem. Soc., 2007, 129, 1532 CrossRef CAS.
- J. X. Qiu, E. J. Petersson, E. E. Matthews and A. Schepartz, J. Am. Chem. Soc., 2006, 128, 11338 CrossRef CAS.
- W. S. Horne, J. L. Price, J. L. Keck and S. H. Gellman, J. Am. Chem. Soc., 2007, 129, 4178 CrossRef CAS.
- R. P. Cheng, S. H. Gellman and W. F. DeGrado, Chem. Rev., 2001, 101, 3219 CrossRef CAS.
- Z.-T. Li, J.-L. Hou and H.-P. Yi, Chem.–Asian J., 2006, 1, 766 CrossRef CAS.
- I. Huc, Eur. J. Org. Chem., 2004, 17 CrossRef CAS.
- B. Gong, Chem.–Eur. J., 2001, 7, 4336 CrossRef CAS.
- Y. Hamuro, S. J. Geib and A. D. Hamilton, J. Am. Chem. Soc., 1996, 118, 7529 CrossRef CAS.
- D. A. P. Delnoye, R. P. Sijbesma, J. A. J. M. Vekemans and E. W. Meijer, J. Am. Chem. Soc., 1996, 118, 8717 CrossRef CAS.
- J. T. Ernst, J. Becerril, H. S. Park, H. Yin and A. D. Hamilton, Angew. Chem., Int. Ed., 2003, 42, 535 CrossRef CAS.
- Z.-Q. Wu, X.-K. Jiang, S.-Z. Zhu and Z.-T. Li, Org. Lett., 2004, 6, 229 CrossRef CAS.
- J. Zhu, X.-Z. Wang, Y.-Q. Chen, X.-K. Jiang, X.-Z. Chen and Z.-T. Li, J. Org. Chem., 2004, 69, 6221 CrossRef CAS.
- J. M. Davis, L. K. Tsou and A. D. Hamilton, Chem. Soc. Rev., 2007, 36, 326 RSC.
- K. Cernovská, M. Kemter, H.-C. Gallmeier, P. Rzepecki, T. Schrader and B. König, Org. Biomol. Chem., 2004, 2, 1603 RSC.
- H. C. Gallmeier and B. König, Eur. J. Org. Chem., 2003, 3473 CrossRef CAS.
- F. Campbell, J. Plante, C. Carruthers, M. J. Hardie, T. J. Prior and A. J. Wilson, Chem. Commun., 2007, 2240 RSC.
- A. Tanatani, A. Yokoyama, I. Azumaya, Y. Takahura, C. Mitsui, M. Shiro, M. Uchiyama, A. Muranaka, N. Kobayashi and T. Yokozawa, J. Am. Chem. Soc., 2005, 127, 8553 CrossRef CAS.
- I. Azumaya, H. Kagechika, K. Yamaguchi and K. Shudo, Tetrahedron, 1995, 51, 5277 CrossRef CAS.
- A. Itai, Y. Toriumi, N. Tomioka, H. Kagechika, I. Azumaya and K. Shudo, Tetrahedron Lett., 1989, 30, 6177 CrossRef CAS.
- C. M. Gothard, N. A. Rao and J. S. Nowick, J. Am. Chem. Soc., 2007, 129, 7272 CrossRef CAS.
- J.-M. Ahn and S.-Y. Han, Tetrahedron Lett., 2007, 48, 3543 CrossRef CAS.
- F. Mohamadi, N. G. J. Richards, W. C. Guida, R. Liskamp, M. Lipton, C. Caufield, G. Chang, T. Henrickson and W. C. Still, J. Comput. Chem., 1990, 11, 440 CrossRef CAS.
- L. A. Estroff, C. D. Incarvito and A. D. Hamilton, J. Am. Chem. Soc., 2004, 123, 2 CrossRef.
Footnotes |
| † Dedicated to Prof. Sir J. Fraser Stoddart on the occasion of his knighthood and 65th birthday. |
| ‡ Electronic supplementary information (ESI) available: CIF files, full details of X-ray crystallographic structure determination and additional NMR spectra. See DOI: 10.1039/b712606a |
| § CCDC reference number 661106. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b712606a |
| This journal is © The Royal Society of Chemistry 2008 |