Molecular rearrangements through thermal [1,3] carbon shifts
John E.
Baldwin
*a and
Phyllis A.
Leber
b
aDepartment of Chemistry, Syracuse University, Syracuse, New York, 13244, USA. E-mail: jbaldwin@syr.edu; Fax: +1-315-443-4070; Tel: +1-315-443-3743
bDepartment of Chemistry, Franklin and Marshall College, Lancaster, Pennsylvania 17604, USA. E-mail: phyllis.leber@fandm.edu; Fax: +1-717-291-4343; Tel: +1-717-291-3801
First published on 11th October 2007
Abstract
Molecular rearrangements through thermal [1,3] carbon shifts, such as vinylcyclopropane-to-cyclopentene and vinylcyclobutane-to-cyclohexene isomerizations, were recognized and exemplified repeatedly from 1960–1964. Serious mechanistic studies of these and related rearrangements over the past 40 years have provided ample grounds for interpreting them as processes taking place by way of conformationally flexible but not statistically equilibrated diradical intermediates. Orbital symmetry theory fails to account for the stereochemical characteristics of [1,3] carbon shifts. For sigmatropic reactions of this class the theory can no longer be retained as a valid basis for mechanistic interpretations, or even as a serious contender for consideration as a mechanistic possibility.
 John E. Baldwin John E. Baldwin | John E. Baldwin, Distinguished Professor of Chemistry and William R. Kenan Jr Professor of Science at Syracuse University, earned degrees from Dartmouth College (AB, 1959) and the California Institute of Technology (PhD, 1963). Following faculty appointments at the University of Illinois (1962–1968) and the University of Oregon (1968–1984) he moved to Syracuse and has concentrated his research on experimental studies of complex isomerizations and fragmentations of simple hydrocarbons. Among his recognitions are Sloan, Guggenheim, and Alexander von Humboldt fellowships and awards. |
 Phyllis A. Leber Phyllis A. Leber | Phyllis A. Leber received a BS in Chemistry from Albright College in 1976 and a PhD from the University of New Mexico in 1981. After a visiting appointment at Pomona College, she joined the faculty at Franklin & Marshall College in 1982, where she is currently the Dr E. Paul and Frances H. Reiff Professor of Chemistry. Her research encompasses kinetic and stereochemical investigations of vinylcyclobutane thermal rearrangements; she has collaborated with Professor Baldwin in this mutual endeavor since 2000. |
Introduction
From 1960 to 1964 several thermal rearrangements involving the migration of a sigma bond from one end of an allylic sytem to the other were discovered and studied. Vinylcyclopropane was found to isomerize thermally to cyclopentene.1 Isopropenylcyclobutane was converted to 1-methylcyclohexene (Scheme 1).2![Early examples of rearrangements through [1,3] carbon shifts.](/image/article/2008/OB/b711494j/b711494j-s1.gif) | ||
| Scheme 1 Early examples of rearrangements through [1,3] carbon shifts. | ||
A degenerate isomerization of this type was demonstrated for bicyclo[3.1.0]hex-2-ene with the aid of deuterium labeling.3 Optically active α-thujene was shown to racemize when heated to 250 °C, revealing the degenerate isomerization through another experimental probe.4 With such precedents the classic instance of the thermal racemization of α-pinene, first recorded in 1927, could be rationalized as following the same pattern (Scheme 2).5
![Degenerate [1,3] carbon shifts.](/image/article/2008/OB/b711494j/b711494j-s2.gif) | ||
| Scheme 2 Degenerate [1,3] carbon shifts. | ||
In 1962 the bicyclic vinylcyclobutane 6-endo-acetoxybicyclo[3.2.0]hept-2-ene was rearranged to a structural isomer, 2-exo-acetoxybicyclo[2.2.1]hept-2-ene, through a [1,3] migration of a sigma bond (Scheme 3).6
![Isomerization of a bicyclo[3.2.0]hept-2-ene to a bicyclo[2.2.1]hept-2-ene.](/image/article/2008/OB/b711494j/b711494j-s3.gif) | ||
| Scheme 3 Isomerization of a bicyclo[3.2.0]hept-2-ene to a bicyclo[2.2.1]hept-2-ene. | ||
These early examples of [1,3] carbon shifts were soon joined by others.3,7–9 More than a dozen reactions corresponding to this paradigm for thermal isomerizations were well established by 1965. Early interpretation of these rearrangements postulated that the reactions take place by way of diradical intermediates. The intermediates were thought to be accessible through thermally activated cleavages of an allylic sigma bond positioned in a cyclopropane or cyclobutane moiety with concomitant relief of ring strain; the diradical intermediate accommodated structurally linked alkyl radical and allylic radical components.
Orbital symmetry theory and [1,3] carbon sigmatropic shifts
Orbital symmetry theory, introduced in 1965, had and continues to have a profound influence on many aspects of organic chemistry. The third of the seminal communications by Woodward and Hoffmann outlined and applied the theory to a general class of transformations defined as sigmatropic reactions.10 It introduced the terms suprafacial and antarafacial (abbreviated s and a) to specify whether one or both faces of a planar π system participated in a specific sigmatropic isomerization. In suprafacial sigmatropic reactions the same face of a π system participates in both sigma bond-breaking and bond-making events. In antarafacial reactions, a π system participates in a sigma bond cleavage from one face and formation of a new sigma bond from the other face. Woodward and Hoffmann provided a general set of orbital symmetry theory based selection rules that correlated reaction stereochemical characteristics with various types of sigmatropic reactions. Some were allowed to occur with preservation of orbital symmetry and others were forbidden to occur in a concerted pericyclic process. They also noted some significant limitations on the relevance of the theory. Antarafacial processes were considered impossible for transformations within small or medium-sized rings. The carbon framework as the transformation progressed could not be contorted through geometrical constraints so as to force a serious impairment of conjugation within the π-electron system of the pericyclic array. Finally, sigmatropic reactions in violation of the selection rules might well take place through multi-step processes involving diradical intermediates, under relatively vigorous conditions.The table of selection rules in the 1965 communication was limited to reactions involving hydrogen migrations: thermal [1,3] sigmatropic shifts of hydrogen demanded antarafacial utilization of the π component to be allowed by symmetry, but an antarafacial participation of an allylic π system was not countenanced. A footnote mentioned that a migrating group might participate in a sigmatropic reaction with inversion of stereochemical configuration if certain conditions were met. For [1,3] sigmatropic shifts of carbon, however, this option was not considered seriously. In a 1966 lecture, Woodward explicitly characterized [1,3] carbon sigmatropic shifts as symmetry forbidden; thermal sigmatropic changes of order [1,3] in general were said to be forbidden.11 Exploratory studies of [1,3] shifts shown by 6-endo-acetoxybicyclo[3.2.0]hept-2-ene were framed using hypotheses based on diradical intermediates.12
An important contribution published in 1967 reported reaction stereochemistry for the [1,3] shift reaction of 6-endo-acetoxybicyclo[3.2.0]hept-2-ene.13,14 The system was prepared with an exodeuterium label at C7; when isomerized at 307 °C in decalin it afforded 5-exo-acetoxy-6-exo-deuteriobicyclo[2.2.1]hept-2-ene. The [1,3] carbon shift products were analyzed and found to have occurred with at least 95% inversion of configuration at the migrating carbon. This stereochemical finding was rationalized in terms of a transition state involving simultaneous bonding of opposite faces of C7 with C1 and C3. In other words, the dominant stereochemistry of the isomerization corresponded to a suprafacial,inversion (si) outcome, and the reaction was considered to be a concerted pericyclic transformation. The minor stereoisomer formed was the suprafacial,retention (sr) product.
This dramatic observation stimulated a complete about-face in interpretation of [1,3] carbon sigmatropic shifts. By 1969, the reaction in Scheme 4 was stated to be a concerted symmetry-allowed suprafacial [1,3] shift with inversion at the migrating center.15 Thus the structures of starting material and product were taken to be sufficient grounds for defining reaction mechanism and the essential bonding characteristics of the transition structure. Further, this precedent, this authoritatively authenticated occurrence of a symmetry-allowed concerted [1,3] carbon shift “despite extraordinarily unfavorable geometric constraints” led on easily to speculation that isomerizations of vinylcyclopropanes to cyclopentenes might take place through orbital-symmetry-stipulated paths.
![First report of a [1,3] carbon shift with a predominant si stereochemical outcome.](/image/article/2008/OB/b711494j/b711494j-s4.gif) | ||
| Scheme 4 First report of a [1,3] carbon shift with a predominant si stereochemical outcome. | ||
The stereochemical options for such isomerizations were set forth by considering a vinylcyclopropane labeled with stereochemically well-defined R substituents at the migration terminus and at C2 and C3 of the cyclopropane ring (Scheme 5). The vinylcyclopropane could isomerize though four possible paths to give three distinguishable products.
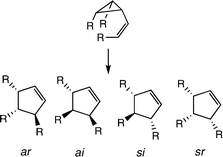 | ||
| Scheme 5 Four outcomes for a vinylcyclopropane isomerization leading to three products. | ||
The cis,trans product shown might be formed in an allowed ar or a forbidden ai way. The stereochemical participation at the migrating carbon is irrelevant, for the enantiomers of trans-3,5-R2-cyclopentene have C2 symmetry and C4 is on the symmetry axis. Both antarafacial paths, initiated by cleavage of C1–C2, would give the same cis,trans R3-substituted enantiomer, while ar and ai reactions when the C1–C3 bond was broken would lead to the other enantiomer. The observable cis,trans product would be racemic. The si outcome following a C1–C2 bond cleavage would give the trans,trans isomer. The third possible product, the cis,cis isomer, would correspond to a forbidden sr reaction. Just how one might define the relative importance of all four possible stereochemical paths, when only three possible products could be quantified, was not immediately obvious.
Stereochemistry and mechanism of vinylcyclopropane rearrangements
Another impediment to determining reaction stereochemistry for vinylcyclopropane-to-cyclopentene isomerizations emerged as Willcott and Cargill found that deuterium-labeled vinylcyclopropanes undergo thermal stereomutations in the three-membered ring much faster than they isomerize to cyclopentenes.16 A third difficulty was posed by the inherent complexity of the reactions involved: even if all secondary kH/kD effects were neglected as a simplifying device, one would have to deal with 28 rate constants (but only 6 independent kinetic parameters) and pay attention to the symmetry considerations associated with equal probabilities of bond cleavage for C1–C2 and C1–C3. And simply tracking the time-dependent concentrations of all three distinguishable 2,3-d2-1-vinylcyclopropanes and all three distinguishable labeled cyclopentenes at very low conversions to the structural isomers could present substantial technical challenges.Not surprisingly, early attempts to investigate and gain stereochemical information on vinylcyclopropane-to-cyclopentene isomerizations depended on more heavily substituted reactants. In 1970 Mazzocchi and Tamburin found that the substituted vinylcyclopropanes shown in Scheme 6 rearranged to cyclopentene products to give stereochemical outcomes inconsistent with the predictions of orbital symmetry theory for concerted reactions.17 Isomerizations of both starting materials took place at 285 °C to afford a preponderance of the same product. From the reactant having the ethoxycarbonyl substituent cis to the propenyl function the (sr + ai) outcomes and the trans,trans isomeric substituted cyclopentene were favored (64 : 36). From the other starting material this trans,trans product was reached through (si + ar) outcomes in preference to the cis,trans isomer (59 : 41).
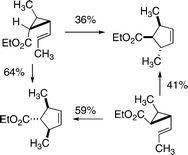 | ||
| Scheme 6 Stereochemistry for vinylcyclopropane isomerizations inconsistent with orbital symmetry theory based expectations. | ||
Doering and Sachdev prepared and followed the thermal stereomutations and the [1,3] carbon shifts of the non-racemic cis and trans isomers of 2-cyano-1-isopropenylcyclopropane (Scheme 7).18 (Here, and elsewhere, the nomenclature convention adopted treats the compound as a substituted 1-alkenylcycloalkane, rather than as a substituted cycloalkane .) The enantiomerization and diastereomerization rate constants for the stereomutations implicated non-concerted reactions involving a common diradical intermediate, a dynamically continuous intermediate providing for reactions leading from one stereoisomeric vinylcyclopropane to the three others as variations on a single type of continuous process. In retrospect, this formulation may be recognized as the direct forerunner of the “caldera” representation of transformations dependent on diradical intermediates and dynamic processes.19
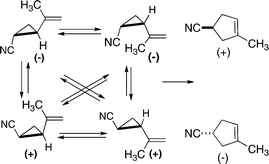 | ||
| Scheme 7 Reactions finding a conflict between theory-based expectations and experimental findings. | ||
Deconvolution of the kinetic complexities associated with time-dependent concentrations of the four stereoisomeric isopropenylcyclopropanes and of the two enantiomeric [1,3] carbon shift products led to rate constants for [1,3] shifts with retention and with inversion of cyano-substituted carbons in cis and trans reactants. Stereochemical information at the migration terminus could not be gained, but the retention versus inversion reaction stereochemical characteristics at the migrating carbon for the four non-racemic reactants were determined securely. The trans starting material isomerized with a ki:kr rate constant ratio, the [1,3] shift with inversion : [1,3] shift with retention ratio, of 70 : 30. The cis starting material gave kinetically controlled products reflecting a ki:kr ratio of 40 : 60!18
The ki and kr rate constants are equivalent to pairs of rate constants labeled according to suprafacial,antarafacial and retention,inversion options (ki = (ksi + kai); kr = (ksr + kar)). Here and elsewhere the rate-constant labeling convention adopted (ksi, kai, ksr, kar) is used to denote stereochemical relationships between the stereochemical specifics of a starting material and of a distinct kinetically controlled [1,3] shift product, whatever the mechanistic course of the isomerization. The convention labels structural relationships, not mechanistic rationalizations.
The kinetic findings demonstrated that the diastereomeric starting materials isomerize with different stereochemical preferences, even though both products formed are enantiomeric and thus of equal heats of formation. Isomerizations of the cis starting materials take place with (ksr + kar) > (ksi + kai). Orbital symmetry theory applied to these vinylcyclopropane isomerizations would lead one to anticipate that the si allowed path would be kinetically dominant and the forbidden ai path would be inconsequential. The experimental observations clearly conflict with this expectation. The continuous diradical formulation so useful for conceptualizing the stereomutations served well to accommodate the kinetically controlled cyclopentene products, pending quadrisection of the rearrangement to gain information on the relative participation of all four stereochemically distinct paths for [1,3] carbon shifts.
The first complete stereochemical study of a vinylcyclopropane-to-cyclopentene isomerization was provided by Andrews in 1976.20 Using two methyl groups as stereochemical markers, and following racemization of the non-racemic starting material as well as the competitive isomerization to the cis-2-methyl-1-(E)-propenylcyclopropane, which quickly reacts further to give 1,4(Z)-heptadiene and thus simplifies the kinetic situation considerably, the distribution of isomeric products was as shown in Scheme 8. The more thermodynamically stable trans enantiomers of the product were favored, and the si outcome was preferred to the ar alternative. The cis enantiomers of the cyclopentene products, formally related to forbidden sr and ai paths, were clearly in evidence, to different extents.
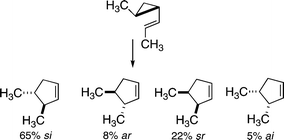 | ||
| Scheme 8 Four stereochemically distinct outcomes quantified for a vinylcyclopropane rearrangement. | ||
The mechanistic implications and even the reliability of these experimental findings were challenged as two further attempts to define stereochemical outcomes for selected vinylcyclopropane-to-cyclopentene isomerizations were published. The uncertainties prompted by these investigations led to further experimental work and to complete reaction stereochemical definitions for an additional seven substituted vinylcyclopropanes.21 All systems having a trans substituent at C2 gave [1,3] carbon shift products through all four stereochemically distinct paths in proportions similar to those reported by Andrews. The subtle problems related to polarimetric data and attempted chromatographic separations that contributed to the two challenging experimental studies were uncovered and the body of experimental data providing a footing for mechanistic assessments was clarified.
The stereochemical findings for deuterium-substituted vinylcyclopropanes were particularly helpful, for they prompted serious calculational efforts to define a potential energy surface for isomerizations to deuterium-labeled cyclopentenes and then dynamics calculations to model reaction outcomes for short-lived diradicals on the caldera energy plateau.
The experimental work of Villarica and the expert NMR analyses secured by Freedberg and Anet to define isomeric distinctions in product mixtures provided information on all four paths by studying the reactions of two isomeric d3-vinylcyclopropanes.22 The two give different pairs of stereochemical outcomes leading to the same racemic product (Scheme 9). Very rapid stereomutations of the vinylcyclopropanes and relatively slow [1,3] shifts restricted data collection to times sufficient for only 1.0 to 2.7% conversions to cyclopentenes.
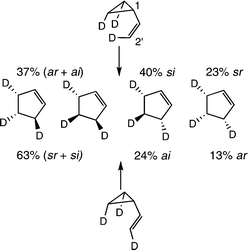 | ||
| Scheme 9 Four stereochemical outcomes for the vinylcyclopropane-to-cyclopentene isomerization. | ||
Calculations providing the potential energy surface (PES) for the isomerization soon followed. The PES included a broad, relatively flat transition region surrounded by four isometric exit channels.23 It led in turn to dynamics calculations to model reaction stereochemistry. Some 34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 trajectories initialized quasi-classically with Boltzmann normal mode sampling at 573 K were followed.24 The projected distribution of stereochemical outcomes corresponded to si:ar:sr:ai ratios of 42 : 10 : 30 : 18, values reasonably close to the experimentally estimated si:ar:sr:ai ratios of 40 : 13 : 23 : 24. The two sets of ratios have reactions that correspond to allowed si and ar paths and forbidden sr and ai paths in 52 : 48 and 53 : 47 proportions. The reaction stereochemical data and detailed theoretical assessments leave no grounds for viewing the vinylcyclopropane-to-cyclopentene isomerization as a process controlled by orbital symmetry. It is clearly a transformation taking place through multi-step processes involving diradical intermediates.
000 trajectories initialized quasi-classically with Boltzmann normal mode sampling at 573 K were followed.24 The projected distribution of stereochemical outcomes corresponded to si:ar:sr:ai ratios of 42 : 10 : 30 : 18, values reasonably close to the experimentally estimated si:ar:sr:ai ratios of 40 : 13 : 23 : 24. The two sets of ratios have reactions that correspond to allowed si and ar paths and forbidden sr and ai paths in 52 : 48 and 53 : 47 proportions. The reaction stereochemical data and detailed theoretical assessments leave no grounds for viewing the vinylcyclopropane-to-cyclopentene isomerization as a process controlled by orbital symmetry. It is clearly a transformation taking place through multi-step processes involving diradical intermediates.
The complete stereochemical quadrisection of isomeric 2-cyano-1-(E)-propenylcyclopropanes achieved by Doering and Barsa provided additional insight on how diastereomeric distinctions at the migrating carbon might influence reaction product ratios (Scheme 10).25 The trans diastereomer gave a preponderance (a 67% preference) of trans enantiomeric products, corresponding to the allowed si and ar paths. The cis diastereomers give a preponderance of the same products (a 74% preference) corresponding to sr and ai forbidden baths. Diradical conformational flexibilities on the transition region caldera allow for some bias toward the more thermodynamically favored products, unconstrained by any orbital symmetry related limitations.
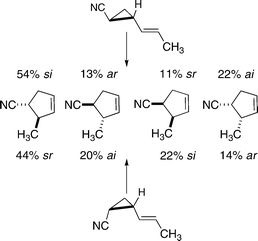 | ||
| Scheme 10 Partitionings among different stereochemical paths for 2-cyano-1-(E)-propenylcyclopropanes. | ||
Stereochemistry and mechanism of vinylcyclobutane rearrangements
The complexities associated with determining reaction stereochemistry for conversions of various vinylcyclobutanes to cyclohexenes crop up again in experimental attempts to uncover rate constants for the four possible paths from some specific monocyclic vinylcyclobutane to four distinguishable cyclohexenes. Relatively rapid stereomutations interconvert the four possible stereoisomeric vinylcyclobutanes as each reacts to form each isomeric cyclohexene product. One must determine the time dependence of all isomers of starting material and all isomers of [1,3] carbon shift products, then deconvolute the raw kinetic data to define rate constants for each possible stereomutation reaction and each stereochemically distinct [1,3] shift.For an exemplary case, that presented by 2-methyl-1-(E)-propenylcyclobutanes, the kinetic situation is typical (Scheme 11).26 The stereomutations involve 12 rate constants but only 5 independent kinetic parameters. Each enantiomer of a trans reactant and each of a cis diasteroisomer could isomerize through [1,3] shifts of a methyl-substituted C2 carbon in four ways. Or, stated otherwise, each stereoisomeric substituted vinylcyclobutane would give, through a particular stereochemical linkage between starting isomer and product isomer, a unique product. In the formulations of Scheme 11 and Scheme 12, rate constants ksr link a trans diastereomer to a product and the k′sr paths originate with cis diastereomers.
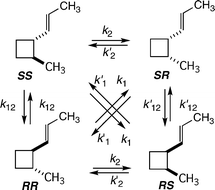 | ||
| Scheme 11 Thermal stereomutations of 2-methyl-1-(E)-propenylcyclobutanes. | ||
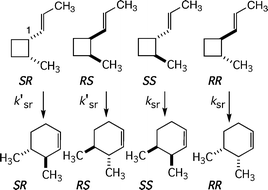 | ||
| Scheme 12 Different isomers of starting materials may give different isomers of products even though all correspond to the same stereochemical path. | ||
The kinetic analysis needs to take into account the two possible kinetically competitive retro-[2 + 2] cycloaddition reactions of each diastereomer leading to ethylene or propene, as well as [1,3] carbon shifts forming stereoisomeric 3,6-dimethylcyclohexenes. The cis diastereomers also react through a homodienyl hydrogen migration to form cis-1,5-octadiene; this isomerization takes place, but it is not so fast that the [1,3] carbon shifts of interest cannot compete. All of which seems very complicated, but the kinetic situation can be mastered through some patience and careful treatments of the relevant differential equations and the related integrated solutions. The more arduous requirements relate to synthetic preparations of specific stereoisomers of starting materials and 3,4-dimethylcyclohexene products, sure assignments of absolute stereochemistry, and developing and validating suitable analytical techniques for gaining reliable information on mole percent concentrations of all isomers in reaction mixtures as functions of reaction times.
The gas phase kinetic investigation of thermal reactions at 275 °C leading from 2-methyl-1-(E)-propenylcyclobutanes to 3,4-dimethylcyclohexenes gave the distribution of relative rate constants shown in Scheme 13.
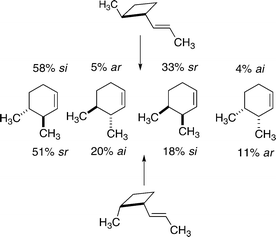 | ||
| Scheme 13 Partitionings among different stereochemical paths for 2-methyl-1-(E)-propenylcyclobutanes. | ||
The trans reactant isomerized through the various stereochemically distinct [1,3] carbon shift paths in proportions quite similar to those observed in 1976 for a non-racemic trans-2-methyl-1-(E)-propenylcyclopropane (65% si, 8% ar, 22% sr, 5% ai).20 The cis diastereoisomer of the substituted vinylcyclobutane provided another set of four rate constants for [1,3] shifts; similar data were not accessible starting from a non-racemic cis-2-methyl-1-(E)-propenylcyclopropane, since [1,3] carbon migrations for this reactant could not compete with a much faster isomerization through a homodienyl hydrogen shift.
The dominant reaction paths from both diastereomers of 2-methyl-1-(E)-propenylcyclobutane favor the thermodynamically more stable products, the trans-3,4-dimethylcyclohexenes: the trans reactant gives allowed si and ar outcomes 63% of the time; the cis reactant leads through [1,3] carbon sigmatropic shifts to forbidden sr and ai outcomes rather than allowed si and ar outcomes by a 71 : 29 margain.
An independent study led to a complete definition of rate constants for all stereomutations and [1,3] carbon shifts in deuterium-labeled racemic 2-cyano-1-(E)-propenylcyclobutanes.27 Different analytical techniques, facilitated by the cis-disposed deuterium labels at C3 and C4, gave the required data, which were processed to yield the relative rate-constant distributions for the [1,3] shifts at 207 °C (Scheme 14).
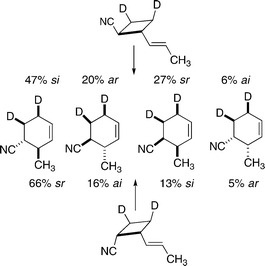 | ||
| Scheme 14 Partitionings among different stereochemical paths for 2-cyano-1-(E)-propenylcyclobutanes. | ||
Again, one sees trans isomers of the products favored by both diastereomers of the substituted vinylcyclobutanes: the orbital symmetry theory forbidden sr and ai paths from the cis reactants account for 82% of the [1,3] shifts. This fact undercuts any temptation to interpret the observation that allowed si and ar paths are favored starting from trans isomers of 2-substituted 1-(E)-propenylcyclobutanes, whether the C2 substituent is methyl or cyano, because of orbital symmetry factors. The preference for trans isomers of products from either cis or trans isomers of the reactants are more plausibly explicated by postulating a common determinant: dynamic effects as conformationally flexible diradical intermediates seek exit channels from the caldera energetic plateau.
Work has been initiated on the stereochemical aspects of more minimally substituted vinylcyclobutanes.28 2-Deuterio-1-(E)-propenylcyclobutanes at 276 °C gave stereomutations and isomerized through [1,3] shifts to 3-methyl-4-deuteriocyclohexenes and 3-methyl-6-deuteriocyclohexenes (Scheme 15). The balance between migrations of C2 through (ksi + kar) and through (ksr + kai) paths was found to be 72 : 28. More detailed kinetic investigations using reactants of very high enantiomeric and diastereomeric ratios, and reliable analytical methods for determining all relevant mole percent concentrations as functions of reaction time, will be necessary to determine all four [1,3] carbon shift rate constants and the relative participation of each in the isomerizations.
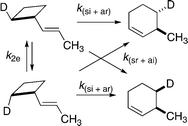 | ||
| Scheme 15 The relative importance of allowed si + ar versus forbidden sr + ai stereochemical outcomes for 1-(E)-propenylcyclobutane is 72 : 28. | ||
Still more challenging will be experiments to obtain all four rate constants for [1,3] carbon shifts from one specific 2,2′-d2-vinylcyclobutane. While the required synthetic chemistry, determinations of absolute stereochemistry for all isomers related by stereomutations and [1,3] shifts of C2, and management of the relevant kinetic expressions should be relatively tractable, the analytical demands of such a project could present serious difficulties. Techniques based on NMR spectroscopy, an obvious approach one might consider, would be complicated by products formed through [1,3] shifts of C4. Yet the data sought would provide invaluable information to compare with insights provided by initial efforts to construct a PES for the isomerizations and in due course calculated distributions of kinetically controlled mixtures of stereoisomeric products formed from one stereoisomer of a 2,2′-d2-vinylcyclobutane (Scheme 16).
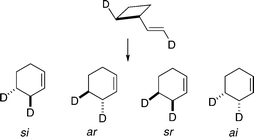 | ||
| Scheme 16 Possible isomerizations of a non-racemic 2,2′-d2-vinylcyclobutane. | ||
Stereochemistry and mechanism of degenerate rearrangements of bicyclo[3.1.0]hex-2-enes
Carbon [1,3] sigmatropic shifts of vinylcyclopropanes and vinylcyclobutanes have counterparts of a degenerate sort. Both bicyclo[3.1.0]hex-2-enes and bicyclo[3.1.1]hept-2-enes are known to isomerize through [1,3] migrations detectable through isotopic labeling experiments or by following thermal racemizations. The former reactions have been investigated far more thoroughly.Stereochemical investigations of thermal rearrangements in bicyclo[3.1.0]hex-2-enes have uncovered three distinguishable modes of isomerizations: [1,3] carbon shifts of sr stereochemistry, [1,3] carbon shifts with ai stereochemistry, and a skeletal inversion or “ring-flip” process. All three modes of isomerization are forbidden according to orbital symmetry considerations.
A detailed investigation of the degenerate isomerizations shown by (–)-α-thujene ((–)-(1S,5S)-2-methyl-5-isopropylbicyclo[3.1.0]hex-2-ene) demonstrated that three processes are of significance.29 The system can undergo enantiomerization of the bicyclic skeleton through a ring-flip process or through a [1,3] shift with sr stereochemistry (Scheme 17). Thus by following the rate of racemization the rate constant combination (k(ring-flip) + ksr) may be readily determined. By following the equilibration of deuterium labels through NMR analyses the rate constant combination (ksr + kai) may be measured. To gain the individual rate constants required determinations of deuterium labeling distributions of specific enantiomers of partially racemized samples of the various α-thujenes in reaction mixtures, or of structural derivatives thereof. This arduous necessity was achieved through classical optical resolutions of isothujyl hydrogen phthalates derived from the thujenes followed by determinations of optical activity and deuterium labels. The relative importance of the three modes of isomerization was estimated to be ksr:k(ring-flip):kai = 56 : 24 : 20, and the activation parameters for racemization in the gas phase were found to be Ea 43.4 kcal mol–1 and logA 14.3.
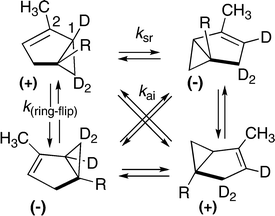 | ||
| Scheme 17 Degenerate thermal isomerizations of optically active and deuterium labeled α-thujenes (R = isopropyl). | ||
Following on the early kinetic work of Cooke and Andrews,30 a more detailed investigation of the equilibrations of the four 4- and 6-d-bicyclo[3.1.0]hex-2-enes (Scheme 18) found that all three types of isomerizations had identical activation parameters (Ea 44.3 kcal mol–1, logA 14.1), indicating that differences in rate constants did not stem from different activation energy barriers. The three reactions participate in the proportions ksr: k(ring-flip):kai = 48 : 36 : 16.31
![Degenerate isomerizations of deuterium-labeled bicyclo[3.1.0]hex-2-enes.](/image/article/2008/OB/b711494j/b711494j-s18.gif) | ||
| Scheme 18 Degenerate isomerizations of deuterium-labeled bicyclo[3.1.0]hex-2-enes. | ||
The theory-based potential energy surface for the isomerizations of bicyclohexene and the conformational options available once the diradical intermediate formed in the transition region followed quickly.32 Dynamics trajectories were calculated based on the PES and gave estimations of the relative importance of the three overall isomerization processes observed: at 498 and 528 K there were (47 ± 1.7)% ksr, (38 ± 1.7)% k(ring-flip), and (15 ± 1.3)% kai.33 The mechanistic model based on short-lived diradical intermediates of some conformational flexibility on the caldera until encountering an exit channel fits the experimental observations and the trajectory-calculated representation of the isomerizations quite well.
Studies of thermal isomerizations of bicyclo[3.1.1]hept-2-enes have been less frequent and less detailed. The degenerate isomerizations of bicyclo[3.1.1]hept-2-ene itself at 226–236 °C through [1,3] carbon shifts were demonstrated by Dietrich and Musso using a deuterium labeling approach (Scheme 19).34
![The interconversion of 1-d- and 3-d-bicyclo[3.1.1]hept-2-ene.](/image/article/2008/OB/b711494j/b711494j-s19.gif) | ||
| Scheme 19 The interconversion of 1-d- and 3-d-bicyclo[3.1.1]hept-2-ene. | ||
So far there has been no reported attempt to obtain information on the stereochemistry of the [1,3] carbon shifts involved. Given the bicyclic skeleton, only si and sr stereochemical outcomes are available, but that limitation does not translate into a very simple kinetic situation. Thermal epimerization at a deuterium-labeled C6 is a definite possibility, and migrations of both C6 and C7 would occur. A complete stereochemical study to define the kinetic parameters kr, ki, and kepim (dependent on C5–C6 bond cleavage) and k13 (for migration of C7) would involve following with good accuracy the time evolution of six degenerate isomers (three pairs of enantiomers). The kinetic scheme would involve 20 rate constants but only four independent kinetic parameters.
The earliest publication now interpretable in terms of a [1,3] carbon shift concerned the degenerate thermal racemization of α-pinene. That work of Smith in 1927, prompted by discussions with G. N. Lewis, may have been the first study involving determinations of reaction rate constants for a gas phase degenerate thermal racemization.5 The rate constants obtained between 184 and 237 °C led to a calculated Ea value of 43.7 kcal mol–1.
Smith's report on the racemization of α-pinene in 1927 was contested by Conant and Carlson in 1929.35 Both papers are well worth reading today, for they provide a vivid reminder of the limitations in experimental methods available at that time. Both experimental approaches included conscientious efforts to make reliable allowances for the complications contributed by other reactions available to α-pinene (2,6,6-trimethylbicyclo[3.1.1]hept-2-ene). Of particular note are retro-ene isomerizations leading to limonene (1-methyl-4-isopropenylcyclohexene) or racemic limonene (dipentene) and retro-[2 + 2] additions giving ocimene (3,7-dimethylocta-1,3,6-triene) and then through a rapid [1,5] hydrogen shift to allocimene (3,7-dimethylocta-2,4,6-triene) (Scheme 20). These types of reactions are often competitive with [1,3] carbon shifts when structural characteristics make them conceivable.
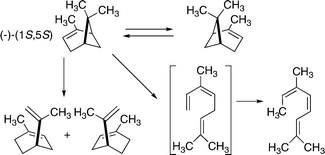 | ||
| Scheme 20 Complexities presented by thermal reactions of α-pinene. | ||
Gajewski and coworkers approached these reactions through careful kinetic studies of the reactions shown by (1S)-(–)-α-pinene and a deuterium-labeled analog.36 Activation parameters for the thermal enantiomerization were found to be logA 14.4 and Ea 45.0 kcal mol–1. At 257 °C the relative rate constants for enantiomerization through a [1,3] shift, a retro-ene isomerization, and the retro [2 + 2] cycloaddition were 1, 2.5, and 3.3. The retro-ene product mixture retained only 5% of its stereochemical integrity; most of the diene was racemic limonene (dipentene). A Cs-symmetric diradical intermediate able to transfer a hydrogen to either end of an allylic unit with almost equal efficiency could well be responsible for this observation.
When syn-6-CD3-(1S)-α-pinene was reacted at 257 °C for 40 min, the rate for loss of α-pinene was diminished by a factor of about 1.15; the conversion to limonene through retro-ene reactions was diminished by about 30% (Scheme 21). Yet transfer of D rather than H was favored by a factor of 2, according to deuterium NMR analyses. Distinctions regarding deuterium placements for the separate enantiomers of limonene were not accessible.
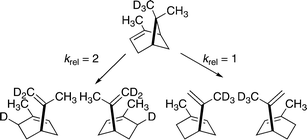 | ||
| Scheme 21 Limonene products from syn-6-CD3-(1S)-α-pinene. | ||
After 60 min the recovered α-pinene was separated into enantiomers by GC on an α-cyclodextrin-packed column. For the S isomer the syn:anti ratio for the C6-CD3 groups was 95 : 5. Little loss of stereochemistry at C6 had occurred; presumably most of the recovered starting material never entered the caldera. In the R enantiomer, the syn:anti ratio was 55 : 45, corresponding to a small preference for an overall [1,3] shift with inversion (Scheme 22).
![Isomerization through a [1,3] carbon shift with a slight preference for inversion.](/image/article/2008/OB/b711494j/b711494j-s22.gif) | ||
| Scheme 22 Isomerization through a [1,3] carbon shift with a slight preference for inversion. | ||
One rationale to account for these observations would postulate Cs-symmetric diradical intermediates, with either the 6-CD3 group or the 6-CH3 group oriented toward the center of the allylic sub-structure. The diradicals might equilibrate to some degree before collapsing to one or another closed shell product, but the ratio would always be ≥1. The 30% reduction of the rate of formation of limonene products would be an expected outcome for a primary kH/kD effect from an initially formed Cs-intermediate having CD3 in position to transfer a deuterium. The slower rate of formation of limonene could lead by default to the modestly lower rate for loss of α-pinene, for the partitioning ratios from the diradical would be modified in that sense. The other conformational isomer could only transfer hydrogen. The factor of 2 preference for deuterium transfer could reflect the time-averaged preponderance of the initially formed and for some time favored diradical conformation. The nearly complete loss of stereochemical integrity in the d3-labeled (1R)-α-pinene product formed through an overall [1,3] carbon shift would be consistent with considerable equilibration between the two postulated diradical conformers.
Thermal reactions of bicyclo[3.2.0]hept-2-enes and bicyclo[4.2.0]oct-2-enes
Given the importance accorded the report in 1967 that 6-endo-acetoxy-7-exo-deuteriobicyclo[3.2.0]hept-2-ene isomerizes through a [1,3] carbon shift with si stereochemistry,13 it was inevitable that more detailed investigations of bicyclo[3.2.0]hept-2-ene and other bicyclic vinylcyclobutanes would be pursued.37 A kinetic investigation of the parent hydrocarbon by Cocks and Frey in 1971 demonstrated that bicyclo[3.2.0]hept-2-ene reacts through three homogeneous first-order processes.38 A direct fragmentation gave cyclopentadiene and ethylene. Bicyclo[2.2.1]hept-2-ene was formed through a [1,3] shift isomerization, and then reacted further, at a rate much faster that the rate of its formation, through a retro-Diels–Alder reaction. A retro-[2 + 2] cycloaddition cleaving C1–C5 and C6–C7 gave 1,3Z,6-heptatriene as a primary product. Rate constants for the reactions leading directly and indirectly to cyclopentadiene and ethylene were subject to large errors, though the sum of these rate constants was well defined.The rate constants k1 and k2 in Scheme 23 were hard to determine individually because the bicyclo[2.2.1]hept-2-ene formed directly through a relatively slow reaction was depleted quickly through a relatively fast subsequent isomerization, and it never amounted to much. It reached a maximum concentration of about 1.5 mol% as the reactions took place at 276 °C.39
![Reaction paths leading from bicyclo[3.2.0]hept-2-ene.](/image/article/2008/OB/b711494j/b711494j-s23.gif) | ||
| Scheme 23 Reaction paths leading from bicyclo[3.2.0]hept-2-ene. | ||
These two rate constants were measured successfully through a dynamic isotope dilution kinetic study.39 While the magnitudes of those rate constants are of little interest here, the study also found that 1,2-d2-ethylene formed from cis-6,7-d2-bicyclo[2.2.1]hept-2-ene was entirely cis, while the 1,2-d2-ethylene formed directly from cis-6,7-d2-bicyclo[3.2.0]hept-2-ene was entirely stereochemically scrambled. A similar result was later confirmed for cis-2,3-d2-vinylcyclobutane: it fragments to give equal amounts of E and Z isomers of 1,2-d2-ethylene.40
Though only minimal amounts of d2-labeled bicyclo[2.2.1]hept-2-enes in reaction mixtures from cis-6,7-d2-bicyclo[3.2.0]hept-2-ene ever built up, two groups were able to isolate some and gain information on reaction stereochemistry.41 Belfield found that the isomerization at 276 °C takes place with an si:ar ratio of about 76 : 24, based on the average of determinations for seven reactions, some with slightly different distributions of deuterium labels. At 312 °C Klärner and co-workers found that the isomerization took place with an 89 : 11 si:sr balance.
An observed thermal epimerization at C7 in a bicyclo[3.2.0]hept-2-ene might be rationalized by postulating a ring inversion or a stereomutation at C7 made possible by a C1–C7 bond cleavage (Scheme 24). The relevance of the two possible paths was probed for the parent bicyclic system with the aid of deuterium labeling at C6, then at C7.42 Isomerization through a ring inversion process was very slow at 275 °C but clearly detectable (k = 4 × 10–8 s–1), while the overall epimerization at C7 through both a ring inversion and a stereomutation at C7 was an order of magnitude faster (k = 5.4 × 10–7 s–1). The difference between the two rate constants, 5 × 10–7 s–1, ascribed to the one-way epimerization at C7, is an order of magnitude smaller that the [1,3] carbon shift reactions (k = 4.4 × 10–6 s–1 at 276 °C).
![Ring inversions and stereomutations at C7 of deuterium-labeled bicyclo[3.2.0]hept-2-enes.](/image/article/2008/OB/b711494j/b711494j-s24.gif) | ||
| Scheme 24 Ring inversions and stereomutations at C7 of deuterium-labeled bicyclo[3.2.0]hept-2-enes. | ||
Racemic epimers of 7-methylbicyclo[3.2.0]hept-2-enes equilibrate thermally at rates competitive with the [1,3] shifts. The epimerizations might result from a ring inversion process triggered by cleavage of the C1–C5 bond or through a one-centered stereomutation at C7 dependent on breaking the C1–C7 bond. Heating 6-endo-methylbicyclo[3.2.0]hept-2-ene at 275 °C led to fragmentation and some 5-exo-methylbicyclo[2.2.1]hept-2-ene; no 6-exo-methylbicyclo[3.2.0]hept-2-ene was detected in any product mixture over 27 h (Scheme 25).43 Hence the epimeric 6-methylbicyclo[3.2.0]hept-2-enes do not suffer a kinetically competitive ring inversion and, presumably, neither do 7-methyl analogs. The epimerizations they exhibit derive from the same diradical intermediates responsible for [1,3] carbon shift reactions.
![endo-6-Methylbicyclo[3.2.0]hept-2-ene does not exhibit a kinetically competitive ring inversion reaction.](/image/article/2008/OB/b711494j/b711494j-s25.gif) | ||
| Scheme 25 endo-6-Methylbicyclo[3.2.0]hept-2-ene does not exhibit a kinetically competitive ring inversion reaction. | ||
Stereochemical outcomes for [1,3] carbon shifts in 7-methylbicyclo[3.2.0]hept-2-enes are strongly dependent on the stereochemistry of the reactant. In the 6-endo-acetoxy-7-exo-methyl system thermal isomerization at 290 °C led to a 9.3 : 1 predominance of the 5-exo-acetoxy-6-exo-methylbicyclo[2.2.1]hept-2-ene isomerization product over the 5-exo-acetoxy-6-endo-methyl alternative (Scheme 26).44 Thus the si:sr ratio, 90 : 10, was consistent with the reaction behaving, mostly, according to stereochemical expectations based on the orbital symmetry generalizations of 1969–1970. The epimeric starting material, the 6-endo-acetoxy-7-endo-methyl bicyclic system, isomerized to 3-exo-methyl versus 3-endo-methyl products with a 12 : 88 ratio, almost a complete reversal favoring the forbidden stereochemical outcome.
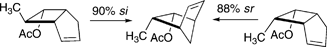 | ||
| Scheme 26 Migrations with reversed stereochemical path preferences. | ||
A still more pronounced preference for an sr [1,3] carbon shift in a bicyclo[3.2.0]hept-2-ene having an endo methyl group at C7 was demonstrated by Forman and Leber in 1986.45endo-7-Methyl-exo-7-vinylbicyclo[3.2.0]hept-2-ene isomerizes at convenient rates at much lower temperatures, thanks to the influence of the vinyl substituent on the strength of the C1–C7 bond. At 150 to 166 °C the reactant gives very little epimerization at C7 and very little endo-5-methyl-exo-5-vinylbicyclo[2.2.1]hept-2-ene, the si product. The dominant product is formed by way of exo-5-methyl-endo-5-vinylbicyclo[2.2.1]hept-2-ene, the structure formed through an sr stereochemical outcome, followed by a rapid Cope rearrangement. The si:sr ratio was estimated to be 0.04, or 4 : 96 (Scheme 27).
![Strong preference for sr stereochemistry in a [1,3] carbon shift.](/image/article/2008/OB/b711494j/b711494j-s27.gif) | ||
| Scheme 27 Strong preference for sr stereochemistry in a [1,3] carbon shift. | ||
Similar stereochemical results were found when the isomerizations shown by the exo and endo isomers of 7-methylbicyclo[3.2.0]hept-2-ene were characterized (Scheme 28).46 The [1,3] carbon shifts of the exo isomer at 275 °C occurred in a si:sr 88 : 12 stereochemical ratio, while the endo reactant gave almost exclusively 6-exo-methylbicyclo[2.2.1]hept-2-ene, the forbidden sr product. The si:sr ratio was close to 0.
![Reactions giving a preponderance of the same [1,3] shift product through different stereochemical paths.](/image/article/2008/OB/b711494j/b711494j-s28.gif) | ||
| Scheme 28 Reactions giving a preponderance of the same [1,3] shift product through different stereochemical paths. | ||
These stereochemical facts can't be interpreted in any consistent fashion through the generalization that orbital symmetry factors dictate that a [1,3] suprafacial carbon shift must occur with inversion. The symmetry-based analysis leading to the generalization is perfectly fine, but the conclusion is not applicable to the reactions in question, for they do not show isomerizations in strict adherence to the rules for conservation of orbital symmetry.
A more plausible model for these [1,3] shifts was advanced by Carpenter in 1995.47,48 Classical molecular dynamics calculations using a semi-empirical potential energy surface concluded that the isomerizations involve a stepwise mechanism and a singlet diradical intermediate. The early stages of C1–C7 bond cleavage do not include much torsional change about the C6–C7 bond. As C1–C7 is broken, and a rotation about C5–C6 begins to slide C7 and its substituents across the five-membered ring, a rotation about C6–C7 comes into play so as to maintain some residual C1–C7 bonding as long as possible. This factor sets the sense of the torsional rotation about C6–C7: in the (1R)-bicyclo[3.2.0]hept-2-ene structures shown, the C6–C7 rotation is counter-clockwise. The residual bonding between C1 and C7, not an incipient bonding between C3 and C7, provides this stereochemical bias. Conservation of angular momentum favors a continuation of this rotation and thus the face of the migrating methylene group corresponding to an inversion outcome is presented to its future bonding partner C3 even as C7 and C3 are relatively far apart.
An 7-exo-methyl substituent does not alter this scenario much, but 7-endo-methylbicyclo[3.2.0]hept-2-ene cannot follow a similar trajectory. The counter-clockwise C6–C7 rotational motion is impeded, for it would tend to bring the methyl group and five-membered ring into serious steric conflict. The C1–C7 bond is cleaved without the facilitating contribution from the C6–C7 counter-clockwise torsion; the cleavage leads to a more extended conformation of the diradical having the face of the migrating group corresponding to a retention outcome positioned for eventual bond formation with C3.
One may question this or that detail of the dynamics calculations, given the recognized computational limitations inherent in the study. Yet at a minimum the findings make a powerful case for not taking an experimental observation of si reaction stereochemistry, corresponding to a Woodward–Hoffmann-allowed [1,3] carbon sigmatropic isomerization, as conclusive evidence against a stepwise mechanism, or as a sure confirmation that the reaction under consideration was concerted and featured a transition structure involving bonding interactions between the migration carbon and both migration origin and migration terminus. The calculated mid-reaction diradical structure for the isomerization of bicyclo[3.2.0]hept-2-ene to bicyclo[2.2.1]hept-2-ene features a rotational barrier about the original C6–C7 bond of 1.2 kcal mol–1.47 The alkyl radical and the allylic radical entities are separate sub-structures within the diradical intermediate; they are not connected through residual or incipient bonding interactions. A preference for migration with inversion rather than retention of stereochemistry in the isomerization of bicyclo[3.2.0]hept-2-ene to bicyclo[2.2.1]hept-2-ene (norbornene) is due to dynamic factors. Conservation of orbital symmetry plays no role.
Other assessments of thermal [1,3] suprafacial carbon shifts leading from bicyclo[3.2.0]hept-2-ene to norbornene were forthcoming in 1998–1999 when Houk and coworkers exercised DFT calculations and complete active space CASSCF and CASPT2 theory to investigate alternative concerted and stepwise diradical-mediated mechanistic understandings of the isomerization.49 Their findings confirmed that the reaction takes place by way of a diradical transition structure. The breaking C1–C7 bond in the transition structure, according to the calculations, is 2.589 Å long, and the length of the bond eventually formed between C3–C7 is 3.694 Å. There is no cyclic conjugation in the transition structure, and the overall reaction stereochemistry, known to exhibit a preference for si over sr outcomes, can be traced to reaction dynamics as diradical conformers traverse a relatively flat energy plateau prior to the formation of norbornene. While many important details of the computational methods, calculated structures and energies, and insights reported by Carpenter and by Houk and co-workers are distinct, the essential conclusions reached are identical. The isomerization involves a diradical intermediate, not an orbital symmetry allowed concerted process. Reaction stereochemistry is defined after the rate-determining step featuring a diradical transition structure. Whether a “significant intermediate” is involved or not depends directly on careful semantic distinctions separating “significant intermediate” from “short-lived intermediate” and “stable intermediate”.
Recent preparations and investigations of the thermal reactions of bicyclo[4.2.0]oct-2-enes have provided an additional basis for judging whether there might be some orbital symmetry allowed and concerted si [1,3] carbon shifts along with diradical-mediated two-step reactions that give the si product for other reasons. In the bicycloheptene, the hypothetical concerted transition structure, having partial bonding on either side of the migrating methylene function with C1 and with C3, would entail “extraordinarily unfavorable geometric constraints”. In a [1,3] carbon shift process starting from bicyclo[4.2.0]oct-2-ene, the hypothetical transition structure would be less strained. The geometric constraints would be to some degree relaxed. Hence higher si:sr ratios could be anticipated for isomerizations of bicyclooctenes relative to similarly substituted bicycloheptenes.
The 8-methylbicyclo[4.2.0]oct-2-enes were prepared and gas phase thermal reactions at 275 °C were followed for each isomer (Scheme 29).50 The 8-exo-methyl isomer isomerized to give 5-exo-methylbicyclo[2.2.2]oct-2-ene and the 5-endo-methyl epimer in a 71 : 29 ratio. The 8-endo-methylbicyclo[4.2.0]oct-2-ene did not form a [1,3] shift product: it gave its epimeric 8-exo-methyl isomer and fragmented to yield 1,3-cyclohexadiene and propylene.
![Stereochemical preferences for [1,3] carbon shifts shown by 8-exo-methyl- and 8-exo-methoxybicyclo[4.2.0]oct-2-ene.](/image/article/2008/OB/b711494j/b711494j-s29.gif) | ||
| Scheme 29 Stereochemical preferences for [1,3] carbon shifts shown by 8-exo-methyl- and 8-exo-methoxybicyclo[4.2.0]oct-2-ene. | ||
Thus 7-exo-methylbicyclo[3.2.0]heptene gives relatively more si [1,3] shift product (88%) than does 8-exo-methylbicyclo[4.2.0]oct-2-ene (71%), a result that does not support the orbital symmetry controlled vision and correlative hypothetical transition structures for these reactions of simple homologs. Similar stereochemical preferences were demonstrated for [1,3] carbon shifts of 8-exo-methoxybicyclo[4.2.0]oct-2-ene: the si:sr ratio for the 5-methoxybicyclo[2.2.2]oct-2-enes formed was 76 : 24.51
A more stringent test of the proposition that more flexible reactants should favor si product outcomes was made with deuterium-labeled bicyclo[4.2.0]oct-2-enes (Scheme 30). A deuterium marker at C7 did not equilibrate with the corresponding epimer at 300 °C over times convenient for measuring rates of conversion to deuterium-labeled bicyclo[2.2.2]octenes and to fragmentation products.
![The epimeric 7-d-bicyclo[4.2.0]oct-2-enes do not exhibit kinetically competitive equilibrations through a ring inversion process.](/image/article/2008/OB/b711494j/b711494j-s30.gif) | ||
| Scheme 30 The epimeric 7-d-bicyclo[4.2.0]oct-2-enes do not exhibit kinetically competitive equilibrations through a ring inversion process. | ||
A sample of 8-d-bicyclo[4.2.0]oct-2-ene (Scheme 31) rich in the endo isomer at 300 °C exhibited a faster epimerization at C8 (keq = 2k8e = 6.1 × 10–5 s–1) than isomerizations through [1,3] carbon shifts (k1,3 = 4.3 × 10–6 s–1).52 The diradical must re-form the starting hydrocarbon structure at least 7 times more often than it gives the [2.2.2] bicyclic product if one assumes that some recombinations must involve no change in configuration.
![Rapid epimerization at C8 of deuterium-labeled bicyclo[4.2.0]oct-2-enes.](/image/article/2008/OB/b711494j/b711494j-s31.gif) | ||
| Scheme 31 Rapid epimerization at C8 of deuterium-labeled bicyclo[4.2.0]oct-2-enes. | ||
The kinetically controlled [1,3] shift product distribution was 58 : 42 favoring the inversion outcome (Scheme 32). Again, and more strikingly, the implication that a more flexible allowed concerted si [1,3] carbon shift transition structure might raise the si:sr ratio proved to be incorrect.
![Isomerizations through [1,3] carbon shifts with nearly comparable si and sr outcomes.](/image/article/2008/OB/b711494j/b711494j-s32.gif) | ||
| Scheme 32 Isomerizations through [1,3] carbon shifts with nearly comparable si and sr outcomes. | ||
Comparing rate constants for fragmentations, epimerizations, and [1,3] shifts in bicycloheptenes (at 275 °C) and bicyclooctenes (at 300 °C) reveals that the largest difference is evident in epimerizations. The rate constant ratios for the C8 to C7 reactants at these two temperatures are about 5 (for fragmentations), 120 (for epimerizations), and 1 (for [1,3] shifts). The conformationally flexible diradical formed as the C1–C8 bond of bicyclo[4.2.0]oct-2-ene is cleaved has the capacity to present either face of the alkyl radical unit to possible reaction partners to similar extents. A suitable direct dynamics calculational investigation could show whether these relatively permissive yet not statistically defined stereochemical preferences are based on different classes of trajectories launched as the diradical is formed, or through relatively long-lived diradical intermediates able to rotate more than 180° about the C7–C8 bond before the singlet diradical intermediate is transformed into a [4.2.0] or a [2.2.2] product.
Conclusions
Over the past 40 years a substantial body of experimental evidence bearing on mechanistic understandings of thermal [1,3] carbon shifts has been compiled. Taken as a whole, it provides a firm basis for interpreting such isomerizations as reactions taking place by way of conformationally flexible but not statistically equilibrated diradical intermediates. Competing reactions such as thermal stereomutations and fragmentations reflect other options open to the diradicals. Kinetically controlled product distributions do not follow anticipations based on orbital symmetry rules. They often reflect some bias in favor of more thermodynamically stable isomers. Often si stereochemical outcomes are favored to some extent, but in other cases the sr outcomes are dominant. These preferences can be traced to structural changes associated with progress along the reaction coordinate leading to the transition region and launching the diradical intermediate onto the caldera energetic plateau as a composite of vibrational states. Exit channels are found and product distributions are determined by dynamic factors. Conservation of orbital symmetry plays no role. Orbital symmetry theory for [1,3] carbon shifts recognizes that they could take place in a concerted fashion with si stereochemistry, which is certainly correct. The theory is fine. But it is not applicable to thermal [1,3] carbon shifts in the real world. A beautiful and powerful theory may in practice prove wanting.This point has been made repeatedly over the years by many commentators. One of the most concise and pellucid formulations of the truism has been enunciated by Yogi Berra: “In theory there is no difference between theory and practice. In practice there is”. Yet a theory can be so attractive, so seductive to one impatient to recognize an understanding, that inappropriate applications of the theory can be all but irresistible.
Much of the progress in physical organic chemistry and the study of reaction mechanisms since the 1930s has depended on considering the behavior of closely related groups of molecules. Correlating variations in experimentally determined structural and reaction characteristics led to mechanistic insights of great utility and aesthetic satisfaction. Neither could have been attained by a generalization based on a single example.
In retrospect, one can surmise that interpretations of thermal [1,3] carbon shifts would have evolved differently, if bicyclo[4.2.0]oct-2-ene systems had been studied thoroughly before experiments based on a single structural template, a substituted bicyclo[3.2.0]hept-2-ene, were rationalized in terms of the principle of orbital symmetry control of concerted chemical changes. But once that rationalization was made, and confirmed in a most authoritative fashion, it became established dogma, at least in many quarters, and dogmas are hard to overturn. In some of the most widely used textbooks of organic chemistry today, and even the leading modern physical organic chemistry text, si [1,3] carbon shifts are represented as concerted, orbital symmetry controlled reactions! So the mechanistic controversy, at least in the secondary literature, still retains a vestigial presence.
Acknowledgements
We thank the donors of the Petroleum Research Fund, administered by the ACS, for support of this research at Franklin & Marshall College, and the National Science Foundation for support at Syracuse University, most recently through CHE-0211120 and CHE-0514376.References
- E. Vogel, Angew. Chem., 1960, 72, 4–26 CrossRef CAS , note 162; E. Vogel, R. Palm and K. H. Ott, unpublished; C. G. Overberger and A. E. Borchert, J. Am. Chem. Soc., 1960, 82, 1007–1008 Search PubMed ; see also: N. P. Neureiter, J. Org. Chem., 1959, 24, 2044–2046 Search PubMed.
- R. J. Ellis and H. M. Frey, Trans. Faraday Soc., 1963, 2076–2079 RSC.
- W. von E. Doering and W. R. Roth, Angew. Chem., Int. Ed. Engl., 1963, 2, 115–122 CrossRef , note 13; W. von E. Doering and W. Grimme, unpublished.
- W. von E. Doering and J. B. Lambert, Tetrahedron, 1963, 19, 1989–1994 CrossRef CAS.
- D. F. Smith, J. Am. Chem. Soc., 1927, 49, 43–50 CrossRef CAS.
- J. A. Berson and J. W. Patton, J. Am. Chem. Soc., 1962, 84, 3406–3407 CrossRef CAS.
- H. M. Frey, Adv. Phys. Org. Chem., 1966, 4, 148–193.
- H. M. Frey and R. Walsh, Chem. Rev., 1969, 69, 103–124 CrossRef CAS.
- M. R. Willcott, R. L. Cargill and A. B. Sears, Prog. Phys. Org. Chem., 1972, 9, 25–98 CAS.
- R. B. Woodward and R. Hoffmann, J. Am. Chem. Soc., 1965, 87, 2511–2513 CrossRef.
- R. B. Woodward, in Aromaticity, Special Publication No. 21, The Chemical Society, London, 1967, pp. 217–249 Search PubMed.
- J. A. Berson and R. S. Wood, J. Am. Chem. Soc., 1967, 89, 1043–1044 CrossRef CAS.
- J. A. Berson and G. L. Nelson, J. Am. Chem. Soc., 1967, 89, 5503–5504 CrossRef CAS.
- G. L. Nelson, Ph.D. Dissertation, University of Wisconsin, 1969 Search PubMed.
- R. B. Woodward and R. Hoffmann, Angew. Chem., Int. Ed. Engl., 1969, 8, 781–853 CrossRef CAS; R. B. Woodward and R. Hoffmann, The Conservation of Orbital Symmetry, Verlag Chemie, Weinheim, 1970 Search PubMed.
- M. R. Willcott and V. H. Cargle, J. Am. Chem. Soc., 1967, 89, 723–724 CrossRef CAS; M. R. Willcott and V. H. Cargle, J. Am. Chem. Soc., 1969, 91, 4310–4311 CrossRef CAS.
- P. H. Mazzocchi and H. J. Tamburin, J. Am. Chem. Soc., 1970, 92, 7220–7221 CrossRef CAS; P. H. Mazzocchi and H. J. Tamburin, J. Am. Chem. Soc., 1975, 97, 555–561 CrossRef CAS.
- W. von E. Doering and K. Sachdev, J. Am. Chem. Soc., 1974, 96, 1168–1187 CrossRef CAS; W. von E. Doering and K. Sachdev, J. Am. Chem. Soc., 1975, 97, 5512–5520 CrossRef.
- W. von E. Doering, J. L. Ekmanis, K. D. Belfield, F.-G. Klärner and B. Krawczyk, J. Am. Chem. Soc., 2001, 123, 5532–5541 CrossRef; W. von E. Doering, J. He and L. Shao, J. Am. Chem. Soc., 2001, 123, 9153–9161 CrossRef CAS.
- G. D. Andrews and J. E. Baldwin, J. Am. Chem. Soc., 1976, 98, 6705–6706 CrossRef CAS.
- J. E. Baldwin, Chem. Rev., 2003, 103, 1197–1212 CrossRef CAS.
- J. E. Baldwin, K. A. Villarica, D. I. Freedberg and F. A. L. Anet, J. Am. Chem. Soc., 1994, 116, 10845–10846 CrossRef CAS.
- E. R. Davidson and J. J. Gajewski, J. Am. Chem. Soc., 1997, 119, 10543–10544 CrossRef CAS; K. N. Houk, M. Nendel, O. Wiest and J. W. Storer, J. Am. Chem. Soc., 1997, 119, 10545–10546 CrossRef CAS; J. E. Baldwin, J. Comput. Chem., 1998, 19, 222–231 CrossRef CAS.
- C. Doubleday, M. Nendel, K. N. Houk, D. Thweatt and M. Page, J. Am. Chem. Soc., 1999, 121, 4720–4721 CrossRef CAS; C. Doubleday, J. Phys. Chem. A, 2001, 105, 6333–6341 CrossRef CAS.
- W. von E. Doering and E. A. Barsa, Proc. Natl. Acad. Sci. U. S. A., 1980, 77, 2355–2357 CrossRef; E. A. Barsa, Ph.D. Dissertation, Harvard University, 1977 Search PubMed; W. von E. Doering and E. A. Barsa, J. Am. Chem. Soc., 2004, 126, 12353–12362 Search PubMed.
- J. E. Baldwin and R. C. Burrell, J. Am. Chem. Soc., 2001, 123, 6718–6719 CrossRef CAS; J. E. Baldwin and R. C. Burrell, J. Am. Chem. Soc., 2003, 125, 15869–15877 CrossRef CAS; J. E. Baldwin and R. C. Burrell, J. Phys. Chem. A, 2003, 107, 10069–10073 CrossRef CAS.
- W. von E. Doering, X. Cheng, K. Lee and Z. Lin, J. Am. Chem. Soc., 2002, 124, 11642–11652 CrossRef.
- J. E. Baldwin and J.-M. Fedé, J. Am. Chem. Soc., 2006, 128, 5608–5609 CrossRef CAS.
- W. von E. Doering and E. K. G. Schmidt, Tetrahedron, 1971, 27, 2005–2030 CrossRef CAS ; see also W. von E. Doering, T.-H. Zhang and E. K. G. Schmidt, J. Org. Chem., 2006, 71, 5688–5693 Search PubMed.
- R. S. Cooke and U. H. Andrews, J. Am. Chem. Soc., 1974, 96, 2974–2980 CrossRef CAS.
- J. E. Baldwin and E. J. Keliher, J. Am. Chem. Soc., 2002, 124, 380–381 CrossRef CAS.
- C. P. Suhrada and K. N. Houk, J. Am. Chem. Soc., 2002, 124, 8796–8797 CrossRef CAS.
- C. Doubleday, C. P. Suhrada and K. N. Houk, J. Am. Chem. Soc., 2006, 128, 90–94 CrossRef CAS.
- K. Dietrich and H. Musso, Chem. Ber., 1974, 107, 731–734 CrossRef CAS.
- J. B. Conant and G. H. Carlson, J. Am. Chem. Soc., 1929, 51, 3464–3469 CrossRef CAS.
- J. J. Gajewski and C. M. Hawkins, J. Am. Chem. Soc., 1986, 108, 838–839 CrossRef CAS; J. J. Gajewski, I. Kuchuk, C. Hawkins and R. Stine, Tetrahedron, 2002, 58, 6943–6950 CrossRef CAS.
- P. A. Leber and J. E. Baldwin, Acc. Chem. Res., 2002, 35, 279–287 CrossRef CAS.
- H. M. Frey and A. T. Cocks, J. Chem. Soc. A, 1971, 2564–2566 RSC.
- J. E. Baldwin and K. D. Belfield, J. Phys. Org. Chem., 1989, 2, 455–466 CrossRef CAS.
- D. K. Lewis, A. Hutchinson, S. J. Lever, E. L. Spaulding, S. J. Bonacorsi and J. E. Baldwin, Isr. J. Chem., 1996, 36, 233–238 CAS.
- J. E. Baldwin and K. D. Belfield, J. Am. Chem. Soc., 1988, 110, 296–297 CrossRef CAS; F.-G. Klärner, R. Drewes and D. Hasselmann, J. Am. Chem. Soc., 1988, 110, 297–298 CrossRef.
- J. E. Baldwin and P. A. Leber, J. Am. Chem. Soc., 2001, 123, 8396–8397 CrossRef CAS.
- J. E. Baldwin and P. A. Leber, Tetrahedron Lett., 2001, 42, 195–197 CrossRef CAS.
- J. A. Berson and G. L. Nelson, J. Am. Chem. Soc., 1970, 92, 1096–1097 CrossRef CAS; J. A. Berson and L. Salem, J. Am. Chem. Soc., 1972, 94, 8917–8918 CrossRef CAS; J. A. Berson, Acc. Chem. Res., 1972, 5, 406–414 CrossRef CAS.
- J. D. Burkey, P. A. Leber and L. S. Silverman, Synth. Commun., 1986, 16, 1363–1370 CrossRef CAS; M. A. Forman and P. A. Leber, Tetrahedron Lett., 1986, 27, 4107–4110 CrossRef CAS.
- J. D. Bender, P. A. Leber, R. R. Lirio and R. S. Smith, J. Org. Chem., 2000, 65, 5396–5402 CrossRef CAS.
- B. K. Carpenter, J. Am. Chem. Soc., 1995, 117, 6336–6344 CrossRef CAS.
- B. K. Carpenter, J. Am. Chem. Soc., 1985, 107, 5730–5732 CrossRef CAS; B. K. Carpenter, Acc. Chem. Res., 1992, 25, 520–528 CrossRef CAS; B. K. Carpenter, J. Org. Chem., 1992, 57, 4645–4648 CrossRef CAS; B. K. Carpenter, J. Am. Chem. Soc., 1996, 118, 10329–10330 CrossRef CAS; B. K. Carpenter, Am. Sci., 1997, 85, 138–149 Search PubMed; B. K. Carpenter, Angew. Chem., Int. Ed., 1998, 37, 3340–3350 CrossRef; B. A. Carpenter, in Reactive Intermediates, ed. R. A. Moss, M. S. Platz and M. Jones, Jr., Wiley-Interscience, Hoboken, NJ, 2004, pp. 925–960 Search PubMed.
- K. N. Houk, S. L. Wilsey, B. R. Beno, A. Kless, M. Nendel and J. Tian, Pure Appl. Chem., 1998, 70, 1947–1952 CAS; B. R. Beno, S. L. Wilsey and K. N. Houk, J. Am. Chem. Soc., 1999, 121, 4816–4826 CrossRef CAS; S. L. Wilsey, K. N. Houk and A. H. Zewail, J. Am. Chem. Soc., 1999, 121, 5772–5786 CrossRef CAS.
- X. S. Bogle, P. A. Leber, L. A. McCullough and D. C. Powers, J. Org. Chem., 2005, 70, 8913–8918 CrossRef CAS.
- P. A. Leber, C. C. Lasota, N. A. Strotman and G. S. Yen, J. Org. Chem., 2007, 72, 912–919 CrossRef CAS.
- J. E. Baldwin, P. A. Leber and D. C. Powers, J. Am. Chem. Soc., 2006, 128, 10020–10021 CrossRef CAS; D. C. Powers, P. A. Leber, S. S. Gallagher, A. T. Higgs, L. A. McCullough and J. E. Baldwin, J. Org. Chem., 2007, 72, 187–194 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2008 |