Synthesis of fluorescein-labelled O-mannosylated peptides as components for synthetic vaccines: comparison of two synthetic strategies
Margaret A.
Brimble
*ac,
Renata
Kowalczyk
ac,
Paul W. R.
Harris
ac,
P. Rod
Dunbar
bc and
Victoria J.
Muir
a
aDepartment of Chemistry, University of Auckland, 23 Symonds St, Auckland 1142, New Zealand. E-mail: m.brimble@auckland.ac.nz; Fax: +64 9 3737422
bSchool of Biological Sciences, University of Auckland, Private Bag 92019, Auckland 1142, New Zealand
cMaurice Wilkins Centre for Molecular Biodiscovery, University of Auckland, Private Bag 92019, Auckland 1142, New Zealand
First published on 26th October 2007
Abstract
Mannose-binding proteins on the surface of antigen-presenting cells (APCs) are capable of recognizing and internalizing foreign agents in the early stages of immune response. These receptors offer a potential target for synthetic vaccines, especially vaccines designed to stimulate T cells. We set out to synthesize a series of fluorescein-labelled O-mannosylated peptides using manual solid phase peptide synthesis (SPPS) on pre-loaded Wang resin, in order to test their ability to bind mannose receptors on human APCs in vitro. A flexible and reliable method for the synthesis of fluorescein-labelled O-mannosylated glycopeptides was desired in order to study their lectin-binding properties using flow cell cytometry. Two synthetic strategies were investigated: incorporation of a fluorescein label into the peptide chain via a lysine side chain ε-amino group at the final stage of standard Fmoc solid phase peptide synthesis or attachment of the fluorescein label to the Nα-amino group of a lysine with further incorporation of a mannosylated peptide unit through the side chain Nε-amino group. The latter strategy proved more effective in that it facilitated SPPS by positioning the growing mannosylated peptide chain further removed from the fluorescein label.
Introduction
The ability of chemical synthesis to provide glycopeptides in homogeneous form plays a pivotal role for the advancement of glycobiology.1 For example there is current interest in the design and synthesis of synthetic vaccine formulations capable of stimulating the immune system for use against diseases caused by infectious agents and for the treatment of cancer. In particular, new vaccines are needed that are capable of strongly stimulating T cells, which recognise unique peptides bound to MHC molecules on the surface of pathogen-infected cells and tumour cells. Peptides are attractive agents for incorporation into vaccines because they can be synthesized and purified without the need for biological processes and because they are flexible and inexpensive to manufacture.2 Nevertheless, peptides often show poor stability in biological fluids3 and are often not targeted efficiently to the cells responsible for initiating an immune response, namely antigen-presenting cells (APCs).APCs capture pathogen- or tumour-derived protein antigens, and process them into MHC-bound peptides that are transported to the cell surface for presentation to T cells.4APCs express receptors capable of recognizing and internalising foreign agents. Several of these receptors are C-type lectins that bind carbohydrates5 and mannose receptors in particular, and are known to take up and present mannosylated antigens to T cells.6 Indeed within this lectin family there are a number of proteins apart from the classically-described mannose receptor (also known as CD206) that are expressed on APCs.7 APC membrane lectins that mediate efficient sugar-specific endocytosis could be potentially exploited for glycotargeting tumour antigens to enhance antigen presentation to different subsets of T cells. Burgdorf et al.8,9 have recently demonstrated that mannose-mediated endocytosis of the model antigen soluble ovalbumin (OVA) targets early endosomes enabling its cross-presentation to CD8+T cells. In order to direct a synthetic vaccine to these receptors, ligands capable of binding to mannose receptors on APC should ideally be incorporated into the vaccine construct. Many high affinity glycopeptide binding ligands for mannose lectins have been produced based on assembly of clustered patches of mannose ligands to enhance binding to the protein receptors relying on the cluster effect.10–12 Most of these constructs have relied on use of mannosylation through the side chain Nε-amino group of a lysine residue.13,14 An additional complication in the field is that although most mannose receptors are probably multimeric, and the highest affinity ligands will probably engage more than one carbohydrate-recognition domain (CRD), the spacing between these CRDs is poorly defined. One high-resolution (1.8 Å) crystal structure15 of the head and neck part of rat serum mannose-binding protein A, a soluble homologue of the mannose receptor, is available. This structure showed that the individual CRDs are separated from each other by 53 Å. This suggests that assembly of a polymannosylated scaffold with the mannose units spaced 53 Å apart provides an avenue for investigation of ligand binding.
We therefore postulate that placement of mannose units carefully positioned at defined distance on a linear peptide scaffold may provide an alternative strategy for targeting the mannose receptor on human APCs. With this idea in mind we have focused on targeting mannose receptors through O-mannosylated serine units dispersed at defined sites along a linear peptide scaffold. Our initial attention has therefore focused on the efficient synthesis of O-mannosylated polyalanine peptide scaffolds of differing chain length and mannose configurations in order to evaluate their specificity for targeting human APCsin vitro. Thus, in the work reported herein a set of glycopeptides containing O-mannosylated serine residues was prepared with the aim of developing a suitable APC-binding motif that can not only be conjugated to antigenic peptides known to stimulate T cells,16 but also subsequently spaced appropriately to allow multivalent binding to more than one carbohydrate recognition domain of mannose receptor multimers. Given that the exact binding requirements for the mannose receptor are as yet unknown our initial intention was to produce a series of fluorescently-labelled O-mannosylated peptides that could be analysed in cellular immunology screens.
Results and discussion
The most efficient and versatile method for the preparation of O-glycopeptides employs protected glycosylated amino acids as building blocks in a sequential assembly of peptides.1 It was decided to establish an O-linked mannosyl linkage by incorporation of a mannosylated serine residue into an alanine based peptide scaffold. Previous reports on the synthesis of peptides containing O-mannosylated serines are rare17 especially given the additional requirement for introduction of a fluorescent label. Moreover, a comparison of the O-mannosylation of serine residues on resin-bound assembled peptides with the incorporation of O-mannosylated serine units into SPPS concluded that the synthesis of glycopeptides containing O-mannosylated serine units via this latter method is in fact the method of choice.18 A fluorescent label could be introduced in the final stages of SPPS via attachment of 5(6)-carboxyfluorescein to a side chain Nε-amino group of a lysine residue that is indirectly attached to Wang resin. Thus our initial synthetic strategy focused on solid phase glycopeptide assembly proceeding via coupling through the Nα-amino group of the lysine residue that was attached to pre-loaded alanine Wang resin. After SPPS that included introduction of the mannosylated serine residue, the orthogonally Mtt-protected Nε-amino group of the lysine residue was deprotected and the 5(6)-carboxyfluorescein label introduced.Preparation of glycosyl amino acid Fmoc-[Man(OBz)4α1-]Ser
The initial synthetic strategy outlined above required access to an Fmoc-protected α-mannosylated serine building block. A brief screening of methods to effect mannosylation of Fmoc-protected serine derivatives with several mannosyl donors led us to the conclusion that the optimum procedure involved mannosylation of Fmoc-protected allyl serine (5) with tetra-O-benzoyl-α-D-mannose trichloroacetimidate (3) using trimethylsilyl trifluoromethanesulfonate (20 mol%) in dichloromethane at –40 °C for 3 h affording Fmoc-[Man(OBz)4α1-]Ser allyl ester (6) in 71% yield (Scheme 1). Deprotection of the allyl ester using Pd(PPh3)4 and phenylsilane in dichloromethane afforded Fmoc-[Man(OBz)4α1-]Ser (7) in 70% yield. Protection of the serine carboxyl group as an allyl ester proved more effective than either the use of a benzyl ester or a pentafluorophenyl ester in that the pentafluorophenyl ester proved more difficult to purify and degraded upon prolonged storage whilst deprotection of a benzyl ester by hydrogenolysis proved unreliable in our hands and the lability of the Fmoc group under hydrogenolysis conditions has been reported.19 Use of an allyl ester offered the advantage of its facile preparation, stability under a variety of glycosylation conditions20 and its small size that does not hinder the glycosylation step.21 The use of a benzoyl-protected mannosyl donor rather than an acetate-protected mannosyl donor was also advantageous in that the benzoyl groups reduced the possibility of orthoester formation.22 Mannosyl trichloroacetimidate 3 was readily prepared via perbenzoylation of mannose to pentabenzoate 1,23 selective hydrolysis of the anomeric benzoate group to tetrabenzoate 224 and finally conversion to trichloroacetimidate 325 upon treatment with trichloroacetonitrile and potassium carbonate. Fmoc-Ser allyl ester (5) was readily prepared by treatment of serine with Fmoc-OSu affording Fmoc-Ser (4)26 in 97% yield followed by allylation of the ester group to Fmoc-Ser allyl ester (5)27 in 87% yield using allyl bromide in dichloromethane under phase transfer conditions.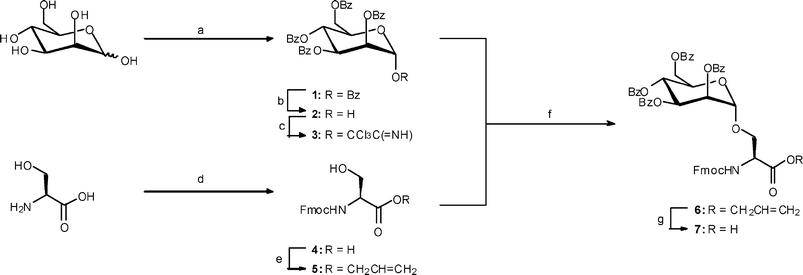 | ||
Scheme 1
Reagents and conditions: (a) BzCl, py, 66%; (b) ethanolamine, THF, 54%; (c) K2CO3, Cl3CCN, CH2Cl2, 94%; (d) Fmoc-OSu, Na2CO3, dioxane, 97%; (e) CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2Br, NaHCO3, Aliquot 336, H2O, CH2Cl2, 24 h, 87%; (f) SiMe3OTf (20%), CH2Cl2, –40 °C, 3 h, 71%; (g) Pd(PPh3)4, PhSiH3, CH2Cl2, 1.5 h, 70%. CHCH2Br, NaHCO3, Aliquot 336, H2O, CH2Cl2, 24 h, 87%; (f) SiMe3OTf (20%), CH2Cl2, –40 °C, 3 h, 71%; (g) Pd(PPh3)4, PhSiH3, CH2Cl2, 1.5 h, 70%. | ||
Synthesis of the mannosylated peptides: H2N-[Man(OH)4α1-]Ser-(Ala)n-[Lys{5(6)-carboxyfluorescein}]-Ala-OH (8) (n = 3), (9) (n = 4) and (10) (n = 6)
The synthesis of the H2N-[Man(OH)4α1-]Ser-(Ala)n-[Lys{5(6)-carboxyfluorescein}]-Ala-OH glycopeptides 8 (n = 3), 9 (n = 4) and 10 (n = 6) was initiated by deprotection of the Fmoc group from Fmoc-Ala-Wang resin using 20% piperidine in DMF followed by HBTU–HOBt mediated coupling of Fmoc-Lys(Mtt)-OH (Scheme 2) and capping of unreacted amino groups using acetic anhydride in NMP. The required number of alanine units (n = 3, 4 or 6) were then introduced in a similar fashion also using HBTU–HOBt as the coupling agent with a capping step to minimize the formation of deletion sequences. Finally the Fmoc-[Man(OBz)4α1-]Ser residue was introduced into the series of alanine-based glycopeptides using the more powerful coupling agent HATU in the presence of HOAt, collidine and DMAP in DMF with a longer reaction time (overnight).![Reagents and conditions: (a) 20% piperidine in DMF, rt; (a*) 6 cycles of 20% piperidine in DMF; (b) Fmoc-Lys(Mtt)-OH, HBTU, HOBt, iPr2EtN, DMF, rt, 1.5 h; (c) cat. HOBt, Ac2O, iPr2EtN, NMP, 2 × 5 min, rt; (d) Fmoc-Ala-OH coupling, HBTU, HOBt, iPr2EtN, DMF, (e) repeat steps (d), (c), (a) twice (n = 3), three times (n = 4) or five times (n = 6); (f) Fmoc-[Man(OH)4α1-]Ser-OH, HATU, HOAt, collidine, DMAP, DMF, rt, overnight; (g) 2% TFA in CH2Cl2, rt; (h) 5(6)-carboxyfluorescein, DIC, HOBt, DMF, rt, overnight; (i) TFA, iPr3SiH, H2O, rt, 2 h; (j) cat. NaOMe in MeOH, rt, pH 11.7–11.8, 3 h.](/image/article/2008/OB/b712926b/b712926b-s2.gif) | ||
| Scheme 2 Reagents and conditions: (a) 20% piperidine in DMF, rt; (a*) 6 cycles of 20% piperidine in DMF; (b) Fmoc-Lys(Mtt)-OH, HBTU, HOBt, iPr2EtN, DMF, rt, 1.5 h; (c) cat. HOBt, Ac2O, iPr2EtN, NMP, 2 × 5 min, rt; (d) Fmoc-Ala-OH coupling, HBTU, HOBt, iPr2EtN, DMF, (e) repeat steps (d), (c), (a) twice (n = 3), three times (n = 4) or five times (n = 6); (f) Fmoc-[Man(OH)4α1-]Ser-OH, HATU, HOAt, collidine, DMAP, DMF, rt, overnight; (g) 2% TFA in CH2Cl2, rt; (h) 5(6)-carboxyfluorescein, DIC, HOBt, DMF, rt, overnight; (i) TFA, iPr3SiH, H2O, rt, 2 h; (j) cat. NaOMe in MeOH, rt, pH 11.7–11.8, 3 h. | ||
The Mtt group was removed from the Nε-amino group of the lysine residue using 2% TFA in dichloromethane and a 5(6)-carboxyfluorescein label was then introduced via DIC-mediated coupling in the presence of HOBt in DMF overnight. Aminolysis of the additional 5(6)-carboxyfluorescein molecules bound to the phenols of the labelled peptide using six cycles of the 20% piperidine deprotection protocol also effected concomitant removal of the Fmoc group from the newly introduced mannosylated serine residue.28 Subsequent cleavage of the mannosylated peptide from the resin using 95% TFA with triisopropylsilane and water as scavengers, concentration in vacuo and trituration with cold diethyl ether gave the crude benzoate-protected glycopeptides. These crude glycopeptides were purified by reverse-phase HPLC before final removal of the mannose benzoate protecting groups with catalytic NaOMe in MeOH. A final purification step using reverse-phase HPLC gave glycopeptides 8, 9 and 10 in 5%, 6% and 5% overall yield respectively.
The structure of glycopeptide 9 was confirmed by MALDI-TOF analysis (observed [MH + Na]+ 1132.4115) and 1H NMR spectroscopy thus supporting the assigned structure. Similar analyses (ESI-MS) supported the successful formation of glycopeptides 8 and 10.
Synthesis of the mannosylated peptides: 5(6)-carboxyfluorescein-Lys-[(Ala)n-{Man(OH)4α1-}Ser]-Gly-OH (11) (n = 3), (12) (n = 4) and (13) (n = 6)
The procedure described above in which the glycopeptide chain is elaborated through the Nα-amino group of lysine was found to be problematic as complete removal of the Nε Mtt group proved difficult even after prolonged treatment with 2% TFA in dichloromethane. The overall yields of the glycopeptides were also very disappointing. Hence, a second strategy was implemented using Dde (1-(4,4-dimethyl-2,6-dioxocyclohexylidene)ethyl) as the Nε protecting group of a lysine residue. In this revised strategy it was also decided to introduce the fluorescein label by initial coupling of 5(6)-carboxyfluorescein through the Nα-amino group of the lysine residue attached to Gly-Wang resin (Scheme 3).28 The mannosylated peptide chain was then attached to this pre-labelled resin (Scheme 4) via the side chain Nε-amino group of the same lysine residue. Using this improved method three additional glycopeptides, with three (11), four (12), and six (13) alanine residues in the peptide chain were successfully prepared.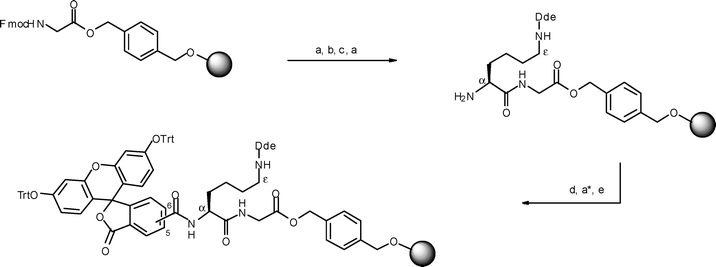 | ||
| Scheme 3 Reagents and conditions: (a) 20% piperidine in DMF, rt; (a*) 6 cycles of 20% piperidine in DMF; (b) Fmoc-Lys(Dde)-OH, HBTU, HOBt, iPr2EtN, DMF, rt, 1.5 h; (c) cat. HOBt, Ac2O, iPr2EtN, NMP, 2 × 5 min, rt; (d) 5(6)-carboxyfluorescein, DIC, HOBt, DMF, rt, overnight; (e) trityl chloride, iPr2EtN, CH2Cl2, rt, overnight. | ||
![Reagents and conditions: (a) 2% hydrazine hydrate in DMF, rt, 2 × 3 min; (b) Fmoc-Ala-OH, HBTU, HOBt, iPr2EtN, DMF, rt, 1.5 h; (c) cat. HOBt, Ac2O, iPr2EtN, NMP, 2 × 5 min, rt; (d) 20% piperidine in DMF, rt; (e) repeat steps (b), (c), (d) twice (n = 3), three times (n = 4) or five times (n = 6), (f) Fmoc-[Man(OBz)4α1-]Ser-OH, HATU, HOAt, collidine, DMAP, DMF, rt, overnight; (g) TFA, iPr3SiH, H2O, rt, 2 h; (h) NaOMe in MeOH, rt, pH 11.7–11.8, 3 h.](/image/article/2008/OB/b712926b/b712926b-s4.gif) | ||
| Scheme 4 Reagents and conditions: (a) 2% hydrazine hydrate in DMF, rt, 2 × 3 min; (b) Fmoc-Ala-OH, HBTU, HOBt, iPr2EtN, DMF, rt, 1.5 h; (c) cat. HOBt, Ac2O, iPr2EtN, NMP, 2 × 5 min, rt; (d) 20% piperidine in DMF, rt; (e) repeat steps (b), (c), (d) twice (n = 3), three times (n = 4) or five times (n = 6), (f) Fmoc-[Man(OBz)4α1-]Ser-OH, HATU, HOAt, collidine, DMAP, DMF, rt, overnight; (g) TFA, iPr3SiH, H2O, rt, 2 h; (h) NaOMe in MeOH, rt, pH 11.7–11.8, 3 h. | ||
This revised method provides a modular approach for the construction of libraries of fluorescently-labelled glycopeptides. The nature of the glycosylated amino acid can be varied as can the composition of the basic peptide framework. For example, introduction of a mannosylated hydroxyproline unit provides access to a peptide chain in which the mannose residue is placed within a turn. More importantly, it was hoped that by positioning the large fluorescein label on the Nα-amino group of the resin-bound lysine unit, with the growing mannosylated peptide chain extending from Nε lysine side chain amino group, the overall yield of the glycopeptides would be improved.
With the above ideas in mind, the synthesis of trityl-protected 5(6)-carboxyfluorescein-Lys(Dde)-Gly-Wang resin was undertaken as outlined (Scheme 3). The procedure was based on the work of Brock et al.28 who extended the applicability of 5(6)-carboxyfluorescein-labelled peptides in SPPS by on-resin introduction of the trityl group as a protecting group strategy for 5(6)-carboxyfluorescein. Introduction of O-trityl protecting groups to the polymer-bound carboxyfluorescein prevented reaction of the hydrazine used for removal of the lysine Dde group with the enone tautomers of the phenolic groups.28 In the present work we have extended this protocol to the synthesis of 5(6)-carboxyfluorescein-labelled mannosylated peptides which can be readily evaluated using flow cell cytometry based assays.
Thus, pre-loaded Fmoc-Gly-Wang resin was Fmoc-deprotected and coupled to Fmoc-Lys(Dde)-OH (HBTU, HOBt in DMF). After a capping step and Nα-Fmoc deprotection, the Nα-amino group of the lysine residue was coupled to 5(6)-carboxyfluorescein by activation with DIC and HOBt in DMF overnight. The additional phenol-linked carboxyfluorescein was then cleaved using 20% piperidine in DMF (six cycles) and the resultant phenols protected as trityl ethers upon treatment with trityl chloride and diisopropylamine in dichloromethane overnight. The trityl groups on the fluorescein were chemically inert towards treatment with 2% hydrazine hydrate that was required for removal of the Dde-protecting group in order to elaborate SPPS through the side chain Nε-amino group of the lysine residue.
The synthesis of mannosylated peptides 11, 12 and 13 was then undertaken starting from the same trityl-protected fluorescein-labelled resin (Scheme 4). Removal of the Dde group allowed coupling to an Fmoc-protected alanine mediated by HBTU and HOBt. After a capping step the SPPS procedure was then repeated two, three or five times to introduce the appropriate number of alanine residues. Finally the mannosylated serine building block was introduced onto the peptide chain using the more powerful coupling agent HATU–HOAt with collidine and DMAP in DMF overnight. Cleavage of the glycopeptide from the resin using 95% TFA, triisopropylsilane and water at room temperature for 2 h also effected cleavage of the trityl groups. Concentration in vacuo and trituration with cold diethyl ether gave the crude benzoate-protected glycopeptides that were purified by reverse-phase HPLC. Finally deprotection of the benzoate esters on the mannose residue using catalytic NaOMe in MeOH and final purification by reverse-phase HPLC gave glycopeptides 11, 12 and 13 in 33%, 15% and 20% overall yield respectively. Pleasingly, the yields obtained for this set of mannosylated peptides (11, 12 and 13) were thus substantially improved compared to the yields obtained for the previous set of mannosylated peptides (8, 9 and 10).
The structure of glycopeptide 11 was confirmed by 1H NMR spectroscopy and MALDI-TOF analysis (observed [MH + Na]+ 1047.7072) thus supporting the assigned structure. Similar analyses supported the successful formation of glycopeptides 12 and 13.
Conclusion
In summary two sets of fluorescein-labelled peptides containing mannosylated serine units incorporated into peptide scaffolds with different numbers of alanines attached to an Nα-amino group or a side chain Nε-amino group of lysine have been successfully synthesized. These mannosylated peptides will be tested in vitro for their ability to bind to mannose receptors on human APC subsets. It was found that incorporation of benzoyl-protected Fmoc-[Man(OBz)4α1-]Ser into a polyalanine peptide chain was more facile and proceeded in better overall yield when the peptide chain was assembled through the side chain Nε-amino of lysine rather than through the Nα-amino of the lysine residue. It is postulated that differential positioning of the fluorescein label relative to the growing peptide chain in the two alternative synthetic strategies together with the associated differences in solid phase reaction dynamics, contributes to a more efficient synthetic method. This strategy also makes use of Fmoc-Lys(Dde) in preference to Fmoc-Lys(Mtt) thus reducing the exposure of the acid-sensitive Wang resin bound glycopeptide to 2% TFA in dichloromethane that is required for removal of the Mtt group. Furthermore, prolonged treatment with 2% TFA can result in cleavage of the peptide from the resin.29 More significantly, adopting this improved strategy allows the initial synthesis of a fluorescein-preloaded Wang resin that can be used for the modular synthesis of diverse peptide scaffolds prepared in an analogous fashion to those reported herein.Experimental
General
All reagents were purchased as reagent grade and used without further purification. Solvents were dried according to standard methods.30 Solvents for RP HPLC were purchased as HPLC grade and used without further purification. Fmoc-Gly-Wang resin and Fmoc-Lys(Dde) were purchased from IRIS Biotech GmbH; Fmoc-Ala-Wang resin, Fmoc-Ala, HOBt and HBTU were purchased from Advanced ChemTech; Fmoc-Lys(Mtt) was purchased from Biochem; HOAt was purchased from Acros Organics; HATU, Fmoc-Ala-TentaGel S PHB resin and 5(6)-carboxyfluorescein (isomeric mixture) were purchased from Fluka. Analytical thin layer chromatography was performed using 0.2 mm plates of Kieselgel F254 (Merck) and compounds were visualised by ultraviolet fluorescence or by staining with 4% sulfuric acid in ethanol or ethanolic ninhydrin solution (0.3% ninhydrin in ethanol + 1% v/v acetic acid), followed by heating the plate for a few minutes. Flash chromatography was carried out on Kieselgel F254 S 0.063–0.1 mm (Riedel de Hahn) silica gel with indicated solvents. Infrared spectra were obtained using a Perkin Elmer Spectrum One Fourier Transform infrared spectrometer as a thin film between sodium chloride plates. Absorption maxima are expressed in wavenumbers (cm–1) with the following abbreviations: s = strong, m = medium, w = weak, br = broad and v = varying.Melting points were determined on an Electrothermal® melting point apparatus and are uncorrected. Optical rotations were determined at the sodium D line (589 nm), at 20 °C with a Perkin-Elmer 341 polarimeter and are given in units of 10–1 deg cm2 g–1. Nuclear magnetic resonance (NMR) spectra were recorded as indicated on either a Bruker AVANCE DRX300 (1H, 300 MHz, 13C, 75 MHz), a Bruker AVANCE DRX400 spectrometer (1H, 400 MHz, 13C, 100 MHz) or on a Bruker AVANCE DRX600 spectrometer (1H, 600 MHz). Chemical shifts are reported in parts per million (ppm) relative to the tetramethylsilane signal recorded at δH 0.00 ppm in CDCl3–SiMe4 solvent, or the residual methanol signal at δH 3.34 ppm in CD3OD solvent, or the residual water signal at δH 4.79 ppm in D2O solvent. The 13C values were referenced to the residual chloroform signal at δC 77.0 ppm in CDCl3–SiMe4 solvent or the residual methanol signal at δC 49.15 ppm in CD3OD solvent. 1H NMR shift values are reported as chemical shift (δH), relative integral, multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br s, broad singlet; dd, doublet of doublets; ddt, doublet of doublet of triplets), coupling constant (J in Hz) and assignments. 13C values are reported as chemical shift (δC), degree of hybridisation and assignment. Assignments were made with the aid of DEPT135, COSY, HSQC and HMBC experiments. The ratio of 5-carboxyfluorescein and 6-carboxyfluorescein regioisomers was assumed to be 60 : 40 for the assignment of 1H NMR data. Low resolution mass spectra were recorded on a VG-70SE mass spectrometer operating at a nominal accelerating voltage of 70 eV (FAB). High resolution mass spectra were recorded using a VG-70SE spectrometer at a nominal resolution of 5000 to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 as appropriate. Major and significant fragments are quoted in the form x (y%), where x is the mass to charge ratio and y is the percentage abundance relative to the base peak. Electrospray ionization mass spectra (ESI-MS) were recorded on a Thermo Finnigan Surveyor MSQ Plus spectrometer. Accurate mass spectra were recorded using Matrix-assisted laser desorption/ionisation-time of flight mass spectrometry (MALDI-TOF MS) on a Voyager-DE spectrometer using α-cyano-4-hydroxycinnamic acid (CHCA) as matrix. Analytical RP HPLC was performed using a Waters 600 System using an analytical column (Phenomenex Jupiter C4, 300 Å, 150 mm × 4.6 mm, 5 µm or Phenomenex Jupiter C18, 300 Å, 150 mm × 4.6 mm, 5 µm) at a flow rate of 1 mL min–1, using a linear gradient of 10% B to 100% B over 35 min with detection at 254 nm; buffer A = 0.1% TFA–H2O, buffer B = 0.1% TFA–CH3CN. Semi-preparative RP HPLC was performed using a Waters 600 System using a semi-preparative column (Waters XTerra® Prep. C18, 300 mm × 19 mm, 10 µm, or Phenomenex Jupiter C4, 300 Å, 250 mm × 10 mm, 5 µm) at a flow rate of 10 mL min–1 or 13 mL min–1, using a linear gradient of 0.1% TFA–H2O (buffer A) and 0.1% TFA–CH3CN (buffer B) with detection at 254 nm. Gradient systems were adjusted according to the elution profiles and peak profiles obtained from the analytical RP HPLC chromatograms.
000 as appropriate. Major and significant fragments are quoted in the form x (y%), where x is the mass to charge ratio and y is the percentage abundance relative to the base peak. Electrospray ionization mass spectra (ESI-MS) were recorded on a Thermo Finnigan Surveyor MSQ Plus spectrometer. Accurate mass spectra were recorded using Matrix-assisted laser desorption/ionisation-time of flight mass spectrometry (MALDI-TOF MS) on a Voyager-DE spectrometer using α-cyano-4-hydroxycinnamic acid (CHCA) as matrix. Analytical RP HPLC was performed using a Waters 600 System using an analytical column (Phenomenex Jupiter C4, 300 Å, 150 mm × 4.6 mm, 5 µm or Phenomenex Jupiter C18, 300 Å, 150 mm × 4.6 mm, 5 µm) at a flow rate of 1 mL min–1, using a linear gradient of 10% B to 100% B over 35 min with detection at 254 nm; buffer A = 0.1% TFA–H2O, buffer B = 0.1% TFA–CH3CN. Semi-preparative RP HPLC was performed using a Waters 600 System using a semi-preparative column (Waters XTerra® Prep. C18, 300 mm × 19 mm, 10 µm, or Phenomenex Jupiter C4, 300 Å, 250 mm × 10 mm, 5 µm) at a flow rate of 10 mL min–1 or 13 mL min–1, using a linear gradient of 0.1% TFA–H2O (buffer A) and 0.1% TFA–CH3CN (buffer B) with detection at 254 nm. Gradient systems were adjusted according to the elution profiles and peak profiles obtained from the analytical RP HPLC chromatograms.
Synthesis of mannosylated building blocks
O), 1601 m, 1584 m (C–C, Ar), 1271 s (C–O), 756 s (C–H, Ar), 713 s (C–C, Ar); δH (400 MHz; CDCl3) 4.52 (1H, dd, JAB 12.3, J6A,5 3.6, H-6A), 4.59–4.63 (1H, m, H-5), 4.72 (1H, dd, JAB 12.3, J6B,5 2.5, H-6B), 5.94 (1H, dd, J2,3 3.1, J2,1 2.0, H-2), 6.10 (1H, dd, J3,4 10.2, J3,2 3.1, H-3), 6.32 (1H, t, J4,3 = J4,5 10.2, H-4), 6.65 (1H, d, J1,2 1.9, H-1), 7.26–7.69 (15H, m, Ph), 7.86–8.22 (10H, m, Ph); δC (100 MHz; CDCl3) 62.3 (CH, C-6), 66.1 (CH, C-4), 69.4 (CH, C-2), 69.9 (CH, C-3), 69.9 (CH, C-5), 91.3 (CH, d, JC-1,H-1 178.7, C-1), 128.3, 128.4, 128.6, 128.7 (CH, Ph), 129.7, 129.9, 130.1 (quat., Ph), 133.0, 133.3, 133.5, 133.6, 134.0 (CH, Ph), 163.7, 165.1, 165.2, 165.6, 165.9 (quat., COOBz). NMR data were in agreement with those reported in the literature.31
O), 1601 m, 1584 m (C–C, Ar), 1267 s (C–O), 708 s (C–C, Ar); δH (400 MHz; CDCl3) 3.50 (1H, br s, OH), 4.45 (1H, dd, JAB 12.2, J6A,5 3.7, H-6A), 4.65–4.69 (1H, m, H-5), 4.77 (1H, dd, JAB 12.2, J6B,5 2.6, H-6B), 5.54 (1H, d, J1,2 1.7, H-1), 5.74–5.75 (1H, m, H-2), 6.01 (1H, dd, J3,4 10.1, J3,2 3.2, H-3), 6.18 (1H, t, J4,3 = J4,5 10.1, H-4), 7.26–7.60 (12H, m, Ph), 7.84–8.13 (8H, m, Ph); δC (100 MHz; CDCl3) 62.7 (CH2, C-6), 66.8 (CH, C-4), 68.9 (CH, C-5), 69.7 (CH, C-3), 70.8 (CH, C-2), 92.4 (CH, d, JC-1,H-1 173.1, C-1), 128.3, 128.5, 128.6 (CH, Ph), 129.0, 129.1, 129.3, 129.8 (quat., Ph), 129.8, 133.1, 133.2, 133.5 (CH, Ph), 165.5, 165.6, 166.3 (quat., COOBz). NMR data were in agreement with those reported in the literature.32
NH), 3054 w, 3032 w (Ar–H), 1730 s (CO), 1679 v (CN), 1601 m, 1584 m (C–C, Ar), 1265 s (C–O), 1106 s (O–C–C), 756 s (C–H, Ar), 708 s (C–C, Ar); δH (400 MHz; CDCl3) 4.51 (1H, dd, JAB 12.3, J6A,5 4.0, H-6A), 4.64 (1H, ddd, J5,4 10.2, J5,6A 4.0, J5,6B 2.3, H-5), 4.74 (1H, dd, JAB 12.3, J6B,5 2.3, H-6B), 5.95 (1H, dd, J2,3 3.3, J2,1 1.9, H-2), 5.99 (1H, dd, J3,4 10.2, J3,2 3.3, H-3), 6.24 (1H, t, J4,3 = J4,5 10.2, H-4), 6.58 (1H, d, J1,2 1.9, H-1), 7.25–7.63 (12H, m, Ph), 7.83–8.11 (8H, m, Ph), 8.87 (1H, s, NH); δC (100 MHz; CDCl3) 60.3 (CH2, C-6), 66.0 (CH, C-4), 68.8 (CH, C-2), 69.8 (CH, C-3), 71.5 (CH, C-5), 90.6 (quat., CCl3C(NH)), 94.6 (CH, C-1), 128.3, 128.4, 128.6, 128.7, 128.8, 128.9 (CH, Ph), 129.7, 129.8, 129.9 (quat., Ph), 133.1, 133.3, 133.5, 133.7 (CH, Ph), 159.8 (quat., CCl3C(NH)), 165.1, 165.3, 165.4, 166.0 (quat., COOBz). NMR data were in agreement with those reported in the literature.25
O), 1261 s, 1056 s (C–O), 759 s, 737 s (C–H, Ar); δH (400 MHz; MeOD) 3.84 (1H, dd, JAB 11.3, JSerβ-HA,Serα-H 4.0, Serβ-HA), 3.90 (1H, dd, JAB 11.3, JSerβ-HB,Serα-H 5.0, Serβ-HB), 4.20 (1H, t, JCH,CH2 6.9, CH Fmoc), 4.27–4.38 (3H, m, CH2 Fmoc and Serα-H), 7.28 (2H, t, J 7.4, CH Fmoc Ph), 7.36 (2H, t, J 7.4, CH Fmoc Ph), 7.65 (2H, d, J 6.8, CH Fmoc Ph), 7.76 (2H, d, J 7.8, CH Fmoc Ph); δC (100 MHz; MeOD) 48.3 (CH, Fmoc), 57.8 (CH, Serα), 63.1 (CH2, Serβ), 68.2 (CH2, Fmoc), 120.9, 126.3, 128.2, 128.8 (CH, Fmoc), 142.6, 145.2, 145.3 (quat., Fmoc), 158.6 (quat., CONH), 173.8 (quat., COOH).
O), 1213 s, 1052 s (C–O), 763 s, 740 s (C–H, Ar); δH (400 MHz; CDCl3) 2.75 (1H, br s, OH), 3.88–4.01 (2H, m, Serβ-H2), 4.20 (1H, t, JCH,CH2 6.9, CH Fmoc), 4.34–4.47 (3H, m, CH2 Fmoc and Serα-H), 4.66 (2H, m, OCH2CHCH2), 5.23 (1H, dd, J 10.5, JAB 1.1, OCH2CHCHAHB), 5.32 (1H, dd, J 17.2, JAB 1.1, OCH2CHCHAHB), 5.83–5.93 (2H, m, OCH2CHCH2 and NH), 7.29 (2H, t, J 7.4, CH Fmoc Ph), 7.38 (2H, t, J 7.4, CH Fmoc Ph), 7.58 (2H, d, J 6.7, CH Fmoc Ph), 7.74 (2H, d, J 7.4, CH Fmoc Ph); δC (100 MHz; CDCl3) 47.0 (CH, Fmoc), 56.1 (CH, Serα), 60.4 (CH2, Serβ), 66.2 (CH2, OCH2CHCH2), 67.1 (CH2, Fmoc), 118.8 (CH2, OCH2CHCH2), 119.9, 125.0, 127.0, 127.7 (CH, Fmoc Ph), 131.3 (CH, OCH2CHCH2), 141.2, 143.6, 143.7 (quat., Fmoc), 156.2 (quat., CONH), 170.2 (quat., COOAllyl). NMR data were in agreement with those reported in the literature.37
O), 1601 m, 1584 m (C–C, Ar), 1266 s (C–O), 759 s, 741 s (C–H, Ar), 710 s (C–C Ar); δH (400 MHz; CDCl3) 4.18 (2H, m, Serβ-H2), 4.25 (1H, t, JCH,CH2 6.9, CH Fmoc), 4.40–4.52 (4H, m, CH2 Fmoc and H-6A and H-5), 4.69–4.85 (4H, m, Serα-H and H-6B and OCH2CHCH2), 5.12 (1H, m, H-1), 5.31 (1H, dd, J 10.3, JAB 1.0, OCH2CHCHAHB), 5.41 (1H, dd, J 17.1, JAB 1.0, OCH2CHCHAHB), 5.69 (1H, m, H-2), 5.88 (1H, dd, J3,4 10.1, J3,2 3.0, H-3), 5.95–6.05 (1H, m, OCH2CHCH2), 6.09–6.16 (2H, m, H-4 and NH), 7.26–8.12 (28H, m, Ph); δC (100 MHz; CDCl3) 47.0 (CH, Fmoc), 54.4 (CH, Serα), 62.6 (CH2, C-6), 66.5 (CH2, OCH2CHCH2), 66.7 (CH, C-4), 67.4 (CH2, Fmoc), 69.4 (CH, C-5), 69.6 (CH2, Serβ), 69.7 (CH, H-3), 70.1 (CH, H-2), 98.5 (CH, d, JC-1,H-1 172.8, C-1), 119.6 (CH2, OCH2CHCH2), 119.9, 125.1, 127.0, 127.6, 128.3, 128.4, 128.6, 128.8, 128.9, 129.0, 129.6, 129.7 (CH, Ph), 131.2 (CH, OCH2CHCH2), 133.0, 133.2, 133.4, 133.5 (quat., Ph), 141.2, 143.7 (quat., Fmoc), 156.0 (quat., CONH), 163.6, 165.2, 165.4, 166.1 (quat., COOBz), 169.4 (quat., COOAllyl); m/z (FAB) 946.3080 (MH+, C55H48NO14 requires 946.3075), 946 (MH+, 3%), 579 (18), 178 (39), 154 (60), 136 (43) 120 (8), 105(100) and 89 (14).
O), 1601 m, 1584 m (C–C, Ar), 1515 w (N–H), 1451 m (C–O–H), 1265 s (C–O), 1109 s (O–C–C), 1095 s (C–O–C), 759 s, 742 s (C–H, Ar); δH (400 MHz; CDCl3) 4.17–4.29 (3H, m, CH Fmoc and Serβ-H2), 4.34–4.38 (2H, m, CH2 Fmoc), 4.45–4.55 (2H, m, H-5 and H-6A), 4.65–4.73 (2H, m, H-6B and Serα-H), 5.17 (1H, m, H-1), 5.74 (1H, m, H-2), 5.99 (1H, dd, J3,4 9.9, J3,2 2.7, H-3), 6.13 (1H, t, J4,3 = J4,5 9.9, H-4), 6.56 (1H, br s, NH), 7.20–8.08 (28H, m, Ph); δC (100 MHz; CDCl3) 47.0 (CH, Fmoc), 54.6 (CH, Serα), 62.7 (CH2, C-6), 66.6 (CH, C-4), 67.3 (CH2, Fmoc), 69.1 (CH2, Serβ), 69.3 (CH, C-5), 70.3 (CH, C-2 and C-3), 98.2 (CH, C-1), 119.8, 125.1, 127.5, 128.3, 128.4, 128.5, 128.6, 128.7, 129.0, 129.7 (CH, Ph), 132.0, 132.2 (quat., Ph), 141.1, 143.8 (quat., Fmoc), 156.2 (quat., CONH), 165.4, 166.0, 166.1 (quat., COOBz), 171.3 (quat., COOH); m/z (FAB) 906.2792 (MH+, C52H44NO14 requires 906.2762), 906 (MH+, 3%), 579 (12), 178 (24), 120 (11), 105 (66) and 89 (22).
General method for the synthesis of glycopeptides38
Glycopeptides were assembled manually using Fmoc solid phase peptide synthesis (SPPS) using a fritted glass reaction vessel. Pre-loaded Fmoc-Ala-Wang resin (0.7 mmol g–1) (for compounds 8 and 9), pre-loaded Fmoc-Ala-TentaGel S PHB resin (0.2 mmol g–1) (for compound 10†) or 5(6)-carboxyfluorescein-Lys-Gly-Wang resin (0.8 mmol g–1) (for compounds 11, 12, and 13) was shaken in DMF for 30 min prior to the synthesis, followed by filtration. Nα-Fmoc protecting group was deprotected with 20% piperidine solution in DMF (1 × 5 min, 1 × 20 min). Coupling of Fmoc-AA was performed using method 1, coupling of Fmoc-[Man(OBz)4α1-]Ser was performed using method 2 and coupling of 5(6)-carboxyfluorescein was performed using method 3.Coupling method 1 (Fmoc-AA)
N α-Protected amino acid (4 eq.) was dissolved in DMF, HOBt (4 eq.) and HBTU (3.9 eq.) were then added and shaken until dissolved. The solution was transferred to the reaction vessel and shaken for 2 min, followed by the addition of iPr2EtN (8 eq.). The mixture was shaken for 1.5 h, filtered, washed with DMF (3×), ethanol (3×) and CH2Cl2 (3×).Coupling method 2 (Fmoc-[Man(OBz)4α1-]Ser)
Fmoc-[Man(OBz)4α1-]Ser (7) (1.5 eq.) was dissolved in NMP, HOAt (1.5 eq.) and HATU (1.45 eq.) were then added and shaken until dissolved. The solution was transferred to the reaction vessel and shaken for 2 min, followed by the addition of collidine (4.5 eq.). The mixture was shaken overnight, filtered, washed with DMF (3×), ethanol (3×) and CH2Cl2 (3×).Coupling method 3 (5(6)-carboxyfluorescein)
5(6)-Carboxyfluorescein (2.5 eq.) was dissolved in DMF. HOBt (2.5 eq.) was added, the mixture was shaken until dissolved and the solution was transferred to the reaction vessel which was then shaken for 2 min, followed by the addition of DIC (2.5 eq.). The mixture was shaken overnight, filtered, washed with DMF (3×) and treated with 20% piperidine solution in DMF 6 times (1 × 5 min, 1 × 20 min) to remove additional ester-bound 5(6)-carboxyfluorescein, washed with DMF (3×), ethanol (3×) and CH2Cl2 (3×).Unreacted amino groups were capped after each coupling step by treatment of the resin with Ac2O (0.5 M in NMP), iPr2EtN (0.125 M in NMP) and a catalytic quantity of HOBt in NMP. For removal of the Mtt group from the Nε-amino group of the lysine residue the glycopeptidyl resin (compounds 8, 9 and 10) was treated with 2% TFA in CH2Cl2. The mixture was washed with 2% TFA in CH2Cl2 (5×), shaken with 2% TFA in CH2Cl2 for 5 min, filtered, and the cycle was repeated (3×), followed by washing with CH2Cl2 (5×). The procedure was repeated until the yellow colour of the filtrate almost disappeared (12×). Removal of the Dde protecting group from the Nε-amino group of the lysine residue (for compounds 11, 12 and 13) was performed using 2% hydrazine hydrate solution in DMF (2 × 3 min). The Kaiser test39 was performed after each coupling and deprotection step. Cleavage of the resin and simultaneous removal of the protecting groups from the glycopeptides was achieved by treatment of the glycopeptidyl resin with 95 : 2.5 : 2.5 (v/v/v) TFA : TIS : H2O for 2 h. Glycopeptides were precipitated from cold Et2O, isolated by centrifuge, lyophilised from CH3CN–H2O and purified by RP HPLC prior to final removal of the mannose benzoate protecting groups.
Removal of benzoate protecting groups
The benzoate groups were removed by dissolution of the glycopeptides in methanol (3 mL) and treatment with 1 M NaOMe solution in methanol in pH 11.7–11.8 for 3 h. When all starting material had disappeared as judged by analytical RP HPLC, the solution was neutralised with a portion of dry ice, the solvent was removed in vacuo, and the crude product was purified by RP HPLC and lyophilised from CH3CN–H2O.5(6)-Carboxyfluorescein(Trt)2-Lys(Dde)-Gly-Wang resin28
Fmoc-Gly-Wang resin (0.8 mmol g–1) was shaken in DMF for 30 min prior to the synthesis. The Nα-Fmoc protecting group was removed using 20% piperidine solution in DMF (1 × 5 min, 1 × 20 min) and 5(6)-carboxyfluorescein was coupled using method 3 as described above, washed with DMF (3×), CH2Cl2 (5×) and dried under a flow of N2. Trityl chloride (12 eq.) was then added, followed by addition of CH2Cl2 and iPr2EtN (12 eq.), the reaction mixture was shaken overnight, filtered, washed with CH2Cl2 (5×), dried under a flow of N2 and a fresh portion of the tritylation reagents was added and the procedure was repeated to yield 5(6)-carboxyfluorescein(Trt)2-Lys(Dde)-Gly-Wang resin which was used for further synthesis (compounds 11, 12 and 13). A sample of the trityl-protected fluorescein-labelled resin (25.0 mg) was Dde deprotected using 2% hydrazine hydrate solution in DMF (2 × 3 min), cleaved using 95 : 2.5 : 2.5 (v/v/v) TFA : TIS : H2O for 2 h, precipitated from cold Et2O, isolated by centrifuge and lyophilised from CH3CN–H2O to yield 5(6)-carboxyfluorescein-Lys-Gly-OH; Rt 13.08 min (Phenomenex Jupiter C18); m/z (ESI-MS) 562.3 ([M + H]+ requires 562.2).Acknowledgements
We thank the Maurice Wilkins Centre for Molecular Biodiscovery for the award of a Doctoral Scholarship to RK.References
- Glycopeptides and Glycoproteins, Synthesis, Structure and Application, Topics in Current Chemistry, ed. V. Wittmann, Springer-Verlag, Berlin and Heidelberg, 2007, vol. 267 Search PubMed.
- S. Kaech, J. Wherry and R. Ahmed, Nat. Rev. Immunol., 2002, 2, 251 CrossRef CAS.
- J. K. Adam, B. Odhav and K. D. Bhoola, Pharmacol. Ther., 2003, 99, 113 CrossRef CAS.
- V. H. Engelhard, Sci. Am., 1994, 271(2), 54 CrossRef CAS.
- C. G. Figdor, Y. van Kooyk and G. J. Adema, Nat. Rev. Immunol., 2002, 2, 77 CrossRef CAS.
- A. J. Engering, M. Cella, D. M. Fluitsma, E. C. M. Hoefsmit, A. Lanzavecchia and J. Pietres, Adv. Exp. Med. Biol., 1997, 417, 183 CAS.
- T. B. H. Geijtenbeek, S. J. van Vliet, A. Engering, B. A. t'Hart and Y. van Kooyk, Annu. Rev. Immunol., 2004, 22, 33 CrossRef CAS.
- S. Burgdorf, A. Kautz, V. Bohnert, P. A. Knolle and C. Kurts, Science, 2007, 216, 612 CrossRef.
- S. Burgdorf, V. Lukacs-Kornek and C. Kurts, J. Immunol., 2006, 176, 6770 CAS.
- K. Drickamer, Nat. Struct. Biol., 1995, 2, 437 CrossRef CAS.
- N. Frison, M. E. Taylor, E. Soilleux, M.-T. Bousser, R. Mayer, M. Monsigny, K. Drickamer and A.-C. Roche, J. Biol. Chem., 2003, 278, 23922 CrossRef CAS.
- P. R. Crocker and T. Feizi, Curr. Opin. Struct. Biol., 1996, 6, 679 CrossRef CAS.
- E. A. Biessen, F. Noorman, M. E. van Teijlingen, J. Kuiper, M. Barrett-Bergshoeff, M. K. Bijsterbosch, D. C. Rijken and T. J. Berkel, J. Biol. Chem., 1996, 271, 28024 CrossRef CAS.
- G. Angyalosi, C. Grandjean, M. Lamirand, C. Auriault, H. Gras-Masse and O. Melnyk, Bioorg. Med. Chem. Lett., 2002, 12, 2723 CrossRef CAS.
- W. E. Weis and K. Drickamer, Structure, 1994, 2, 1227 CrossRef.
- P. van der Bruggen and B. J. van den Eynde, Curr. Opin. Immunol., 2006, 18, 98 CrossRef CAS.
- G. Kragol and L. Otvos, Jr., Tetrahedron, 2001, 57, 957 CrossRef CAS.
- D. M. Andrews and P. W. Seale, Int. J. Pept. Protein Res., 1993, 42, 165 CAS.
- J. Martinez, J. C. Tolle and M. Bodansky, J. Org. Chem., 1979, 44, 3596 CrossRef CAS.
- H. W. Kunz, Angew. Chem., Int. Ed. Engl., 1984, 23, 71 CrossRef.
- K. Horst, Angew. Chem., Int. Ed. Engl., 1987, 26, 294 CrossRef.
- T. K. Lindhorst, M. Dubber, U. Krallmann-Wenzel and S. Ehlers, Eur. J. Org. Chem., 2000, 2027 CrossRef CAS.
- R. K. Ness, H. G. Fletcher and C. S. Hudson, J. Am. Chem. Soc., 1950, 72, 2200 CrossRef CAS.
- J. Fiandor, M. T. Garcia-Lopez, F. G. de Las Heras and P. P. Mendez-Castrillon, Synthesis, 1985, 1121 CrossRef CAS.
- B. Becker, R. H. Furneaux, F. Reck and O. A. Zubkov, Carbohydr. Res., 1999, 315, 148 CrossRef CAS.
- M. A. Blaskovich and G. A. Lajoie, J. Am. Chem. Soc., 1993, 115, 5021 CrossRef CAS.
- W. F. Veldhuyzen, Q. Nguyen, G. McMaster and D. S. Lawrence, J. Am. Chem. Soc., 2003, 125, 13358 CrossRef CAS.
- R. Fischer, O. Mader, G. Jung and R. Brock, Bioconjugate Chem., 2003, 14, 653 CrossRef CAS.
- L. Bourel, O. Carion, H. Gras-Masse and O. Melnyk, J. Pept. Sci., 2000, 6, 264 CrossRef CAS.
- D. D. Perrin, D. R. Perrin and W. L. F. Amarego, Purification of Laboratory Chemicals, Pergamon Press Ltd., Oxford, 2nd edn, 1980 Search PubMed.
- E. A. Ivlev, L. V. Backinowsky, P. I. Abronina, L. O. Kononov and N. K. Kochetkov, Pol. J. Chem., 2005, 79, 275 CAS.
- R. Saksena, J. Zhang and P. Kováč, J. Carbohydr. Chem., 2002, 21, 453 CrossRef CAS.
- F. Bien and T. Ziegler, Tetrahedron: Asymmetry, 1998, 9, 781 CrossRef CAS.
- K. J. Jensen, M. Meldal and K. Bock, J. Chem. Soc., Perkin Trans. 1, 1993, 17, 2119 RSC.
- C. D. Chang, M. Waki, M. Ahmad, J. Meienhofer, E. O. Lundell and J. D. Haug, Int. J. Pept. Protein Res., 1980, 15, 59 CAS.
- A. Paquet, Can. J. Chem., 1982, 60, 976 CAS.
- M. D. Gieselman, L. Xie and W. A. van der Donk, Org. Lett., 2001, 3, 1331 CrossRef CAS.
- W. C. Chan and P. D. White, Fmoc Solid Phase Peptide Synthesis: a Practical Approach, Oxford University Press, 2000 Search PubMed.
- E. Kaiser, R. L. Colescott, C. D. Bossinger and P. I. Cook, Anal. Biochem., 1970, 34, 595 CrossRef CAS.
Footnote |
| † Synthesis of glycopeptide 10 using Fmoc-Ala-Wang (loading 0.66 mmol g–1) was unsuccessful hence Fmoc-Ala-TentaGel S PHB resin (0.2 mmol g–1) was used. |
| This journal is © The Royal Society of Chemistry 2008 |