Kinetic studies on the formation of DNA triplexes containing the nucleoside analogue 2′-O-(2-aminoethyl)-5-(3-amino-1-propynyl)uridine
David A. Ruslinga, Victoria J. Broughton-Heada, Alex Tucka, Hannah Khairallaha, Sadie D. Osborneb, Tom Brownb and Keith R. Fox*a
aSchool of Biological Sciences, University of Southampton, Bassett Crescent East, Southampton, SO16 7PX, UK. E-mail: k.r.fox@soton.ac.uk; Fax: +44 23 8059 4459; Tel: +44 23 8059 4374
bSchool of Chemistry, University of Southampton, Highfield, Southampton, SO17 1BJ, UK
First published on 1st November 2007
Abstract
We have examined the kinetics of triple helix formation of oligonucleotides that contain the nucleotide analogue 2′-O-(2-aminoethyl)-5-(3-amino-1-propynyl)uridine (bis-amino-U, BAU), which forms very stable base triplets with AT. Triplex stability is determined by both the number and location of the modifications. BAU-containing oligonucleotides generate triplexes with extremely slow kinetics, as evidenced by 14 °C hysteresis between annealing and melting profiles even when heated at a rate as slow as 0.2 °C min−1. The association kinetics were measured by analysis of the hysteresis profiles, temperature-jump relaxation and DNase I footprinting. We find that the slow kinetics are largely due to the decreased rate of dissociation; BAU modification has little effect on the association reaction. The sequence selectivity is also due to the slower dissociation of BAU from AT than other base pairs.
Introduction
Triplex-forming oligonucleotides (TFOs) bind within the major groove of duplex DNA forming Hoogsteen hydrogen bonds with exposed groups on the base pairs.1,2 Their unique base–base recognition properties make them ideal candidates as gene-recognition agents for exploitation in medicine and biotechnology.2–7 The base composition of the oligonucleotide dictates its binding orientation and selectivity; those composed of pyrimidine bases bind in a parallel orientation to the purine-strand of the duplex, generating T.AT and C+.GC triplets.2 The application of these compounds is currently restricted by several intrinsic limitations. Under physiological pH and ionic conditions the binding of a TFO is weak, predominantly due to the requirement for cytosine protonation and charge repulsion between the three negatively charged strands. TFOs are also restricted to binding to oligopurine.oligopyrimidine sequences as there is no method for recognising TA or CG base pairs (pyrimidine inversions) using natural nucleotides. We and others have prepared nucleotide analogues to overcome these restrictions and have used these in combination to generate oligonucleotides which form stable triplexes at pH 7.0 at target sites which contain pyrimidine interruptions.8–13As well as improving the overall stability of triplexes, the kinetic details of the binding of these modified TFOs is extremely important. The biological activities of these molecules will not only depend on increasing their affinity, but on their individual association and dissociation rates.14 Ideally TFOs should bind rapidly to their target sites and dissociate very slowly, while their interaction with non-targeted sites must be much weaker and should be characterised by fast dissociation. In this manner they may be able to compete with, and selectively disrupt, the interaction of cellular DNA binding proteins, such as transcription factors and polymerases. It is known that the rate of triplex formation is very slow, about three orders slower than duplex formation.15–19 It is also thought to proceed via a nucleation-zipper mechanism, dependent on the formation of a quasi-stable intermediate consisting of a few productive triplets, before a ‘zippering’ of the remainder of the third strand around the duplex helix.20,21 The apparent rate of triplex association therefore decreases with temperature, as lower temperatures stabilise this intermediate. Triplex dissociation is also slow, with reports suggesting half lives of between 30 minutes and several days.15–17,22–24
In this study we have determined how the nucleoside analogue 2′-O-(2-aminoethyl)-5-(3-amino-1-propynyl)uridine (bis-amino-U; BAU: Fig. 1A) affects the kinetics of DNA triplex formation. Previous studies have shown that bis-amino-U binds to AT base pairs with a higher affinity and specificity than T.25–27 Here we use fluorescence melting and DNase I footprinting experiments to assess the kinetic details of the interaction of TFOs containing this analogue with their intended target sites, and with sites that differ by a single base pair.
![(A) Chemical structures of the T.AT and BAU.AT triplets. (B) Sequences of the oligonucleotides used in fluorescence melting and temperature jump experiments. The TFOs were labelled at the 5′-end with methyl red while the 5′-end of the purine-containing strand of each duplex was labelled with fluorescein. TFOs -2, -3, -4 and -5 contain one or two substitutions with BAU (B). The duplex is boxed and ZY corresponds to each of the four base pairs in turn. (C) Sequence of the oligonucleotides used in the footprinting experiments. TFOs -7, -8 and -9 contain one, two or three substitutions with BAU (B). The duplex target is boxed and is found within the 110 bp tyrT(43–59) DNA fragment. This fragment was labelled at the 3′-end with [α-32P]dATP.](/image/article/2008/OB/b713088k/b713088k-f1.gif) | ||
| Fig. 1 (A) Chemical structures of the T.AT and BAU.AT triplets. (B) Sequences of the oligonucleotides used in fluorescence melting and temperature jump experiments. The TFOs were labelled at the 5′-end with methyl red while the 5′-end of the purine-containing strand of each duplex was labelled with fluorescein. TFOs -2, -3, -4 and -5 contain one or two substitutions with BAU (B). The duplex is boxed and ZY corresponds to each of the four base pairs in turn. (C) Sequence of the oligonucleotides used in the footprinting experiments. TFOs -7, -8 and -9 contain one, two or three substitutions with BAU (B). The duplex target is boxed and is found within the 110 bp tyrT(43–59) DNA fragment. This fragment was labelled at the 3′-end with [α-32P]dATP. | ||
Results
BAU.AT triplets
| TFO | Tm/°C | Hysteresis | Temperature-jump | ||||
|---|---|---|---|---|---|---|---|
| Eoff/kJ mol−1 | LnA | 37k−1/s−1 | 37t1/2/s | Eoff/kJ mol−1 | LnA | ||
| 1 | 63.8 (63.4) | 362 ± 4 | 125 ± 3 | 1.9 × 10−7 | 3.6 × 106 | 427 ± 12 | 148 ± 5 |
| 2 | 70.4 (64.6) | 340 ± 12 | 114 ± 4 | 1.7 × 10−8 | 4.2 × 107 | 418 ± 6 | 140 ± 2 |
| 3 | 78.4 (64.1) | 332 ± 12 | 108 ± 4 | 9.1 × 10−10 | 7.6 × 108 | ||
| 4 | 77.8 (64.8) | 300 ± 11 | 97 ± 4 | 3.8 × 10−9 | 1.8 × 108 | ||
| 5 | 73.0 (64.9) | 291 ± 14 | 95 ± 4 | 1.7 × 10−8 | 4.1 × 107 | ||
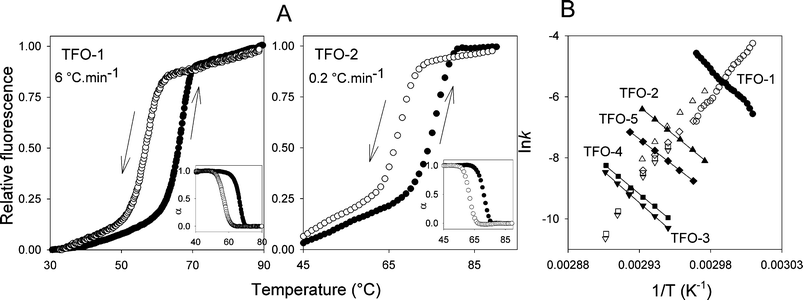 | ||
| Fig. 2 (A) Representative annealing (down arrow, open symbols) and heating (up arrow, filled symbols) profiles for the interaction of TFO-1 and TFO-2 with the duplex target shown in Fig. 1B, ZY = AT. The profiles for TFO-1 were obtained at a rate of temperature change of 6 °C min−1 whilst the profiles for TFO-2 were determined at 0.2 °C min−1. Fraction folded (α) plots for each triplex are included in the insets. (B) Representative Arrhenius plots for the association (k1) and dissociation (k−1) constants for TFOs 1–5. | ||
The effect of including a second BAU substitution was examined using TFOs 3–5. The results are shown in Fig. 2 and Table 1. When the two BAU residues are close together or separated by two nucleotides (TFO-3 and TFO-4 respectively) the Tm is increased further and the hysteresis becomes more pronounced. The Arrhenius plots for their association parameters lie on the same line as for TFO-1 and TFO-2, confirming that these modifications have little or no effect on the association reaction. In contrast the dissociation rates are even slower. Surprisingly the activation energies are slightly lower than for TFO-1 and TFO-2 (which alone would lead to a faster dissociation rate) and the main effect is in the pre-exponential factor (A). Separating the two BAU residues by seven nucleotides (TFO-5) does not increase the Tm relative to TFO-2; the activation energy is lowered as too is the pre-exponential factor. In this case these two effects cancel each other and TFO-2 and TFO-5 display similar dissociation half lives. The faster dissociation of TFO-5 compared to TFO-3 and TFO-4 could either be due to the greater separation of the two BAU residues or the nature of the flanking triplets. In TFO-4 each BAU.AT triplet is flanked by T.AT and C+.GC, while in TFO-5 both BAU.AT triplets are flanked by C+.GC on each side.
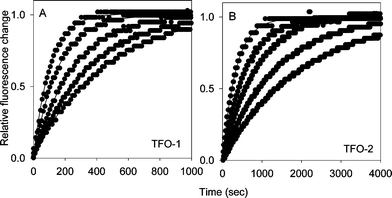 | ||
| Fig. 3 Representative temperature-jump profiles for the interaction of TFO-1 (panel A) and TFO-2 (panel B) with the target site containing a central AT base pair. The TFO and target duplex concentrations were both 0.25 μM. Each curve was obtained by rapidly increasing the temperature by 5 °C, measuring the time-dependent increases in fluorescence. The final temperatures were 60, 61, 62, 63 and 64 °C for TFO-1 and 66, 67, 68, 69 and 70 °C for TFO-2, each increasing from right to left. | ||
Similar experiments with TFO-3, TFO-4 and TFO-5 confirm the results from the hysteresis experiments. Representative temperature-jump relaxation curves for all five oligonucleotides at 70 °C are presented in Fig. 4B. These show similar profiles for TFO-3 and TFO-4, which are both slower than TFO-2, while TFO-5, in which the two BAU residues are separated by a greater distance and flanked by C+.GC triplets has similar properties to TFO-2. Arrhenius plots derived from the relaxation profiles with 3 μM TFO-3–5 are shown in Fig. 4A. It can be seen that the linear portions of these plots (at high temperatures) have similar slopes (activation energies), but are in different positions on the graph. TFO-3 (inverted diamonds) and TFO-4 (squares) are very similar, while TFO-5 (diamonds) shows faster dissociation.
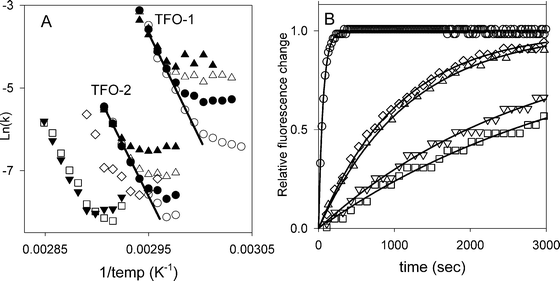 | ||
| Fig. 4 (A) Arrhenius plots showing the temperature dependence of the relaxation rate constants for TFO-1–5. The concentrations of TFO-1 and TFO-2 were 0.25 μM (open circles), 1 μM (filled circles), 3 μM (open triangles) 10 μM (filled triangles). For TFO-3 (inverted triangles), TFO-4 (squares) and TFO-5 (diamonds) data for 3 μM oligonucleotide are presented. (B) Representative temperature-jump profiles for the interaction of TFOs 1–5 with the target site containing a central AT base pair. The TFO concentration was 0.25 μM in each case and the temperature was rapidly increased from 65 to 70 °C. TFO-1 (circles), TFO-2 (triangles), TFO-3 (inverted triangles), TFO-4 (squares), TFO-5 (diamonds). | ||
BAU triplet mismatches
We have previously demonstrated that BAU retains the selectivity for AT base pairs and has enhanced discrimination against CG and TA base pairs.25 We therefore examined whether this discrimination results from changes in the association or dissociation rates and performed hysteresis and temperature-jump experiments with TFO-1 and TFO-2 using duplex targets, in which the central base pair was exchanged for each base pair in turn. Arrhenius plots for the complexes containing these triplex mismatches are presented in Fig. 5. A full quantitative analysis of these data is not possible since, as we have previously noted, the melting curves for complexes that contain triplex mismatches are biphasic,25 as the triplex melts at much lower temperatures than the duplex. The estimated kinetic parameters therefore contain large systematic errors and so can only be used for a qualitative comparison of the different complexes. Once again the association kinetics (derived from the hysteresis profiles) show negative activation energies and, although the individual curves do not overlap, they are in similar regions (unlike the dissociation profiles). For TFO-1 the association plots have similar slopes and those for T.CG and T.TA are almost contiguous. Since the Arrhenius plots for the association reaction are clearly not measuring primary kinetic events they were not analysed further. In contrast the Arrhenius plots for the dissociation reactions show clear differences between the targets, with similar results obtained by the two techniques. Looking first at the results for TFO-1 it can be seen that T.TA has a faster dissociation than T.GC or T.CG, which are faster than T.AT. The slower dissociation of T.CG may not be surprising since this triplet has been used as a (poor) means of recognising CG inversions. The slow dissociation of T.GC is also consistent with the reported Tm values of TFOs that contain this triplet, which are similar to those containing T.CG.25,28 For TFO-2, which contains a central BAU residue, the association plots are again almost contiguous, though with different slopes. As for TFO-1 the dissociation plots for the four targets have different rates. BAU.AT is much slower than the other three, though GC is considerably slower than both BAU.TA and BAU.CG, which have similar profiles. It is clear that, at these elevated temperatures around the Tm of each complex, the major differences between the matched and mismatched complexes arise from changes in the dissociation, rather than the association rates.![Arrhenius plots for the interaction of TFO-1 and TFO-2 with target sites containing ZY as each base pair in turn. Open symbols correspond to data obtained from hysteresis experiments (TFO concentration 3 μM), whilst closed symbols correspond to data obtained from temperature-jump experiments (TFO concentration 0.25 μM). ZY = CG (triangles); GC (diamonds) and TA (circles). The data from T.AT and B.AT are taken from Fig. 3 and 4. The dissociation data lie on lines with negative slopes, while the association data show positive slopes. The association data correspond to the measured pseudo first-order rate constant k* = k1[TFO].](/image/article/2008/OB/b713088k/b713088k-f5.gif) | ||
| Fig. 5 Arrhenius plots for the interaction of TFO-1 and TFO-2 with target sites containing ZY as each base pair in turn. Open symbols correspond to data obtained from hysteresis experiments (TFO concentration 3 μM), whilst closed symbols correspond to data obtained from temperature-jump experiments (TFO concentration 0.25 μM). ZY = CG (triangles); GC (diamonds) and TA (circles). The data from T.AT and B.AT are taken from Fig. 3 and 4. The dissociation data lie on lines with negative slopes, while the association data show positive slopes. The association data correspond to the measured pseudo first-order rate constant k* = k1[TFO]. | ||
DNase I footprinting
| TFO | k1/M−1 s−1 | k−1/s−1 |
|---|---|---|
| *6 | 2.4 × 103 | 2.4 × 10−4 |
| *7 | 3.8 × 103 | †3.6 × 10−4 |
| #6 | 0.7 × 103 | |
| #7 | 0.6 × 103 | |
| #8 | 2.0 × 103 | |
| #9 | 1.0 × 104 |
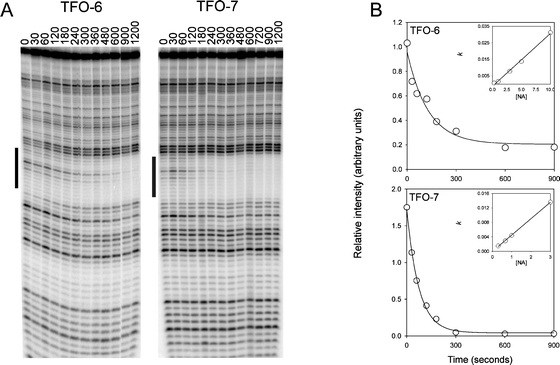 | ||
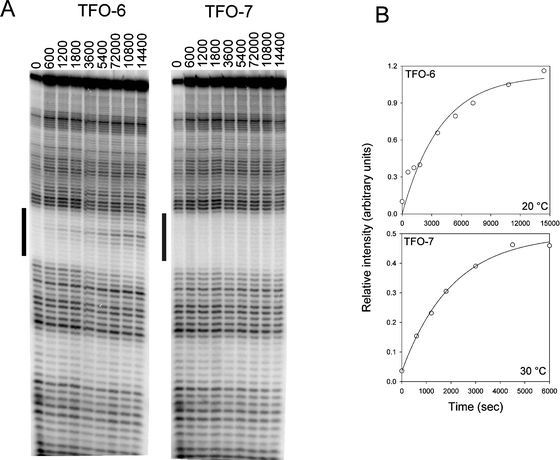
| Fig. 6 Determination of TFO association rates by DNase I footprinting. (A) DNase I cleavage patterns of the tyrT(43–59) DNA fragment in the presence of 3 μM of TFO-6 and 7 at different times after adding the TFOs. The experiments were performed at 20 °C. The time (sec) after adding the TFO is indicated at the top of each gel lane. The black bars show the TFO target site. (B) Plots of relative band intensity within the footprint against time for each TFO. The insets show the concentration dependence of the apparent rate constants, from which the association rate constants, listed in Table 4, were derived. | ||
 | ||
| Fig. 7 Determination of TFO dissociation rates by DNase I footprinting. (A) DNase I cleavage patterns of the tyrT(43–59) DNA fragment in the presence of 3 μM of TFO-6 and 7. The complexes were equilibrated overnight at 20 °C before adding 30 μM of the complementary oligonucleotide 3′-AGAGAAAAAAGA. The time (sec) after adding the complementary oligonucleotides is shown at the top of the gel lanes. The black bars indicate the TFO target site. (B) Plots of relative band intensity within the footprint against time for each TFO. The data for TFO-6 were obtained at 20 °C, while those for TFO-7 correspond to 30 °C. | ||
Discussion
These results demonstrate that the enhanced stability of triplexes containing the BAU.AT triplet relative to T.AT is largely due to its slower dissociation rather than changes in the association reaction. Surprisingly we find that the activation energy for the dissociation reaction is largely unaffected by introducing a single BAU modification and the slower dissociation is mainly due to a decrease in the pre-exponential factor (A). The similar values of Ea for TFOs 1 and 2 may not be surprising as they only differ by a single modification at the centre of the oligonucleotides and this region may not be involved in the primary dissociation event. Since the pre-exponential factor is a function of the entropy of the activated state, this suggests that the enhanced stability of BAU.AT may arise from the conformational organisation of the third strand.These conclusions relate to a given set of experimental conditions (200 mM NaCl and pH 5.0). The difference between the BAU and T bases may be altered at different salt concentrations or pH. Since BAU provides two more positive charges than T the differences may be magnified at lower ionic strengths, but reduced at higher pH.
TFO-3 and TFO-4, in which the two BAU residues are adjacent or separated by two nucleotides, show similar properties, though the dissociation half-life for TFO-3 is longer. This is consistent with studies with psoralen-linked TFOs that contain 2′-aminoethoxy-T, which produce more stable complexes when the modifications are close together,14,29 but contrasts to our previous work which suggested that the most stable complexes are produced when these modifications are evenly distributed along the third strand.26 Further increasing the distance between the two BAU residues (TFO-5) generates less stable complexes (equivalent to the TFO containing only one BAU residue), though this may also be due to the presence of C+.GC triplets adjacent to BAU.AT in TFO-5. Although the stability of T.AT triplets is enhanced by the presence of adjacent C+.GC triplets,15,30 analogues that bear positively charged groups can be inhibited by adjacent C+.GC triplets.31
The results with targets that generate triplet mismatches provide further information on the stringency of BAU. This base analogue is highly selective for AT relative to other base pairs, though it binds better to GC than CG or TA.25 In contrast T.GC and T.CG are more stable than T.TA. It is not clear why BAU stabilises the interaction with GC but not CG, especially since the T.CG triplet is known to provide the best means for recognizing CG inversions using natural bases.32,33 It is possible that the additional contacts or structural changes provided by BAU are not compatible with the single hydrogen bond that is formed in the T.CG triplet. Once again it is clear that discrimination between the different base pairs is a consequence of changes in the dissociation, rather than association, rate constants.
It is not possible to compare the results from footprinting with those from hysteresis and temperature-jump experiments as they used different length oligonucleotides (12-mers and 18-mers). The ratio of k−1–k1 derived from the footprinting experiments gives a value of 0.1 μM, which is similar to the dissociation constant of this oligonucleotide.
Effective DNA triple helix formation in vivo will require not only strong equilibrium binding, which we have achieved by using the positively charged nucleotide analogue BAU, but fast association (competing with various DNA binding proteins) and long dissociation half-lives. The results presented in this paper demonstrate that stability of the BAU.AT triplet is achieved by slowing the dissociation reaction, with little effect on the association kinetics. Different nucleic acid derivatives, or the inclusion of many more modifications, will be required to improve on the slow association kinetics.
Experimental
Oligonucleotides
All oligonucleotides were synthesized on an Applied Biosystems ABI 394 automated DNA/RNA synthesizer on the 0.2 μmol or 1 μmol scale using the standard cycles of acid-catalysed detritylation, coupling, capping and iodine oxidation procedures. Phosphoramidite monomers and reagents were purchased from Applied Biosystems or Link Technologies. The phosphoramidite for BAU was prepared as previously reported.25,27 The oligonucleotides were deprotected for 24 h in 2 ml of 30% aqueous methylamine in the presence of phenol (5 mg). The deprotected oligonucleotides were purified by reverse-phase HPLC on a Brownlee Aquapore column (C8) using a gradient of acetonitrile in 0.1 M ammonium acetate. Purified oligonucleotides were analysed by MALDI-TOF mass spectrometer in positive ion mode using internal Tn standards.34 The sequences of the oligonucleotides used in this work are shown in Fig. 1.Fluorescence melting experiments
Kinetic parameters for the intermolecular triplexes shown in Fig. 1B were obtained using fluorescence melting and temperature-jump experiments as previously described.13,15,28,35 TFOs were labelled at the 5′-end with methyl red and the 5′-end of the purine-containing strand of each duplex was labelled with fluorescein. The triplexes were prepared in 50 mM sodium acetate buffer containing 200 mM sodium chloride at pH 5.0. Melting experiments were carried out in a total volume of 20 μl and each assay contained 0.25 μM duplex and 3 μM third strand (unless otherwise stated).To ensure correct annealing, the triplexes were initially heated to 95 °C and then slowly cooled to room temperature. Although the slowest rate of continuous temperature change by the LightCycler is 0.1 °C sec−1, slower rates were obtained by increasing/decreasing the temperature in 1 °C steps, leaving the samples to equilibrate for a suitable length of time (typically 5 min). Hysteresis curves were then obtained by heating the samples at a fast rate (typically 6 °C min−1) to 95 °C, leaving the samples to equilibrate for 5 min, and then cooling the samples to 35 °C at the same rate. Recordings were taken during both the heating and cooling cycles. The rate of temperature change was adjusted for each triplex so as to obtain optimal separation between the melting and annealing curves. Tm values were determined from the first derivatives of the melting profiles using the software provided. If αc and αh are the fractions of the duplex that are occupied by the third strand in the cooling and heating curves respectively, then d(αc)/dT = d(αc)/dt× (dT/dt)−1 and d(αh)/dT = d(αh)/dt× (dT/dt)−1, where t is time and T is temperature. If k1 and k−1 are the triplex association and dissociation rate then d(αc)/dt = k1[TFO](1 −αc) −k−1αc and d(αh)/dt = k1[TFO](1 −αh) −k−1αh. By measuring d(αc)/dT, d(αh)/dT, αc and αh, the individual rate constants can be estimated at each temperature.17,36,37 Since the third strand concentration is in excess in these experiments it effectively remains constant during the reaction (3 μM), yielding a pseudo first order process with a rate constant k* given by k1[TFO]. In several instances the melting curves revealed a biphasic profile corresponding to dissociation of the duplex (with a decrease in fluorescence intensity) at higher temperatures than dissociation of the third strand as previously noted.25,35 This was especially noticeable with complexes that contained a triplex mismatch. These were fitted by assuming a coupled equilibrium in which the third strand dissociates first yielding a species with high fluorescence, followed by dissociation of the underlying duplex producing a random coil with a lower fluorescence.
Quantitative DNase I footprinting experiments
Acknowledgements
This work was funded by the BBSRC and EU grant LSHB-CT-2004-005204. Alex Tuck was supported by a Wellcome Vacation Studentship.References
- G. Felsenfeld, D. R. Davies and A. Rich, J. Am. Chem. Soc., 1957, 79, 2023–2024 CrossRef CAS.
- H. E. Moser and P. B. Dervan, Science, 1987, 238, 645–650 CrossRef CAS.
- S. Buchini and C. J. Leumann, Curr. Opin. Chem. Biol., 2003, 7, 717–726 CrossRef CAS.
- J. Y. Chin, E. B. Schleifman and P. M. Glazer, Front. Biosci., 2007, 12, 4288–4297 CrossRef CAS.
- K. R. Fox, Curr. Med. Chem., 2000, 7, 17–37 CAS.
- C. Helene and J. J. Toulme, Biochim. Biophys. Acta, 1990, 1049, 99–125 CrossRef CAS.
- M. M. Seidman and P. M. Glazer, J. Clin. Invest., 2003, 112, 487–494 CrossRef CAS.
- S. Buchini and C. J. Leumann, Angew. Chem., Int. Ed., 2004, 43, 3925–3928 CrossRef CAS.
- K. R. Fox and T. Brown, Q. Rev. Biophys., 2005, 38, 311–320 CrossRef CAS.
- D. M. Gowers and K. R. Fox, Nucleic Acids Res., 1999, 27, 1569–1577 CrossRef.
- J. S. Li, F. X. Chen, R. Shikiya, L. A. Marky and B. Gold, J. Am. Chem. Soc., 2005, 127, 12657–12665 CrossRef CAS.
- J. Robles, A. Grandas, E. Pedroso, F. J. Luque, R. Eritja and M. Orozco, Curr. Org. Chem., 2002, 6, 1333–1368 CrossRef CAS.
- D. A. Rusling, V. E. C. Powers, R. T. Ranasinghe, Y. Wang, S. D. Osborne, T. Brown and K. R. Fox, Nucleic Acids Res., 2005, 33, 3025–3032 CrossRef CAS.
- M. M. Seidman, N. Puri, A. Majumdar, B. Cuenoud, P. S. Miller and R. Alam, Ann. N. Y. Acad. Sci., 2005, 1058, 119–127 CrossRef CAS.
- P. L. James, T. Brown and K. R. Fox, Nucleic Acids Res., 2003, 31, 5598–5606 CrossRef CAS.
- H. M. Paes and K. R. Fox, Nucleic Acids Res., 1997, 25, 3269–3274 CrossRef CAS.
- M. Rougee, B. Faucon, J. L. Mergny, F. Barcelo, C. Giovannangeli, T. Garestier and C. Helene, Biochemistry, 1992, 31, 9269–9278 CrossRef CAS.
- H. Shindo, H. Torigoe and A. Sarai, Biochemistry, 1993, 32, 8963–8969 CrossRef CAS.
- L. E. Xodo, Eur. J. Biochem., 1995, 228, 918–926 CrossRef CAS.
- P. Alberti, P. B. Arimondo, J. L. Mergny, T. Garestier, C. Helene and J. S. Sun, Nucleic Acids Res., 2002, 30, 5407–5415 CrossRef CAS.
- D. Porschke and M. Eigen, J. Mol. Biol., 1971, 62, 361–381 CrossRef CAS.
- C. Ellouze, F. Piot and M. Takahashi, J. Biochem., 1997, 121, 521–526.
- L. J. Maher, P. B. Dervan and B. J. Wold, Biochemistry, 1990, 29, 8820–8826 CrossRef CAS.
- E. Protozanova and R. B. Macgregor, Anal. Biochem., 1996, 243, 92–99 CrossRef CAS.
- S. D. Osborne, V. E. C. Powers, D. A. Rusling, O. Lack, K. R. Fox and T. Brown, Nucleic Acids Res., 2004, 32, 4439–4447 CrossRef CAS.
- D. A. Rusling, L. Le Strat, V. E. C. Powers, V. J. Broughton-Head, J. Booth, O. Lack, T. Brown and K. R. Fox, FEBS Lett., 2005, 579, 6616–6620 CrossRef CAS.
- M. Sollogoub, R. A. J. Darby, B. Cuenoud, T. Brown and K. R. Fox, Biochemistry, 2002, 41, 7224–7231 CrossRef CAS.
- D. A. Rusling, T. Brown and K. R. Fox, Biophys. Chem., 2006, 123, 134–140 CrossRef CAS.
- N. Puri, A. Majumdar, B. Cuenoud, P. S. Miller and M. M. Seidman, Biochemistry, 2004, 43, 1343–1351 CrossRef CAS.
- R. W. Roberts and D. M. Crothers, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 4320–4325 CrossRef CAS.
- D. M. Gowers, J. Bijapur, T. Brown and K. R. Fox, Biochemistry, 1999, 38, 13747–13758 CrossRef CAS.
- I. Radhakrishnan and D. J. Patel, J. Mol. Biol., 1994, 241, 600–619 CrossRef CAS.
- K. Yoon, C. A. Hobbs, J. Koch, M. Sardaro, R. Kutny and A. L. Weis, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 3840–3844 CAS.
- G. J. Langley, J. M. Herniman, N. L. Davies and T. Brown, Rapid Commun. Mass Spectrom., 1999, 13, 1717–1723 CrossRef CAS.
- R. A. J. Darby, M. Sollogoub, C. McKeen, L. Brown, A. Risitano, N. Brown, C. Barton, T. Brown and K. R. Fox, Nucleic Acids Res., 2002, 30, e39 CrossRef.
- E. Bernal-Mendez and C. J. Leumann, Biochemistry, 2002, 41, 12343–12349 CrossRef CAS.
- A. De Cian, L. Guittat, M. Kaiser, B. Sacca, S. Amrane, A. Bourdoncle, P. Alberti, M. P. Teulade-Fichou, L. Lacroix and J. L. Mergny, Methods, 2007, 42, 183–195 CrossRef CAS.
- N. M. Brown, P. A. Rachwal, T. Brown and K. R. Fox, Org. Biomol. Chem., 2005, 3, 4153–4157 RSC.
- E. E. Merkina and K. R. Fox, Biophys. J., 2005, 89, 365–373 CrossRef CAS.
- P. M. Brown, C. A. Madden and K. R. Fox, Biochemistry, 1998, 37, 16139–16151 CrossRef CAS.
- M. C. Fletcher and K. R. Fox, FEBS Lett., 1996, 380, 118–122 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2008 |