Chiral recognition in noncovalent bonding interactions between helicenes: right-handed helix favors right-handed helix over left-handed helix
Ryo
Amemiya
and
Masahiko
Yamaguchi
*
Department of Organic Chemistry, Graduate School of Pharmaceutical Sciences, Tohoku University, Aoba, Sendai 980-8578, Japan. E-mail: yama@mail.pharm.tohoku.ac.jp; Fax: +81-22-795-6811
First published on 2nd November 2007
Abstract
Our studies of helicenes are summarized in regard to chiral recognition phenomenon in noncovalent bonding interactions. The interactions between helical molecules show a tendency for pairs of the same configuration of the helicenes to form more stable complexes than pairs of enantiomeric helicenes. The observations are made in charge transfer complexation, crystallization, homocoupling reaction, layer structure formation, self-aggregation, and double helix formation. The interactions between a helicene and a right-handed helical polymer, double strand DNA, are also described.
 Ryo Amemiya | Ryo Amemiya is an assistant professor at Tohoku University. He was born in Yamanashi in 1976, and received his BSc degree (1999) from Health Sciences University of Hokkaido. He received his MSc. (2001) and PhD degrees (2006) from Tohoku University. In 2002, he was appointed an assistant professor in the Graduate School of Pharmaceutical Sciences, Tohoku University. His research interests are in the area of organometallic chemistry and functionally interesting compounds. |
 Masahiko Yamaguchi | Masahiko Yamaguchi is a professor of Tohoku University. He was born in Fukuoka in 1954, and received his BSc (1977) and PhD degrees (1982) from the University of Tokyo. He joined the Department of Industrial Chemistry, Kyushu Institute of Technology, in 1982 as assistant professor and was promoted to associate professor in 1985. He became a member of the Department of Chemistry at Tohoku University in 1991. From 1987 to 1988 he worked as a post doctoral fellow at Yale University with Professor S. Danishefsky. In 1997, he was appointed professor in the Faculty of Pharmaceutical Sciences of Tohoku University. He received the Chemical Society of Japan Award for Young Chemists in 1986. His research interests are in the area of synthetic methodology and functionally interesting compounds. |
Introduction
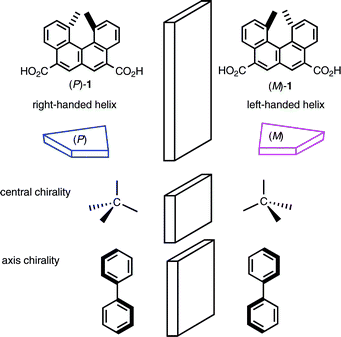
Chirality is an important concept in organic chemistry: Any molecule that is not superimposable on its mirror image is chiral, and possesses two stereoisomers, called enantiomers, with very similar but different properties. Organic molecules having carbon atoms with four nonidentical ligands (central chirality) represent the largest class of chiral molecules. As exemplified by substituted allenes and biaryls, molecules having axis chirality are another important class. According to the Cahn–Ingold–Prelog convention, R and S are assigned to each chiral center. The stereochemical properties of organic molecules with central chirality or axis chirality have extensively been studied.Molecules with helical structures are also chiral; in these molecules the structure is based on the advance of a continuous curve that winds round a central axis. Natural polymers such as DNA, RNA, proteins, and sugars are known as well as certain synthetic polymers.1 Chiral structures with right-handed or left-handed helicity are named the P and M-configuration, respectively. In relation to the studies on such helical polymers, studies on low molecular weight compounds with helical structures are also interesting. We considered that organic molecules with P/M chirality could be as important as those with R/S chirality (central or axis chirality), and started studies on P/M chiral molecular systems of low molecular weight compounds.
Helicenes are a group of ortho-condensed polycyclic aromatic compounds possessing a nonplanar helical π-electron system. Because of the severe steric repulsions between their terminal groups, the aromatic compounds form strained structures, and therefore possess chiral helical structures with either right-handed or left-handed helicity.2 The chirality of helicene was discovered by Newman et al. in the 1950s,3,4 and a variety of derivatives have been prepared.5,6 Their properties,7 however, have not been well investigated except for the work by Katz et al. on metal complexation,8 aggregation,9 and asymmetric catalysis.10 In 1996, we started a project to explore the chemistry of a helicene, 1,12-dimethylbenzo[c]phenanthrene,11a the chirality of which was previously noted by Newman et al. This is one of the configurationally stable helicenes possessing the least number of benzene rings, and does not racemize below 200 °C.4b We developed a method to prepare the optically pure dicarboxylic acid 1 in multigram quantities (Scheme 1),11,12 synthesized various derivatives containing the helicene, and examined their chemical and physical properties.
 | ||
| Scheme 1 | ||
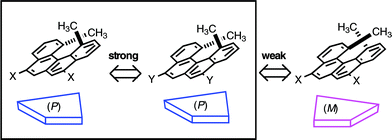
During our studies, the noncovalent bond interactions between helicenes were found to play important roles in their properties. Spectroscopic analyses, X-ray analyses, and calculations indicated the face-to-face orientation of helicenes in such interactions, the origin of which was ascribed to π–π interactions. It appears to us that nonplanar π–π interactions of helicenes are stronger than those of planar π-systems. Chiral recognition in interactions between helicenes therefore has become a subject of interest raising a question: which interactions are more favorable right-handed/right-handed helix or right-handed/left-handed helix (Scheme 2). It turned out that pairs of the same configuration of helicenes form more stable complexes in the P/M chiral molecular systems, a property not observed in the R/S chiral molecular systems. Summarized in this review are the results of our studies on this subject.
 | ||
| Scheme 2 | ||
1. Charge transfer (CT) complexation
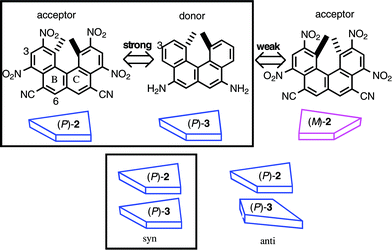
Charge transfer (CT) complexation is a noncovalent bond interaction observed between an electron-rich π-compound (donor) and an electron-deficient π-compound (acceptor). When a chiral donor and an acceptor are employed, a difference in affinity in CT complexation appears between the stereoisomers. Chiral recognition in the complexation of a helicene and compounds with central chirality was examined in relation to the development of chromatographic resolution of racemic compounds.3,13 However, the interactions between chiral helicenes were not reported.Electron-deficient 2,4,9,11-tetranitrohelicenes (P)-2 and (M)-2 were synthesized by the tetranitration of the corresponding helicene-5,8-dinitriles,14 which were obtained from (P)-1 and (M)-1, respectively (Scheme 3).15 The electron-rich (M)-5,8-diaminohelicene (M)-3 was prepared from (M)-1 in two steps including the Curtius rearrangement. The helicenes 2 and 3 form a CT complex in THF, as indicated by the CT absorption band at 500–800 nm.15b The 1H-NMR (THF-d8, 24 °C) signals due to the 6-H of (P)-2 and (M)-2 shifted to higher magnetic fields upon the addition of (M)-3, and the curve fitting assuming 1 : 1 complexation provided the binding constants K; complex of (M)-2 and (M)-3, 12.2 M−1; complex of (P)-2 and (M)-3, 10.2 M−1. The results showed that the same configuration of the helicenes formed the more stable CT complex than that of the enantiomeric helicenes. An NOE was observed between the 3-H of (P)-2 and the 3-H of (P)-3, which suggested a face-to-face structure with a syn-conformation of the CT complex. The syn-configuration was defined in this study as the face-to-face structure with the same direction of the 1,12-dimethyl groups and the anti-configuration with the opposite direction. Next our interest was directed to the generality of the phenomenon that the same configuration of the helicenes form a more stable complex.
 | ||
| Scheme 3 | ||
2. Crystallization
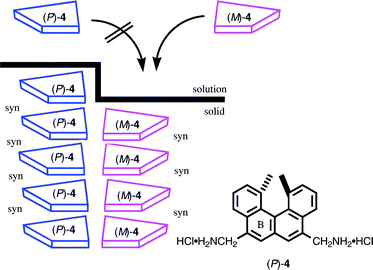
The crystallization of molecules from solution involves a molecular recognition phenomenon. Molecules exit from solution to a cleft on the bulk solid in such a way that the resulting crystals have dense packing. In the crystallization of racemic compounds, enantiomeric compounds compete in crystallization, and chiral recognition phenomenon is observed. If a helicene derivative crystallizes in the face-to-face arrangement, a columnar structure results possessing a layered structure with the combination of either P/P helicene or P/Mhelicene. It may reasonably be concluded that, in the former case, a helicene attaches to a helicene of the same configuration and in the latter that of an antipode.The helicenediamine dihydrochloride (±)-4 crystallized in a columnar structure with a syn-configuration of the helicene, and (M)-4 and (P)-4 formed separate columns with the B-rings stacked on each other (Scheme 4).14b It should be the result of the chiral recognition during crystallization that the helicene molecule favored the helicenes of the same configuration. Other groups have obtained analogous results in X-ray studies of racemic helicenes.16 Thus, the chiral recognition phenomenon noted above is a general trend in crystallization.
 | ||
| Scheme 4 | ||
A related phenomenon was observed in CT crystals of the racemic helicene 2 and pyrene.15b The compound (P)-2 forms a CT complex with pyrene in organic solvents, as indicated by the CT band by UV-VIS at ca. 500 nm. The 1H-NMR (THF-d8, 24 °C) peaks of (P)-2 at methyl and aromatic protons shifted to a higher field upon addition of pyrene. Complexation in a 1 : 1 ratio was confirmed by the Job plots, and a binding constant K = 2.0 M−1 was obtained. The X-ray analysis of the CT complexes of (±)-2 and pyrene indicated the formation of columnar structures with alternating pyrene and 2: separate (P)-2·pyrene and (M)-2·pyrene columns were formed (Scheme 5). One-pitch of the column contained four molecules each of 2 and pyrene, and a molecule of 2 was sandwiched between two pyrenes at the BC rings. The formation of a column containing the same configuration of 2 should be the result of the chiral recognition between helicenes during crystal formation: A molecule of (P)-2 in solution favored interaction with a pyrene molecule upon (P)-2 rather than that upon (M)-2. Related phenomena were observed for other CT crystals.17
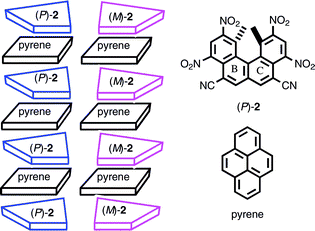 | ||
| Scheme 5 | ||
3. Homocoupling reaction
One of our approaches to study the helicene is to regard it as a chiral equivalent of meta-phenylene or 2,7-naphthylene (Scheme 6). The substitution of the benzene or naphthalene moiety with chiral helicene converts achiral aromatic compounds to chiral compounds without markedly changing the structures of the molecules. Since many functionally interesting compounds possess such partial structures, we considered that manipulation would provide novel chiral aromatic compounds with properties different from those of the original achiral compounds.18 In addition, if the original compound possesses more than one meta-phenylene moiety, the manipulation provides a number of stereoisomers, which can be used to fine-tune or improve the properties. Bihelicenols 6 are the compounds obtained by this manipulation of binaphthol 5, which is widely used as a chiral ligand in asymmetric synthesis.19 Thus it was considered interesting to compare stereoselectivity between 6 with 5 in asymmetric synthesis. Six stereoisomers, (P,P,R)-6, (P,P,S)-6, (P,M)-6, and antipodes, are available for 6, and the comparison of the stereoisomers is another subject of interest.20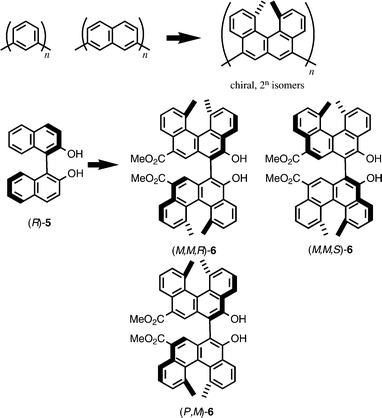 | ||
| Scheme 6 | ||
The homocoupling reaction of racemic helicene derivatives provides an opportunity to study chiral recognition phenomena. The reaction can provide racemic (P*,P*)-compounds and (P*,M*)-compounds, and the former is formed by the coupling between the same enantiomers and the latter the antipodes. The ratio of (P*,P*)-compounds and (P*,M*)-compounds obtained in the homocoupling reaction provides information on the chiral recognition at the transition state.
Bihelicenols (P,M,R)-6 and (P,M,S)-6 were synthesized by the coupling of helicenol (P)-7 and (M)-7 (Scheme 7).20a When racemic helicenol (±)-7 was treated with oxygen in the presence of a copper catalyst, racemic (P*,P*,Z)-8 and (P*,M*,E)-8 were obtained in 60% and 30% yield, respectively. (P*,M*,E)-8 was then converted to (P*,M*)-6, which was resolved by a diastereomer method giving (P,M,R)-6 and (P,M,S)-6. The chiral recognition observed in oxidative coupling is that (P)-7 favored (P)-7 as the coupling counterpart rather than (M)-7. The results show that the same configuration of the helicenes are favored in this homocoupling reaction of a racemic helicene.
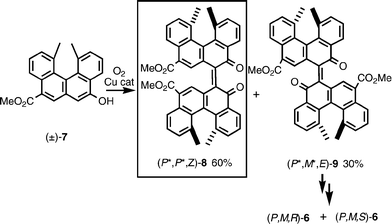 | ||
| Scheme 7 | ||
Bihelicenols (M,M,R)-6 and (M,M,S)-6 were prepared by the oxidative coupling of the optically pure helicenol (M)-7. When (M)-7 was treated with oxygen in the presence of a copper catalyst, an olefin dimer (M,M,Z)-8 was obtained. The hydrogenation of (M,M,Z)-8 led to the formation of (M,M,R)-6, and heating in toluene caused epimerization to give a mixture with (M,M,S)-6, which was separated by chromatography.
The asymmetric reactions using bihelicenol ligands were affected by axis chirality as well as by helical chirality.20b An example is the hydrogenation reaction using bihelicenol l-menthyl phosphite ligand (M,M,S,l)-9 (Scheme 8). In the presence of a catalyst formed from [Rh(cod)2]BF4 (cod = 1,5-cyclooctadiene) (1 mol%) and (M,M,S,l)-9 (3 mol%), dimethyl itaconate was treated with 90 atm hydrogen giving methyl succinate (S)-10 in 90% ee. Switching the ligand to (M,M,S,d)-9 gave (S)-10 in 85% ee indicating that (M,M,S)-bihelicenyl moiety is decisive for stereoselectivity, and that the chirality of menthol is unimportant. In contrast, (R)-10 was obtained in less than 10% ee, when (M,M,R,l)-9 was used. The combination of (M)-helicene and the (S)-axis of 9 represents a matched pair for this reaction. Helicity is important in the asymmetric induction, and the axis chirality is not the only factor that controls the asymmetric induction of the bihelicenol ligand.
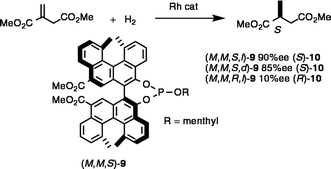 | ||
| Scheme 8 | ||
4. Layer structure formation of helicenediamine oligomers in water
In water, hydrophobic groups aggregate by expulsion of water molecules from the hydration shell (hydrophobic interactions). Since the polycyclic aromatic part of helicene is hydrophobic, water-soluble helicenes possessing polar substituents form a folded structure in water. It was considered interesting to study chiral recognition in intramolecular folding by comparing the structures of compounds possessing two helicene parts. The helicenediamine dimer 11, containing two helicene moieties, was selected for this purpose. The amine moieties are protonated in neutral water providing polar and water soluble substances, and 11 is expected to take a folded structure with a face-to-face orientation at the helicene moiety (Scheme 9).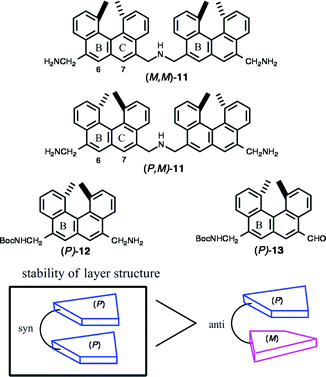 | ||
| Scheme 9 | ||
The dimers (M,M)-11 and (P,M)-11 were synthesized by the reductive amination of the corresponding N-monoprotected helicenediamine 12 and N-protected aminohelicenaldehyde 13.14b Both (M,M)-11 and (P,M)-11 formed folded structures in water, while they possessed a random-coil structure in organic solvents. The aromatic 1H-NMR protons of (M,M)-11 in CD3OD appeared at > δ 7.4, and all shifted to a high field in D2O. In particular, two singlet protons of (M,M)-11 at 6-H and 7-H shifted by more than 1 ppm. The 1H-NMR (D2O) spectra are concentration independent between 0.1 mM and 5 mM indicating the intramolecular nature of the phenomenon. The hypochromism exhibited in UV, CD, and fluorescence spectra of 11 when the solvent was changed from methanol to water, suggested the formation of a layered structure in water. Solvophobic and π–π interactions are considered to play important roles in this folding.
When the UV absorption coefficients ε at 290 nm of the isomeric (M,M)-11 and (P,M)-11 were plotted against the solvent composition of water and methanol at 25 °C, sigmoidal curves were obtained. The free energy difference ΔG in water between the folded structure and the unfolded structure was calculated: ΔG = −1.7 kcal mol−1 for (M,M)-11; ΔG = −1.4 kcal mol−1 for (P,M)-11. The results indicated that 11 with the same configuration of helicenes formed a more stable folded structure than that with the enantiomeric helicenes.
The structures of (M,M)-11 and (P,M)-11 in water were obtained by Amber calculations. Both isomers stack at the B ring of the helicene moiety, and the structures are consistent with the high field shifts of 11 at 6-H and 7-H on folding. The syn-conformation was obtained for (M,M)-11 and the anti-configuration for (P,M)-11. The calculations also indicate the higher stability of the folded structure of (M,M)-11 over (P,M)-11 by 1.7 kcal mol−1, which is in fair agreement with 0.3 kcal mol−1 obtained by the experiments.
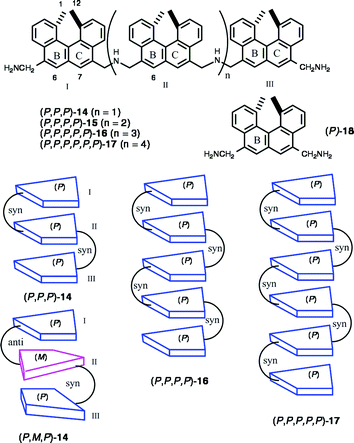
Higher oligoamines up to hexamer (P,P,P)-14, (P,P,P,P)-15 (P,P,P,P,P)-16, and (P,P,P,P,P,P)-17 were synthesized by a two-directional method,14d which involved the reductive amination of a diamine 18 with 2 equivalents of (P)-13. The oligomers formed multilayer structures in water–methanol, and random coil structures in methanol (Scheme 10). The UV, CD, and fluorescence spectroscopies in water–methanol suggested the formation of a π-stacked structure of the aromatic moieties. The aromatic 1H NMR protons of (P,P,P)-14, particularly the three singlet signals 6-HI, 7-HI and 6-HII, shifted upfield in D2O–CD3OD (4 : 1) compared with those in CD3OD. HI and HII indicate the protons of the terminal (first) helicene I and the internal (second) helicene II of (P,P,P)-14. The ROESY examinations revealed that (P,P,P)-14 had a triple-layer structure stacked at the BC ring of the helicene moiety with a syn-conformation. The tetramer (P,P,P,P)-15, pentamer (P,P,P,P,P)-16, and hexamer (P,P,P,P,P,P)-17 also possess analogous multilayer structures.
 | ||
| Scheme 10 | ||
In order to know the effect of helicene stereochemistry on folded structure, a diastereomer (P,M,P)-14 was compared with (P,P,P)-14. In contrast to the dimer 11, appreciable difference was not observed in thermodynamic stability between the diastereomers. However, the folded structures differed as indicated by NMR: a ROESY correlation was observed between 6-HI/7-HI and 1-CH3III/12-CH3III in (P,M,P)-14. The observations were reasonably explained to be related to the anti-conformation of the helicene I and helicene II of (P,M,P)-14. The chirality at the helicene moiety considerably affects multilayer structures.
5. Self-aggregation of [3 + 3]cycloalkynes
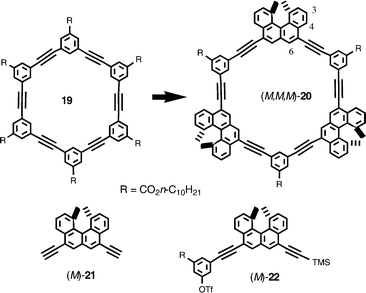
Cyclic aromatic compounds such as porphyrin,21aphthalocyanine,8e,21 and oligoacetylenes22,23 are known to form face-to-face aggregates in solution and in the solid state, which are explained by π–π interactions, the interactions between π electron systems. Helicenes turned out to be a notable group exhibiting such intermolecular forces, and we propose that nonplanar π-systems of helicenes can exert stronger π–π interactions compared with planar π-systems. Although its mechanism is not still clear, this phenomenon provides another opportunity to examine the chiral recognition of helicenes.The achiral cyclic hexamer 19,23 which was known to form aggregates in solution, was converted to chiral [3 + 3]cycloalkynes 20 by substituting three of the m-phenylene moieties with a helicene (Scheme 11).18 The aggregate formation of 19 in solution was compared with that of four stereoisomers (P,P,P)-20, (M,M,M)-20, (P,M,P)-20, and (M,P,M)-20, which was considerably affected by the stereochemistry of the helicene moiety.
 | ||
| Scheme 11 | ||
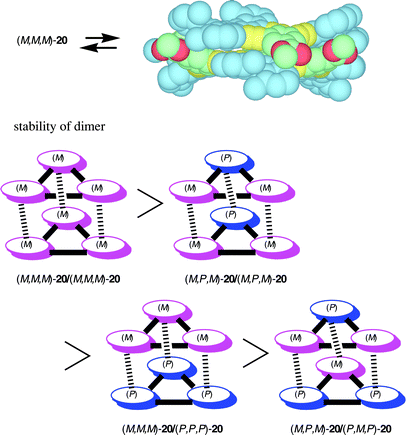
Compound (M,M,M)-20 was synthesized by the Sonogashira coupling of dialkyne (M)-21 and a building block (M)-22 followed by the cyclization with m-diiodobenzene.18a The CD spectra of (M,M,M)-20 in chloroform showed changes at 1 × 10−3 M, when the concentration was increased. Vapor pressure osmometry (VPO) revealed dimer formation at a concentration higher than 2 × 10−3 M, while monomeric formation occurred below this concentration. Higher aggregation was not observed at the higher concentrations. The aggregate formation of (M,M,M)-20 appears to be stronger than for 19, since 19 aggregated only at higher concentrations.23 The 1H-NMR (CDCl3) chemical shifts of (M,M,M)-20 change at concentrations between 1 × 10−4 to 1 × 10−2 M, being consistent with the above observations. Larger shifts were observed at the 3-H, 4-H, and 6-H of the helicene moiety as well as at aromatic protons of the spacer moiety, which suggested a face-to-face orientation. Another interesting feature of (M,M,M)-20 is dimer formation without forming higher aggregates, which is contrasted to 19 which forms higher aggregates with increasing concentration.23 The helicene moiety of (M,M,M)-20 plays an important role in the selective dimerization: calculation indicated the fitting of m-phenylene to the grove of the helicene (Scheme 12).
 | ||
| Scheme 12 | ||
Three other stereoisomers (P,P,P)-20, (P,M,P)-20, and (M,P,M)-20 were also synthesized by the same method, and racemic (M*,M*,M*)-20 and (M*,P*,M*)-20 were prepared by mixing equal amounts of the enantiomers. The VPO studies indicated that the dimerization of the diastereomeric 20 occurred at above a certain concentration in chloroform without forming higher aggregates. The compound (M,P,M)-20 aggregates at 1.5 × 10−2 M, which was the concentrations higher than (M,M,M)-20. Racemic (M*,M*,M*)-20 formed dimer above 2.0 × 10−2 M and racemic (M*,P*,M*)-20 above 3.0 × 10−2 M, which were higher than (M,M,M)-20 and (M,P,M)-20, respectively. The results reflected the stronger homoaggregation than the heteroaggregation: (M,M,M)-20–(M,M,M)-20 complexation is stronger than (M,M,M)-20–(P,P,P)-20; (M,P,M)-20–(M,P,M)-20 complexation is stronger than (M,P,M)-20–(P,M,P)-20. The magnitude of the complex formation between isomeric 20 therefore is summarized as follows: (M,M,M)-20–(M,M,M)-20 > (M,P,M)-20–(M,P,M)-20 > (M,M,M)-20–(P,P,P)-20 > (M,P,M)-20–(P,M,P)-20. Chiral recognition by helicene occurred in the self-aggregation of 20 in organic solvents, and a pair of the same configuration of the helicene formed more stable complexes.
6. Double helix formation of oligo(ethynyl-helicene)s
A double helix is an interesting molecular structure comprising two linear molecules, and possesses three-dimensional structural variations in terms of diameter, length, pitch, and chirality, in addition to the one-dimensional arrangement of atoms.24,25 The structure is formed by several noncovalent bond interactions such as hydrogen bonding, electrostatic interactions, van der Waals interactions, charge transfer interactions, π–π interactions, and CH–π interactions, both in the intramolecular and intermolecular modes. Another notable feature is its potential to reversibly change the structure in response to changes in the environment, and the diversity of the structural features of a double helix makes its structural change extremely interesting. DNA offers an excellent example of a structural change between a double helix and a random coil. However, little was known about the structural change of a synthetic double helix until recently.25 It was found in our study that acyclic ethynylhelicene oligomers form a double helix and a random-coil in solution.18dA series of acyclic ethynylhelicene oligomers from dimer (P,P)-23 to nonamer (P,P,P,P,P,P,P,P,P)-30 possessing two to nine helicenes were synthesized by a two-directional method from (P)-22 (Scheme 13). The CD (CHCl3, 25 °C, 5 × 10−6 M) spectra of (P,P)-23 to (P,P,P,P,P,P)-27 possessing less than seven helicenes showed a monotonic increase in Δε in accordance with the number of helicenes. In contrast, the CD spectra of higher homologs (P,P,P,P,P,P,P)-28, (P,P,P,P,P,P,P,P)-29, and (P,P,P,P,P,P,P,P,P)-30 possessing more than six helicenes markedly changed: An extremely large Δε with an inverted sign of the Cotton effect was observed between 300 and 400 nm. This was ascribed to the formation of highly ordered structures of higher oligomers, most probably helical structures. The vapor pressure osmometry (VPO) analysis indicated the heptamer (P,P,P,P,P,P,P)-28 to possess a dimeric structure or a double helix. The nonplanar π–π interactions between helicenes are considered important for ordered structure formation.
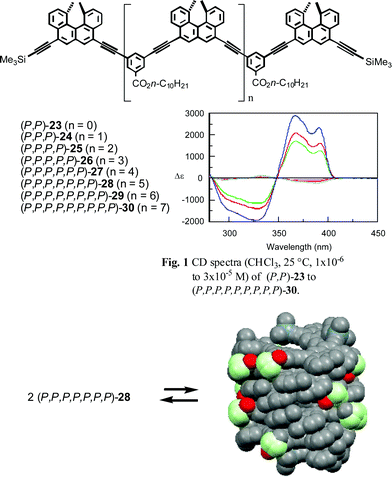 | ||
| Scheme 13 | ||
In chloroform (5 × 10−3 M), the Cotton effect of (P,P,P,P,P,P,P)-28 gradually decreased at 25 °C, and a steady state was reached after 24 h. The resulting CD spectrum was similar to those of (P,P)-23 to (P,P,P,P,P,P)-27 except in terms of intensity. These observations were explained by the slow transition of the double helix of (P,P,P,P,P,P,P)-28 to a random coil. The helix–coil transition examined in several substituted benzenes revealed a large solvent dependence.18d When (P,P,P,P,P,P,P)-28 was dissolved in iodobenzene at 25 °C (5 × 10−6 M), the unfolding was completed within 1 min, and low temperature experiments and the Arrhenius plots provided a rate constant at 25 °C, k = 28 min−1. The unfolding in trifluoromethylbenzene was extremely slow, and provided a rate constant at 25 °C, k < 10−6 min−1. The type of benzene substituent changed the rate constants k by 7 orders of magnitude (Table 1). Such a large aromatic solvent effect in the chemical reaction was not known before.
| Solvent | k/min−1 |
|---|---|
| Iodobenzene | 28 |
| Styrene | 9.3 |
| Thioanisole | 4.6 |
| Bromobenzene | 2.9 |
| Benzonitrile | 1.3 |
| Anisole | 9.0 × 10−1 |
| Chlorobenzene | 5.1 × 10−1 |
| Phenylacetylene | 9.3 × 10−2 |
| Ethylbenzene | 9.2 × 10−2 |
| Ethyl benzoate | 7.1 × 10−2 |
| Toluene | 1.9 × 10−2 |
| Chloroform | 5.7 × 10−3 |
| Pyridine | 4.1 × 10−3 |
| Benzene | 3.6 × 10−4 |
| Fluorobenzene | 5.6 × 10−5 |
| m-Difluorobenzene | 5.8 × 10−6 |
| Trifluoromethylbenzene | <10−6 |
The value of log k exhibited a good correlation with the absolute hardness η,26 which was obtained by Pearson employing ionization potential and electron affinity. The rate constant k decreased with an increase in the η of the solvents, which suggested the soft nature of nonplanar π–π interactions of helicenes (Scheme 14). The HSAB principle was found to be related to π–π interactions.18d
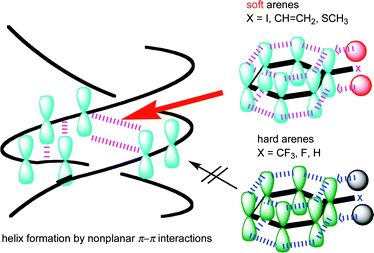 | ||
| Scheme 14 | ||
Folding of (P,P,P,P,P,P,P)-28 from a random coil to a double helix, however, was not observed in chloroform at 5 × 10−6 M, and the helix dimer was regenerated only when the solution was concentrated to a small volume.18d The folding, a bimolecular reaction, should be accelerated at higher concentrations without considerably affecting the unfolding process, a monomolecular reaction. Accordingly, the thermal switching of (P,P,P,P,P,P,P)-28 between a double helix and random coil monomers proceeded at a higher concentration 1 × 10−3 M.18g The process being highly reproducible exhibited an extremely large change in the intensity of the Cotton effect (Fig. 2). Moreover, various patterns of Δε–time profiles were obtained as output against thermal input using (P,P,P,P,P,P,P)-28 depending on changes in concentration, temperature, and solvent type. The diversity reflects the difference of thermodynamic stability and kinetic of the folding and unfolding processes.18g
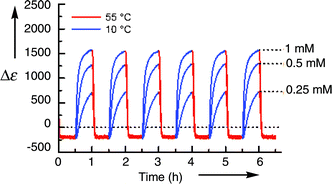 | ||
| Fig. 2 Δε–time profiles of (P,P,P,P,P,P,P)-28 in toluene at 1 mM, 0.5 mM, 0.25 mM for repeating cycles of heating at 55 °C and cooling 10 °C every 30 min. | ||
The effect of the stereochemistry at the helicene moiety was examined by comparing the double helix formation of (P,P,P,P,P,P,P)-28 with diastereomers (M,P,P,P,P,P,M)-28 and (P,M,P,P,P,M,P)-28.27 When (P,M,P,P,P,M,P)-28 was dissolved in chloroform (25 °C, 5 × 10−6 M), CD exhibited the formation of random-coil structure with no symptoms of helix formation. The dissolution of (M,P,P,P,P,P,M)-28 in chloroform showed a rapid decrease in Δε at nm, and unfolding was completed within 30 min. The rates were much faster than (P,P,P,P,P,P,P)-28, unfolding of which required about 24 h under the same conditions. Thus, the double helix stability of the stereoisomers is as follows: (P,P,P,P,P,P,P)-28 > (M,P,P,P,P,P,M)-28 > (P,M,P,P,P,M,P)-28 (Scheme 15). The results indicate that the same configuration of helicenes form more stable complexes in the double helix formation.
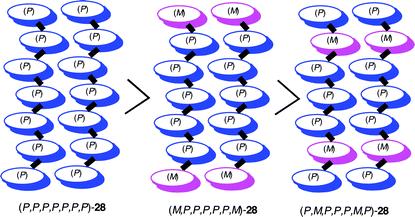 | ||
| Scheme 15 Stability of double helix. | ||
7. Binding of helicene and double strand DNA
As noted in the previous section, a double helix structure is constructed by a number of intermolecular and intramolecular interactions, and possesses a diversity of three-dimensional structural variations. That the double helical oligomers or polymers are able to form right-handed and left-handed helices led us to study their interactions with low molecular weight helical compounds, helicenes. Double strand DNA is one of the most well known double helical molecules, and the binding of helicene was not studied before.28 An interesting subject is which enantiomer of the helicene can preferentially bind to this natural double helical polymer with right handed helicity.14aWhen calf thymus DNA was added to a buffered solution (pH 7) of helicenediamine (P)-18 or (M)-18, The UV, fluorescence, and CD spectra changed indicating complex formation. The fluorescence spectra (1.0 × 10−5 M, 25 °C) were used to obtain the binding constant K employing the McGhee–Hippel method: K = 1.4 × 10−4 M−1 for (P)-18; K = 1.2 × 10−4 M−1 for (M)-18. The isothermal titration calorimetry provided the thermochemical information on the binding of 18 to DNA (pH 7.6, 5 × 10−4 M, 25 °C). In regard to the chiral recognition, the results of the fluorescence titration experiments were confirmed; the binding constant K = 5.7 × 105 M−1 of (P)-18 to DNA was larger than K = 3.6 × 105 M−1 of (M)-18 (Table 2). The binding of 18 is enthalpy driven, and chiral recognition was largely affected by the entropy: the entropy for (P)-18 binding was positive and that for (M)-18 negative. The chiral recognition thus driven by the entropy difference suggested considerably different binding structures between the enantiomeric 18. It is shown that the helical polymer with the right-handed helicity binds to a helicene with a right-handed helical structure (Scheme 16).
| (P)-18 | (M)-18 | |
|---|---|---|
| K /×105 M−1 | 5.7 | 3.6 |
| ΔG/kcal mol−1 | −7.9 | −7.6 |
| ΔH/kcal mol−1 | −6.0 | −8.1 |
| ΔS/cal mol−1K−1 | 6.3 | −1.9 |
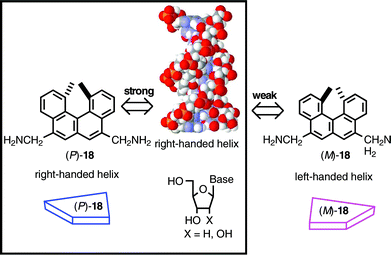 | ||
| Scheme 16 | ||
Chiral recognition in the complexation of double strand DNA and 18 was compared with that of nucleosides. This experiment was conducted to deduce the relationship between chiral recognition in the complexation of a monomeric unit and the corresponding polymer. The 1H-NMR (D2O, pD 5.7, 23 °C) titration experiments of racemic (±)-18 with deoxyribonucleosides (dA, dT, dG, and dC) or ribonucelosides (A, U, G, C) were used to obtain the binding constants K assuming 1 : 1 complexation (Table 3). In cases other than dC and G, the aromatic peaks of (±)-18 separated indicating chiral recognition in the complexation. The complexation experiments of dA, A, and U showed appreciable differences in K, and the binding of (P)-18 was stronger than that of (M)-18. The differences in binding constants K for dT, dG, and C were marginal with slight preferences for (P)-18. As was the double strand DNA, nucleosides also showed higher affinity to right-handed helical (P)-18 (Scheme 16). This is an interesting chiral recognition phenomenon, in which the behaviors of monomeric and polymeric compounds are related.
| Nucleoside | K /M−1 | ||
|---|---|---|---|
| (P)-18 | (M)-18 | ||
| dA | 48 | 45 | |
| dT | 1.7 | 1.6 | |
| dG | 44 | 43 | |
| dC | 6.8 | ||
| A | 36 | 32 | |
| U | 5.2 | 4.5 | |
| G | 37 | ||
| C | 7.6 | 7.4 | |
Chiral recognition phenomenon in the complexation of a double strand DNA and the helical low molecular weight compound 18 was examined: The same configuration of the compounds showed higher affinity than a combination of antipodes. Notably, Sugiyama et al. obtained a contradictory results in the binding of a thiahelicene with Z-DNA possessing the left-handed helicity:29 The right-handed helicene formed a more stable complex with a left-handed helical polymer. The generality of this observation is a subject that needs to be clarified.
Conclusions
Noncovalent bonding interactions with face-to-face orientations play important roles in the chemistry of helicenes, and chiral recognition in preference of the same configuration of helicene appears to be the general trend. To study further examples to confirm the generality and to find exceptions to this rule may be a subject in the future. A comparison of the chemistry of helical chirality, a P/M molecular system, with the chemistry of central chirality, a R/S molecular system, will be another interesting subject.Acknowledgements
This work was supported by grants from the Japan Society of Promotion of Science.Notes and references
- Recent reviews for synthetic helical polymers. (a) D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893 CrossRef CAS; (b) T. Nakano and Y. Okamoto, Chem. Rev., 2001, 101, 4013 CrossRef CAS; (c) L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071 CrossRef CAS; (d) V. G. Machado, P. N. W. Baxter and J.-M. Lehn, J. Braz. Chem. Soc., 2001, 12, 431 CAS; (e) M. Fujiki, J. R. Koe, K. Terao, T. Sato, A. Teramoto and J. Watanabe, Polym. J. (Tokyo), 2003, 35, 297 Search PubMed; (f) I. Huc, Eur. J. Org. Chem., 2004, 17 CrossRef CAS; (g) R. P. Cheng, Curr. Opin. Chem. Biol., 2004, 14, 512 CAS; (h) D. B. Amabilino, J.-L. Serrano, T. Sierra and J. Veciana, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 3161 CrossRef CAS; (i) J. G. Rudick and V. Percec, New J. Chem., 2007, 31, 1083 RSC.
- Reviews of helicenes. (a) R. H. Martin, Angew. Chem., Int. Ed. Engl., 1974, 13, 649 CrossRef; (b) A. Urbano, Angew. Chem., Int. Ed., 2003, 42, 3986 CrossRef CAS; (c) S. K. Collins and M. P. Vachon, Org. Biomol. Chem., 2006, 4, 2518 RSC.
- (a) M. S. Newman, W. B. Lutz and D. Lednicer, J. Am. Chem. Soc., 1955, 77, 3420 CrossRef CAS; (b) M. S. Newman and D. Lednicer, J. Am. Chem. Soc., 1956, 78, 4765 CrossRef CAS.
- (a) M. S. Newman and M. Wolf, J. Am. Chem. Soc., 1952, 74, 3225 CrossRef CAS; (b) M. S. Newman and R. M. Wise, J. Am. Chem. Soc., 1956, 78, 450 CrossRef CAS; Also see. (c) M. S. Newman and W. B. Wheatley, J. Am. Chem. Soc., 1948, 70, 1913 CrossRef CAS.
- (a) C. Goedicke and H. Stegemeyer, Tetrahedron Lett., 1970, 11, 937 CrossRef; (b) D. A. Ligtner, D. T. Hefelfinger, T. W. Powers, G. W. Frank and K. N. Trueblood, J. Am. Chem. Soc., 1972, 94, 3492 CrossRef CAS; (c) R. H. Martin and M. J. Marchant, Tetrahedron, 1974, 30, 347 CrossRef CAS; (d) Y. H. Kim, A. Balan, A. Tishbee and E. Gil-Av, J. Chem. Soc., Chem. Commun., 1982, 1336 RSC; (e) K.-H. Yamada, H. Nakagawa and H. Kawazura, Bull. Chem. Soc. Jpn., 1986, 59, 2429 CAS; (f) N. D. Willmore, L. Liu and T. J. Katz, Angew. Chem., Int. Ed. Engl., 1992, 31, 1093 CrossRef; (g) K. Tanaka, H. Osuga and H. Suzuki, Tetrahedron: Asymmetry, 1993, 4, 1843 CrossRef CAS; (h) R. E. Abed, F. Aloui, J.-P. Genêt, B. B. Hassine and A. Marinetti, Tetrahedron Lett., 2007, 692, 1156.
- Helical low molecular weight compounds other than helicene. (a) M. Wittek, F. Vögtle, G. Stühler, A. Mannschreck, B. M. Lang and H. Irngartinger, Chem. Ber., 1983, 116, 207 CAS; (b) K. Airola, S. Bartram and K. Rissanen, J. Chem. Soc., Perkin Trans. 1, 1995, 895 RSC; (c) X. Qiao, D. M. Ho and R. A. Pascal, Jr., Angew. Chem., Int. Ed. Engl., 1997, 36, 1531 CrossRef CAS; (d) T. Fujita, S. Kuwahara and N. Harada, Eur. J. Org. Chem., 2005, 4533 CrossRef CAS; (e) S. Kuwahara, T. Fujita and N. Harada, Eur. J. Org. Chem., 2005, 4544 CrossRef CAS; (f) M. K. J. ter Wiel, M. G. Kwit, A. Meetsma and B. L. Feringa, Org. Biomol. Chem., 2007, 5, 87 RSC.
- Other examples. (a) M. Nakazaki, K. Yamamoto, T. Ikeda, T. Kitsuki and Y. Okamoto, J. Chem. Soc., Chem. Commun., 1983, 787 RSC; (b) M. T. Reetz, E. W. Beuttenmüller and R. Goddard, Tetrahedron Lett., 1997, 38, 3211 CrossRef CAS; (c) I. Sato, R. Yamashima, K. Kadowaki, J. Yamamoto, T. Shibata and K. Soai, Angew. Chem., Int. Ed., 2001, 40, 1096 CrossRef CAS.
- (a) T. J. Katz and J. Pesti, J. Am. Chem. Soc., 1982, 104, 346 CrossRef CAS; (b) A. Sudhaker, T. J. Katz and B.-W. Yang, J. Am. Chem. Soc., 1986, 108, 2790 CrossRef; (c) Y. Dai, T. J. Katz and D. A. Nichols, Angew. Chem., Int. Ed. Engl., 1996, 35, 2109 CrossRef CAS; (d) Y. Dai and T. J. Katz, J. Org. Chem., 1997, 62, 1274 CrossRef CAS; (e) J. M. Fox, T. J. Katz, S. V. Elshocht, T. Verbiest, M. Kauranen, A. Persoons, T. Thongpanchang, T. Krauss and L. Brus, J. Am. Chem. Soc., 1999, 121, 3453 CrossRef CAS.
- (a) C. Nuckolls, T. J. Katz and L. Castellanes, J. Am. Chem. Soc., 1996, 118, 3767 CrossRef CAS; (b) A. J. Lovinger, C. Nuckolls and T. J. Katz, J. Am. Chem. Soc., 1998, 120, 264 CrossRef CAS; (c) C. Nuckolls, T. J. Katz, T. Verbiest, S. V. Elshocht, H.-G. Kuball, S. Kiesewalter, A. J. Lovinger and A. Persoons, J. Am. Chem. Soc., 1998, 120, 8656 CrossRef CAS; (d) T. Verbist, S. V. Elshocht, M. Kauranen, L. Hellemans, J. Snauwaert, C. Nuckolls, T. J. Katz and A. Persoons, Science, 1998, 282, 913 CrossRef CAS; (e) K. E. S. Phillips, T. J. Katz, S. Jockusch, A. J. Lovinger and N. J. Turro, J. Am. Chem. Soc., 2001, 123, 11899 CrossRef CAS.
- (a) S. D. Dreher, T. J. Katz, K.-C. Lam and A. L. Rheingold, J. Org. Chem., 2000, 65, 815 CrossRef CAS; (b) D. J. Weix, S. D. Dreher and T. J. Katz, J. Am. Chem. Soc., 2000, 122, 10027 CrossRef CAS; (c) D. Z. Wang and T. J. Katz, J. Org. Chem., 2005, 70, 8497 CrossRef CAS.
- (a) M. Yamaguchi, H. Okubo and M. Hirama, Chem. Commun., 1996, 1771 RSC; (b) H. Okubo, M. Yamaguchi and C. Kabuto, J. Org. Chem., 1998, 63, 9500 CrossRef CAS; H. Okubo, M. Yamaguchi and C. Kabuto, J. Org. Chem., 2002, 67, 3540 CrossRef CAS; (c) F. Feng, T. Miyashita, H. Okubo and M. Yamaguchi, J. Am. Chem. Soc., 1998, 120, 10166 CrossRef CAS; (d) H. Okubo, F. Feng, D. Nakano, T. Hirata, M. Yamaguchi and T. Miyashita, Tetrahedron, 1999, 55, 14855 CrossRef CAS; (e) H. Okubo and M. Yamaguchi, Heterocycles, 2000, 52, 863 CrossRef CAS; (f) H. Okubo and M. Yamaguchi, J. Org. Chem., 2001, 66, 824 CrossRef; (g) Also see, synthesis and structure of 1,12-diethyl- and 1,12-diisopropyl-benzo[c]phenanthrenes: H. Sugiura, D. Sakai, H. Otani, K. Teranishi, Y. Takahira, R. Amemiya and M. Yamaguchi, Chem. Lett., 2007, 36, 72 Search PubMed.
- Chiral recognition of 1 with linear and cyclic dextrin: (a) K. Kano, S. Negi, R. Takaoka, H. Kamo, T. Kitae, M. Yamaguchi, H. Okubo and M. Hirama, Chem. Lett., 1997, 715 CrossRef CAS; (b) K. Kano, S. Negi, H. Kamo, T. Kitae, M. Yamaguchi, H. Okubo and M. Hirama, Chem. Lett., 1998, 151 CrossRef CAS; (c) K. Kano, H. Kamo, S. Negi, T. Kitae, R. Takaoka, M. Yamaguchi, H. Okubo and M. Hirama, J. Chem. Soc., Perkin Trans. 2, 1999, 15 RSC.
- (a) Y. H. Kim, A. Tishbee and E. Gil-Av, J. Am. Chem. Soc., 1980, 102, 5915 CrossRef CAS; (b) A. Balan and H. E. Gottlieb, J. Chem. Soc., Perkin Trans. 2, 1981, 350 RSC; (c) Y. H. Kim, A. Tishbee and E. Gil-Av, Science, 1981, 213, 1379 CrossRef CAS; (d) W. J. C. Prinsen and W. H. Laarhoven, J. Chromatogr., 1987, 393, 377 CrossRef CAS.
- (a) S. Honzawa, H. Okubo, S. Anzai, M. Yamaguchi, K. Tsumoto and I. Kumagai, Bioorg. Med. Chem., 2002, 10, 3213 CrossRef CAS; (b) S. Honzawa, H. Okubo, K. Nakamura, S. Anzai, M. Yamaguchi and C. Kabuto, Tetrahedron: Asymmetry, 2002, 13, 1043 CrossRef CAS; (c) S. Honzawa, S. Chiba, H. Okubo and M. Yamaguchi, Heterocycles, 2002, 57, 1091 CrossRef CAS; (d) J. Mizukami, H. Sugiura, M. Yamaguchi and K. Mushiake, Bull. Chem. Soc. Jpn., 2006, 79, 317 CrossRef CAS.
- (a) H. Okubo, D. Nakano, M. Yamaguchi and C. Kabuto, Chem. Lett., 2000, 1316 CAS; (b) H. Okubo, D. Nakano, S. Anzai and M. Yamaguchi, J. Org. Chem., 2001, 66, 557 CrossRef CAS; H. Okubo, D. Nakano, S. Anzai and M. Yamaguchi, J. Org. Chem., 2001, 66, 7560 CrossRef CAS.
- (a) M. K. Lakshman, P. L. Kole, S. Chaturvedi, J. H. Saugier, H. C. J. Yeh, J. P. Glusker, H. L. Carrell, A. K. Katz, C. E. Afshar, W.-M. Dashwood, G. Kenniston and W. M. Baird, J. Am. Chem. Soc., 2000, 122, 12629 CrossRef CAS; (b) S. Han, A. D. Bond, R. L. Disch, D. Holmes, J. M. Schulman, S. J. Teat, K. P. C. Vollhard and G. D. Whitener, Angew. Chem., Int. Ed., 2002, 41, 3223 CrossRef CAS; (c) R. Pathak, K. Vandayar, W. A. L. van Otterlo, J. P. Michael, M. A. Fernandes and C. B. d e Koning, Org. Biomol. Chem., 2004, 2, 3504 RSC; (d) D. C. Harrowven, I. L. Guy and L. Nanson, Angew. Chem., Int. Ed., 2006, 45, 2242 CrossRef CAS.
- CT complex of helicene in the solid state: (a) J. Bernstein and H. Regev, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1977, 33, 1716 CrossRef; (b) M. Konno, Y. Saito, K. Yamada and H. Kawazura, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1980, 36, 1680 CrossRef; (c) H. Nakagawa, K.-I. Yamada and H. Kawazura, J. Chem. Soc., Chem. Commun., 1989, 1378 RSC; (d) N. Thorup, M. Hjorth, J. Larsen and K. Bechgaard, Acta Crystallogr., Sect. C, 1992, 48, 949 CrossRef; (e) J. Larsen, A. Dolbecq and K. Bechgaard, Acta Chem. Scand., 1996, 50, 83 CrossRef CAS; (f) K.-I. Yamada, E. Oguma, H. Nakagawa, H. Kwazura and H. Miyamae, Acta Chem. Scand., 1996, 50, 438 CrossRef CAS.
- (a) K. Nakamura, H. Okubo and M. Yamaguchi, Org. Lett., 2001, 3, 1097 CrossRef CAS; (b) Y. Saiki, K. Nakamura, Y. Nigorikawa and M. Yamaguchi, Angew. Chem., Int. Ed., 2003, 42, 5190 CrossRef CAS; (c) Y. Saiki, H. Sugiura, K. Nakamura, M. Yamaguchi, T. Hoshi and J. Anzai, J. Am. Chem. Soc., 2003, 125, 9268 CrossRef CAS; (d) H. Sugiura, Y. Nigorikawa, Y. Saiki, K. Nakamura and M. Yamaguchi, J. Am. Chem. Soc., 2004, 126, 14858 CrossRef CAS; (e) H. Sugiura, Y. Takahira and M. Yamaguchi, J. Org. Chem., 2005, 70, 5698 CrossRef CAS; (f) Y. Takahira, H. Sugiura and M. Yamaguchi, J. Org. Chem., 2006, 71, 763 CrossRef CAS; (g) H. Sugiura and M. Yamaguchi, Chem. Lett., 2007, 36, 58 CrossRef CAS.
- For reviews of binaphtol ligand in asymmetric synthesis. (a) C. Rosini, L. Franzini, A. Raffaelli and P. Salvadori, Synthesis, 1992, 503 CrossRef CAS; (b) L. Pu, Chem. Rev., 1998, 98, 2405 CrossRef CAS; (c) Y. Chen, S. Yekta and A. K. Yudin, Chem. Rev., 2003, 103, 3155 CrossRef CAS; (d) J. M. Brunel, Chem. Rev., 2005, 105, 857 CrossRef CAS; (e) M. Shibasaki and S. Matsunaga, J. Organomet. Chem., 2006, 691, 2089 CrossRef CAS.
- (a) D. Nakano, R. Hirano, M. Yamaguchi and C. Kabuto, Tetrahedron Lett., 2003, 44, 3683 CrossRef CAS; (b) D. Nakano and M. Yamaguchi, Tetrahedron Lett., 2003, 44, 4969 CrossRef CAS.
- Reviews. (a) J. A. A. W. Elemans, R. van Hameren, R. J. M. Nolte and A. E. Rowan, Adv. Mater., 2006, 18, 1251 CrossRef CAS; (b) Recent examples. A. de la Escosura, M. V. Martínez-Díaz, P. Thordarson, A. E. Rowan, R. J. M. Nolte and T. Torres, J. Am. Chem. Soc., 2003, 125, 12300 Search PubMed; (c) R. A. Hatton, N. P. Blanchard, V. Stolojan, A. J. Miller and S. R. P. Silva, Langmuir, 2007, 23, 6424 CrossRef CAS.
- For examples. (a) J. Zhang, D. J. Pesak, J. L. Ludwick and J. S. Moore, J. Am. Chem. Soc., 1994, 116, 4227 CrossRef CAS; (b) D. W. J. McCallien and J. K. M. Sanders, J. Am. Chem. Soc., 1995, 117, 6611 CrossRef CAS; (c) Z. Wu and J. S. Moore, Angew. Chem., Int. Ed. Engl., 1996, 35, 297 CrossRef CAS; (d) T. Kawase, N. Ueda, H. R. Darabi and M. Oda, Angew. Chem., Int. Ed. Engl., 1996, 35, 1556 CrossRef CAS; (e) R. B. Boese, A. J. Matzger and K. P. C. Vollhardt, J. Am. Chem. Soc., 1997, 119, 2052 CrossRef CAS; (f) M. M. Haley, M. L. Bell, J. J. English, C. A. Johnson and T. J. R. Weakley, J. Am. Chem. Soc., 1997, 119, 2956 CrossRef CAS; (g) Y. Tobe, N. Nakagawa, K. Naemura, T. Wakabayashi, T. Shida and Y. Achiba, J. Am. Chem. Soc., 1998, 120, 4544 CrossRef CAS; (h) Y. Rubin, T. C. Paker, S. J. Pastor, S. Jalisatgi, C. Boulle and C. L. Wilkins, Angew. Chem., Int. Ed., 1998, 37, 1226 CrossRef CAS.
- (a) A. S. Shetty, J. Zhang and J. S. Moore, J. Am. Chem. Soc., 1996, 118, 1019 CrossRef CAS; (b) Also see: Y. Tobe, N. Utsumi, A. Nagano and K. Naemura, Angew. Chem., Int. Ed., 1998, 37, 1285 Search PubMed.
- Synthetic double helix compounds. (a) J.-M. Lehn, A. Rigault, J. Siegel, J. Harrowfield, B. Chevrier and D. Moras, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 2565 CAS; (b) W. Zarges, J. Hall and J.-M. Lehn, Helv. Chim. Acta, 1991, 74, 1843 CrossRef; (c) T. M. Garrett, U. Koert and J.-M. Lehn, J. Phys. Org. Chem., 1992, 5, 529 CAS; (d) R. Krämer, J.-M. Lehn and A. Marquis-Rigault, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5394 CAS; (e) T. W. Bell and H. Jousselin, Nature, 1994, 367, 441 CrossRef CAS; (f) H. Schlaad, T. Krasia and M. Antonietti, J. Am. Chem. Soc., 2004, 126, 11307 CrossRef CAS; (g) Y. Tanaka, H. Katagiri, Y. Furusho and E. Yashima, Angew. Chem., Int. Ed., 2005, 44, 3867 CrossRef CAS; (h) Y. Furushio, Y. Tanaka and E. Yashima, Org. Lett., 2006, 8, 2583 CrossRef CAS; (i) M. Ikeda, Y. Tanaka, T. Hasegawa, Y. Furusho and E. Yashima, J. Am. Chem. Soc., 2006, 128, 6806 CrossRef CAS; (j) H. Katagiri, T. Miyagawa, Y. Furusho and E. Yashima, Angew. Chem., Int. Ed., 2006, 45, 1741 CrossRef CAS.
- Structural change of synthetic double helix compounds. (a) W. Berl, I. Huc, R. G. Khoury, M. J. Krische and J.-M. Lehn, Nature, 2000, 407, 720 CrossRef CAS; (b) V. Berl, I. Huc, R. G. Khoury and J.-M. Lehn, Chem.–Eur. J., 2001, 7, 2810 CrossRef CAS; (c) M. Barboiu, G. Vaughan, N. Kyritsakas and J.-M. Lehn, Chem.–Eur. J., 2003, 9, 763 CrossRef CAS; (d) H.-C. Yang, S.-Y. Lin, H.-C. Yang, C.-L. Lin, L. Tsai, S.-L. Huang, I. W.-P. Chen, C.-h. Chen, B.-Y. Jin and T.-Y. Luh, Angew. Chem., Int. Ed., 2006, 45, 727; (e) C. Zhang, J.-M. Léger and I. Huc, Angew. Chem., Int. Ed., 2006, 45, 4625 CrossRef; (f) D. Haldar, H. Jiang, J.-M. Légr and I. Huc, Angew. Chem., Int. Ed., 2006, 45, 5483 CrossRef CAS; (g) H. Goto, H. Katagiri, Y. Furusho and E. Yashima, J. Am. Chem. Soc., 2006, 128, 7176 CrossRef CAS.
- R. G. Pearson, J. Org. Chem., 1989, 54, 1423 CrossRef CAS.
- Y. Saiki, PhD Thesis, Tohoku University, 2004.
- For examples of chiral recognition between DNA and compounds with central chirality. (a) J. K. Barton, A. T. Danishefsky and J. M. Goldberg, J. Am. Chem. Soc., 1984, 106, 2172 CrossRef CAS; (b) H. Kigoshi, Y. Imamura, K. Mizuta, K. Niwa and K. Yamada, J. Am. Chem. Soc., 1993, 115, 3056 CrossRef CAS; (c) T. Morii, A. Murakami and K. Makino, Tetrahedron Lett., 1994, 35, 1219 CrossRef CAS; (d) D. L. Boger, D. S. Johnson and W. Yun, J. Am. Chem. Soc., 1994, 116, 1635 CrossRef CAS; (e) D. M. Herman, E. E. Baird and P. B. Dervan, J. Am. Chem. Soc., 1998, 120, 1382 CrossRef CAS; (f) A. Tatibouët, M. Demeunynck, C. Andraud, A. Collet and J. Lhomme, Chem. Commun., 1999, 161 RSC; (g) M. S. Tichenor, J. D. Trzupek, D. B. Kastrinsky, F. Shiga, I. Hwang and D. L. Boger, J. Am. Chem. Soc., 2006, 128, 15683 CrossRef CAS; (h) L. Zahng, M. Song, Q. Tian and S. Min, Sep. Purif. Technol., 2007, 55, 388 CrossRef.
- Y. Xu, Y. X. Zhang, H. Sugiyama, T. Umano, H. Osuga and K. Tanaka, J. Am. Chem. Soc., 2004, 126, 6566 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2008 |