Efficient C2 functionalisation of 2H-2-imidazolines
Received
24th August 2007
, Accepted 22nd October 2007
First published on 8th November 2007
Abstract
Alkylation and oxidation of 2H-2-imidazolines, followed by regioselective deprotection, thionation and microwave-assisted Liebeskind–Srogl reaction, efficiently led to 2-aryl-2-imidazolines as new analogues of p53-hdm2 interaction inhibitors (Nutlins).
Introduction
The 2-imidazoline scaffold is found in many biologically active small molecules that target numerous pharmaceutically relevant binding sites and receptors.1 Although substitution patterns are diverse, most biologically active 2-imidazolines are substituted at C2. For example, the Nutlins, which are highly functionalised 2-imidazolines that shown both in vitro and in vivo antitumor activity, all contain a C2 aryl moiety that is believed to be essential.2 The Nutlins are strong inhibitors of hdm2, a protein that negatively modulates the transcriptional activity and stability of the p53 tumor suppressor protein .3 C2-Substituted imidazolines can be prepared by the condensation of a diamine with an imidate, but variation of the substituents is not straightforward.4 Alternatively, a trimethylsilyl chloride mediated multicomponent reaction (MCR) between oxazolones, aldehydes and amines can be employed.5 However, this leads to the formation of C2-functionalised 2-imidazolines with trans-oriented C4/C5 phenyl groups. Although some reports exist of methods that give cis-oriented C4/C5 phenyl groups,5 these seem less efficient for the synthesis of Nutlin analogues, which require cis-oriented C4/C5 phenyl groups.
Recently, we reported a versatile MCR involving amines , aldehydes and isocyanoacetates to access 2H-2-imidazolines.6 All substituents can be varied easily by the choice of readily available reagents. Furthermore, the reaction generally favours formation of the diastereomer with cis-oriented aryl functionalities at C4/C5. Consequently, our method could be exploited to synthesise new, Nutlin-type, p53-hdm2 interaction inhibitors. The approach renders analogues containing an additional carboxylate that may serve to enhance water solubility. However, the 2H-2-imidazolines that result from our MCR should be arylated at C2. Here, we present an efficient synthetic strategy to achieve this.
Results and discussion
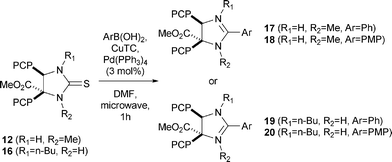
Four relevant 2H-imidazolines (Scheme 1, 4–7) were selected as starting materials for our synthetic study. The two cis-oriented p-chlorophenyl (PCP) groups in the backbone of analogues 4 and 5 are also found in Nutlins, where they seem crucial for the interaction with the Trp23 and Leu26 pockets of hdm2.2 The imidazolines 6 and 7, containing a 5-indolyl substituent, are also highly relevant, since, according to NMR studies of Nutlins bound to hdm2,4 these may improve binding to the Trp23 domain of hdm2. Thus, application of the MCR using isocyanoacetate 17 in combination with two different aldehydes (2) and amines (3) provides the corresponding 2H-2-imidazolines 4–7 in high yields.
 |
| | Scheme 1 2H-2-Imidazolines 4–7 obtained from a 3-component reaction (3-CR). | |
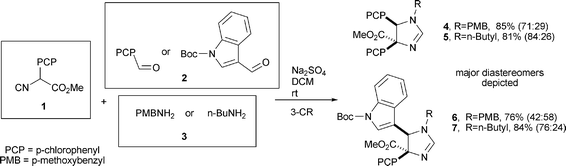
Direct C2 functionalisation of 4–7 proved not to be straightforward, and a procedure for selective C2 arylation had to be developed. Recently, Liebeskind and Srogl reported the Pd(0)-catalyzed, Cu(I)-mediated coupling of thioether -type species with boronic acids under neutral conditions.8 The high thiophilicity of the soft Cu(I) carboxylate cofactor facilitates selective C–C coupling with isothioureas even in the presence of a Suzuki-active bromide.9 This elegant procedure has been used to directly arylate dihydropyrimidine-2-thiones under microwave irradiation.10 We envisioned the mild and easy sulfoxidation of 2H-2-imidazolinium salts through the reaction of in situ-generated N-heterocyclic carbenes with elemental sulfur as an appropriate method to synthesise precursors for Liebeskind–Srogl reactions (Scheme 2).11 Strategic choice of R1 and R3 groups should allow selective deprotection of N1 or N3, leading to Nutlin analogues.
 |
| | Scheme 2 Possible route for C2 functionalisation of 2H-2-imidazolines. | |
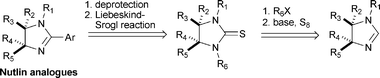
Methylation and sulfoxidation of imidazoline 4 affords the cyclic thiourea 9 in good yield (Scheme 3). Chromatographic separation of the diastereomers was achieved but cleavage of the p-methoxybenzyl (PMB) group could not be realised. Both oxidative and acidic conditions cause decomposition, probably because of the sensitive thiocarbonyl group. Therefore, an alternative route was considered. Imidazolidin-2-thiones can be prepared by the thionation of imidazolidin-2-ones using Lawesson's reagent.12 For this, oxidation of 2H-2-imidazolines was required. Although (low-yielding) oxidations of benzimidazoles with mCPBA are known,13 this procedure proved unsuitable for the direct oxidation of imidazolines. In contrast, mCPBA-mediated oxidation of imidazolinium salt 8 affords cyclic urea 10 as a mixture of diastereomers, in a combined yield of 82% (Scheme 3). For a clean reaction it proved important to add the mCPBA to a cooled solution of 8. The diastereomers of 10 are conveniently separated by chromatography .
 |
| | Scheme 3 Oxidation and thionation of C2. | |
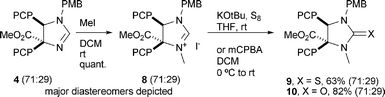
Treatment of trans-10 with TFA (to cleave off the PMB-group) and subsequent thionation provides the Liebeskind–Srogl precursor 12 with a C5 ester function in very high yields (Scheme 4).14
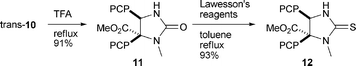 |
| | Scheme 4 Synthesis of Liebeskind–Srogl precursor 12. | |
With the above-described methodology available, we now turned to the synthesis of Liebeskind–Srogl precursors containing the ester functionality at C4. Clean formation of imidazolinium salt 13 from 5 can be achieved using PMBBr and NaI under Finkelstein conditions (Scheme 5). Oxidation then gives cyclic urea 14 in reasonable yield, and the diastereomers could be separated chromatographically. Deprotection and subsequent thionation of trans-14 both went smoothly, affording efficiently the desired Liebeskind–Srogl precursor 16.14
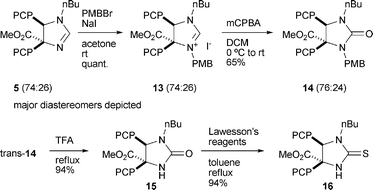 |
| | Scheme 5 Synthesis of Liebeskind–Srogl precursor 16. | |
With 12 and 16 in hand, we could now perform the Liebeskind–Srogl reactions (Table 1). Reactions were run in sealed vessels with controlled single-mode microwave heating. Initially, coupling reactions between boronic acids and 12 or 16 were performed under conditions reported by Kappe et al. (PhB(OH)2, Pd(PPh3)4, Cu(I) thiophene-2-carboxylate, THF, microwave, 100 °C, 30 min).10 These conditions, however, gave only traces of C2-arylated products. Conditions were further refined with respect to the solvent, and the reactions proceeded much better in DMF instead of THF. Also, the reaction temperature appears crucial and is important for achieving good conversions to the desired cross-coupling products. Although not fully optimized yet, running the reaction at 130 °C for 1 h ultimately led to efficient Liebeskind–Srogl cross-coupling of 12 and 16 with two different boronic acids , and the corresponding 2-aryl-2-imidazolines 17–20 were isolated in good yields.
Table 1 Liebeskind–Srogl arylation of 12 and 16
| Product |
T/°C |
Isolated yield (%) |
|
17
|
100 |
14 |
|
17
|
130 |
51 |
|
18
|
100 |
24 |
|
18
|
130 |
41 |
|
19
|
100 |
34 |
|
19
|
130 |
65 |
|
20
|
100 |
6 |
|
20
|
130 |
55 |
Conclusion
In conclusion, a versatile route toward C2-functionalized 2-imidazolines containing Nutlin-like backbones has been developed. Besides the 3-component reaction to access 2H-2-imidazolines, key steps involve the oxidation at C2 and the Liebeskind–Srogl coupling of cyclic thioureas with boronic acids . The analogues 17–20 have been prepared in good overall yields (23% to 35%) starting from commercially available aldehydes and amines . The high overall yields and the flexibility make this procedure amenable to library synthesis of potential p53-hdm2 interaction inhibitors.
Experimental
General
All reactions were carried out under atmospheric conditions, unless stated otherwise. Standard syringe techniques were applied for transfer of air-sensitive reagents and dry solvents. Melting points were measured using a Stuart Scientific SMP3 melting point apparatus and are uncorrected. Infrared (IR) spectra were obtained from CHCl3 films on NaCl tablets (unless noted otherwise), using a Matteson Instuments 6030 Galaxy Series FT-IR spectrophotometer, and wavelengths (ν) are reported in cm−1. 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance 400 (400.13 MHz and 100.61 MHz respectively) or a Bruker Avance 250 (250.13 MHz and 62.90 MHz respectively) with chemical shifts (δ) reported in ppm downfield from tetramethylsilane. MS and HRMS spectral data were recorded on a Finnigan Mat 900 spectrometer or in the Laboratory of Organic Chemistry of the Wageningen University (NL) on a Finnigan MAT95 spectrometer. Chromatographic purification refers to flash chromatography using the indicated solvent (mixture) and Baker 7024-02 silica gel (40 µ, 60 Å). Thin layer chromatography was performed using silica plates from Merck (Kieselgel 60 F254 on aluminium with fluorescence indicator. Compounds on TLC were visualised by UV-detection unless stated otherwise. THF was dried and distilled from sodium benzophenone ketyl prior to use. DCM was dried and distilled from CaH2 prior to use. Toluene was distilled from sodium prior to use. DMF was dried and distilled from phenylzinc iodide prior to use. Other commercially available reagents were used as purchased.
Microwave experiments
Microwave-assisted reactions were performed in a Discover (CEM Corporation) single-mode microwave instrument producing controlled irradiation at 2450 MHz, using standard sealed microwave glass vials. Reaction temperatures were monitored with an IR sensor on the outside wall of the reaction vials. Reaction times refer to hold times at the indicated temperatures, not to total irradiation times.
1-(4-Chlorophenyl)-2-methoxy-2-oxoethanaminium chloride.
p-Chlorophenylglycine (8.62 g, 46.5 mmol) was dissolved in MeOH (110 mL). The solution was cooled to 0 °C and thionyl chloride (6.8 mL, 93 mmol) was added dropwise. The reaction mixture was heated under reflux for 3 h. Cooling to rt followed by concentration in vacuo afforded the title compound as a white solid (10.98 g, quant.) 1H NMR (250 MHz, D2O) δ (ppm) 7.54 (d, J = 8.6 Hz, 2H), 7.49 (d, J = 8.6 Hz, 2H), 5.30 (s, 1H), 3.82 (s, 3H).
Methyl 2-amino-2-(4-chlorophenyl)acetate
.
1-(4-Chlorophenyl)-2-methoxy-2-oxoethanaminium chloride (3.33 g, 14.1 mmol) was suspended in EtOAc (100 mL). Saturated NaHCO3 (aq.) (80 mL) was added and the suspension was stirred until the organic layer was clear and slightly orange. The layers were separated and the aqueous layer was extracted twice with EtOAc. The organic layers were combined, washed with brine and dried with Na2SO4. Filtration and concentration in vacuo afforded the title compound as a yellow–orange solid (2.79 g, 99%) 1H NMR (250 MHz, CDCl3) δ (ppm) 7.36 (s, 4H), 4.67 (s, 1H), 3.73 (s, 3H), 2.60 (br s, 2H).
Methyl 2-(4-chlorophenyl)-2-formamidoacetate.
Methyl 2-amino-2-(4-chlorophenyl)acetate (2.79 g, 14.0 mmol) was dissolved in ethyl formate (80 mL), and a small crystal of pTSA was added. The reaction mixture was refluxed overnight, cooled to rt and the solvent was evaporated. The product was dissolved in DCM, washed with water, dried with Na2SO4, and concentrated in vacuo to afford the title compound as a yellow solid (3.15 g, 99%) 1H NMR (200 MHz, CDCl3) δ (ppm) 8.22 (s, 1H), 7.31 (s, 4H), 6.86 (br s, 1H), 5.62 (d, J = 7.2 Hz, 1H), 3.72 (s, 3H); 13C NMR (63 MHz, CDCl3) δ (ppm) 176.6 (C), 170.6 (C), 160.5 (CH), 134.6 (C), 129.2 (2 × CH), 129.0 (2 × CH), 54.4 (CH3), 53.2 (CH); HRMS (EI, 70 eV) calculated for C10H10ClNO3 (M+) 227.0349, found 227.0346.
Methyl 2-(4-chlorophenyl)-2-isocyanoacetate 1.
This reaction was carried out under an inert atmosphere of dry nitrogen. Methyl 2-(4-chlorophenyl)-2-formamidoacetate (910 mg, 4.0 mmol) was dissolved in DCM (10 mL) and cooled to −30 °C. Triphosgene (504 mg, 1.7 mmol) and N-methylmorpholine (1.57 mL, 14.3 mmol) were added slowly. The solution turned darker orange, and after 30 minutes at −30 °C the temperature was raised to −5 °C and kept at this temperature for an additional 3 hours, during which time the solution slowly turned darker. The reaction mixture was quenched in 20 mL of ice-water. The layers were separated, and the aqueous layer was extracted with Et2O. The organic layers were combined, washed with brine and dried with Na2SO4. Concentration in vacuo followed by flash column chromatography (cyclohexane–ethyl acetate = 4 : 1), afforded 1 as an orange oil (640 mg, 77%). 1H NMR (250 MHz, CDCl3) δ (ppm) 7.42 (br s, 4H), 5.35 (s, 1H), 3.80 (s, 3H); 13C NMR (63 MHz, CDCl3) δ (ppm) 165.6 (C), 162.2 (C), 135.8 (C), 130.2 (C), 129.4 (2 × CH), 128.0 (2 × CH), 59.6 (C), 53.9 (CH3); IR (neat) 2954 (m), 2148 (s), 1753 (s), 1493 (s), 1435 (m), 1250 (m), 1211 (m), 1092 (s), 1014 (s); HRMS (EI, 70 eV) calculated for C10H8ClNO2 (M+) 209.0244, found 209.0248.
General Procedure I for the synthesis of 2-imidazolines
Reactions were carried out under an inert atmosphere of dry nitrogen at a concentration of 1 M of aldehyde 2, 1 M of amine 3, and 0.5 M of isocyanide 1 in dry DCM or MeOH. Na2SO4 and the aldehyde were added, at rt, to a stirred solution of the amine . After the mixture was stirred for 2 h, the isocyanide was added and the reaction mixture was stirred at rt for an additional 18 h. The reaction mixture was filtered and concentrated in vacuo. The crude product was purified by flash column chromatography (c-hexane–EtOAc–Et3N = 2 : 1 : 0.01, gradient, unless stated otherwise).
Methyl 4,5-bis(4-chlorophenyl)-1-(4-methoxybenzyl)-4,5-dihydro-1H-imidazole-4-carboxylate
4
.
According to General Procedure I, reaction between p-methoxybenzylamine (2.74 g, 20 mmol), p-chlorobenzaldehyde (2.80 g, 20.0 mmol) and isocyanoacetate 1 (2.07 g, 9.9 mmol) in DCM, followed by column chromatography (EtOAc–Et3N = 1 : 0.01), afforded 4 (3.96 g, 85%) as a 71 : 29 mixture of diastereomers as a yellow solid. 1H NMR (250 MHz, CDCl3) δ (ppm) 7.47 (d, J = 43.0 Hz, 1H), 7.36–7.21 (m, 3H + 4H), 7.10–6.98 (m, 4H + 4H), 6.89–6.76 (m, 5H + 5H), 5.28 (s, 1H), 4.64 (s, 1H), 4.36–4.31 (m, 1H + 1H), 3.82 (s, 3H), 3.74 (s, 3H), 3.83–3.74 (m, 4H + 1H), 3.28 (s, 3H); 13C NMR (63 MHz, CDCl3) δ (ppm) 173.5 (C), 170.6 (C), 159.4 (C), 156.7 (CH), 155.9 (CH), 141.6 (C), 135.9 (C), 134.9 (C), 134.4 (C), 133.7 (2 × C), 133.6 (2 × C), 133.2 (C), 120.0 (2 × CH), 129.4 (2 × CH), 129.3 (2 × CH), 129.0 (2 × CH), 128.8 (2 × CH), 128.3 (2 × CH), 128.2 (2 × CH), 128.08 (2 × CH), 128.06 (2 × CH), 127.8 (2 × CH), 127.1 (C), 126.9 (C), 114.1 (2 × CH), 114.2 (2 × CH), 84.7 (C), 83.8 (C), 72.4 (CH3), 68.5 (CH3), 55.1 (CH3 + CH3), 53.1 (CH), 52.1 (CH), 48.6 (CH2), 48.1 (CH2); IR (neat) 1730 (s), 1602 (s); HRMS (EI, 70 eV) calculated for C25H22Cl2N2O3 (M+) 468.1007, found 468.1002.
Methyl 1-butyl-4,5-bis(4-chlorophenyl)-4,5-dihydro-1H-imidazole-4-carboxylate
5
.
According to General Procedure I, reaction between n-butylamine (730 mg, 10.0 mmol), p-chlorobenzaldehyde (1.40 g, 10.0 mmol) and isocyanoacetate 1 (990 mg, 4.74 mmol) in DCM, followed by column chromatography (EtOAc–Et3N = 1 : 0.01), afforded 5 (1.61 g, 84%) as a 74 : 26 mixture of diastereomers as a pale yellow oil. 1H NMR (250 MHz, CDCl3) δ (ppm) 7.69 (d, J = 8.7 Hz, 2H), 7.39–7.34 (m, 4H), 7.30–7.26 (m, 3H + 1H), 7.05–7.01 (m, 2H + 2H), 6.90–6.86 (m, 2H + 2H), 5.51 (s, 1H), 4.81 (s, 1H), 3.77 (s, 3H), 3.28 (s, 3H), 3.20–3.08 (m, 1H + 1H), 2.89–2.78 (m, 1H + 1H), 1.54–1.13 (m, 4H + 4H), 0.92–0.79 (m, 3H + 3H); 13C NMR (100.6 MHz, CDCl3) δ (ppm) 173.8 (C), 170.6 (C), 156.8 (CH), 156.1 (CH), 141.9 (C), 136.1 (C), 135.2 (C), 134.4 (C), 134.0 (C), 133.6 (C), 133.5 (C), 133.1 (C), 129.8 (2 × CH), 129.2 (2 × CH), 128.8 (2 × CH), 128.4 (2 × CH), 128.17 (2 × CH), 128.15 (2 × CH), 128.08 (2 × CH), 127.8 (2 × CH), 84.65 (C), 83.9 (C), 73.5 (CH3), 68.7 (CH3), 53.1 (CH), 52.1 (CH), 44.8 (CH2), 44.5 (CH2), 30.2 (CH2), 30.0 (CH2), 19.8 (CH2), 19.7 (CH2), 13.6 (CH3), 13.5 (CH3); IR (neat) 1729 (s), 1599 (s); HRMS (EI, 70 eV) calculated for C21H22Cl2N2O2 (M+) 404.1058, found 404.1046.
tert-Butyl 3-(4-(4-chlorophenyl)-1-(4-methoxybenzyl)-4-(methoxycarbonyl)-4,5-dihydro-1H-imidazol-5-yl)-1H-indole-1-carboxylate
6
.
According to General Procedure I, reaction between p-methoxybenzylamine (384 mg, 2.8 mmol), N-Boc-indolecarboxaldehyde (690 mg, 2.8 mmol) and isocyanoacetate 1 (419 mg, 2.0 mmol) in DCM, followed by column chromatography , afforded 6 (364 mg, 76%) as a 58 : 42 mixture of diastereomers as an orange–pink solid. 1H NMR (400 MHz, DMSO-d6, 360 K): δ (ppm) 8.09 (d, J = 8.3 Hz, 1H), 7.90 (d, J = 8.3 Hz, 1H), 7.63 (s, 1H), 7.61–7.56 (m, 3H + 3H), 7.54 (s, 1H), 7.48 (d, J = 7.8 Hz, 1H), 7.38–7.32 (m, 2H + 2H), 7.25–7.20 (m, 1H + 1H), 7.09–7.01 (m, 1H), 7.03 (d, J = 8.6 Hz, 2H), 6.97 (br s, 1H + 1H), 6.91 (d, J = 8.6 Hz, 2H), 6.84 (d, J = 8.7 Hz, 2H), 6.73 (d, J = 8.7 Hz, 2H), 5.58 (s, 1H), 4.99 (s, 1H), 4.46 (d, J = 15.1 Hz, 1H), 4.39 (d, J = 14.9 Hz, 1H), 3.86–3.82 (m, 1H + 1H), 3.75 (s, 3H), 3.70 (s, 3H), 3.64 (s, 3H), 3.07 (s, 3H), 1.67 (s, 9H), 1.59 (s, 9H); 13C NMR (100.6 MHz, DMSO-d6, 360 K) δ (ppm) 172.5 (C), 170.2 (C), 158.4 (C), 158.3 (C), 156.8 (CH), 156.1 (CH), 148.5 (C), 148.2 (C), 142.2 (C + C), 137.3 (C + C), 134.8 (C + C), 131.7 (C), 131.2 (C), 128.72 (4 × CH), 128.67 (4 × CH), 128.3 (CH), 128.1 (C + C), 128.1 (2 × CH), 127.4 (2 × CH), 126.4 (CH), 125.4 (CH), 124.0 (CH), 123.6 (CH), 122.0 (CH), 121.8 (CH), 119.6 (CH), 119.4 (C), 115.8 (C), 114.3 (CH), 114.0 (CH), 113.54 (2 × CH), 113.46 (2 × CH), 83.8 (C + C), 83.4 (C + C), 65.1 (CH), 61.4 (CH), 54.8 (CH3), 54.7 (CH3), 52.0 (CH3), 50.8 (CH3), 47.5 (CH2), 47.3 (CH2), 27.4 (3 × CH3), 27.3 (3 × CH3); IR (KBr) 1733 (s), 1599 (s), 1512 (s), 1452 (s), 1370 (s), 1248 (s), 1153 (s), 1089 (s); HRMS (EI, 70 eV) calculated for C32H32ClN3O5 (M+) 573.2030, found 573.2036.
tert-Butyl 3-(1-butyl-4-(4-chlorophenyl)-4-(methoxycarbonyl)-4,5-dihydro-1H-imidazol-5-yl)-1H-indole-1-carboxylate
7
.
According to General Procedure I, reaction between n-butylamine (1.10 g, 15.0 mmol), N-Boc-indolecarboxaldehyde (3.68 g, 15.0 mmol) and isocyanoacetate 1 (2.89 g, 13.8 mmol) in MeOH, followed by column chromatography , afforded 7 (5.9 g, 84%) as a 76 : 24 mixture of diastereomers as a pale yellow solid. 1H NMR (400 MHz, CDCl3, 328 K) δ (ppm) 8.14 (d, J = 8.4 Hz, 1H), 7.95 (d, J = 8.3 Hz, 1H), 7.71 (d, J = 8.6 Hz, 1H), 7.62 (s, 1H), 7.57 (d, J = 7.9 Hz, 1H), 7.35–6.99 (m, 6H + 5H), 6.88 (d, J = 8.3 Hz, 2H + 2H), 5.83 (s, 1H), 5.04 (s, 1H), 3.74 (s, 3H), 3.16 (s, 3H), 3.13–3.04 (m, 1H + 1H), 2.90–2.84 (m, 1H + 1H), 1.69 (s, 9H), 1.63 (s, 9H), 1.52–1.45 (m, 2H), 1.37–1.12 (m, 2H + 4H), 0.85 (t, J = 7.3 Hz, 3H), 0.77 (t, J = 7.3 Hz, 3H); 13C NMR (100.6 MHz, CDCl3) δ (ppm) 174.0 (C), 156.4 (CH), 156.1 (CH), 149.4 (C), 149.3 (C), 136.9 (2 × C), 133.6 (C), 128.1 (3 × CH + 5 × CH), 127.3 (2 × CH), 125.5 (CH), 124.1 (CH + CH), 122.7 (CH + CH), 120.1 (CH), 115.6 (C), 115.3 (CH), 114.9 (CH), 84.3 (C), 83.9 (C), 67.9 (CH), 62.6 (CH), 53.2 (CH3), 52.1 (CH3), 44.8 (CH2), 44.3 (CH2), 30.3 (CH2), 30.2 (CH2), 28.2 (3 × CH3), 28.1 (3 × CH3), 19.8 (CH2), 19.7 (CH2), 13.6 (CH3), 13.5 (CH3); the aliphatic CH signals could only be found with gs-HSQC measurements at 328 K; the quaternary carbons of the minor diastereomer of 7 could not be detected; IR (KBr) 2957 (s), 1734 (s), 1599 (s), 1570 (m), 1452 (s), 1370 (s), 1257 (s), 1155 (s), 1089 (s); HRMS (EI, 70 eV) calculated for C28H32ClN3O4 (M+) 509.2081, found 509.2082.
4,5-Bis(4-chlorophenyl)-1-(4-methoxybenzyl)-4-(methoxycarbonyl)-3-methyl-4,5-dihydro-1H-imidazolium iodide 8.
Methyl iodide (447 mg, 3.15 mmol) was added to a solution of imidazoline 4 (1.408 g, 3 mmol) in DCM (20 mL). The reaction mixture was stirred at rt for 18 h and concentrated in vacuo to afford 8 (1.83 g, quant.) as a 68 : 32 mixture of diastereomers as a pale yellow solid. 1H NMR (250 MHz, CDCl3) δ (ppm) 10.65 (s, 1H), 10.52 (s, 1H), 7.48–7.29 (m, 4H + 2H), 7.20–7.17 (m, 4H + 6H), 7.01–6.91 (m, 2H + 2H), 6.91–6.83 (m, 2H + 2H), 5.73 (s, 1H), 5.45 (d, J = 14.1 Hz, 1H), 5.31 (d, J = 14.4 Hz, 1H), 5.14 (s, 1H), 4.23–4.13 (m, 1H + 1H), 3.96 (s, 3H), 3.80 (s, 3H + 3H), 3.56 (s, 3H), 3.34 (s, 3H), 3.26 (s, 3H); 13C NMR (63 MHz, CDCl3) δ (ppm) 168.9 (C), 165.8 (C), 160.5 (C), 160.2 (C), 160.1 (CH), 158.4 (CH), 136.9 (2 × C), 135.8 (C), 135.6 (C), 132.9 (C), 131.3 (2 × CH), 131.0 (4 × CH), 130.5 (2 × CH), 130.0 (2 × CH), 129.9 (C), 129.8 (2 × CH), 129.3 (2 × CH), 129.1 (2 × CH), 128.9 (2 × CH), 128.88 (2 × CH), 128.84 (C), 123.4 (2 × C), 123.1 (C), 114.7 (2 × CH), 114.6 (2 × CH), 80.6 (C), 80.2 (C), 74.1 (CH3), 70.2 (CH3), 55.4 (2 × CH3), 54.6 (CH), 53.2 (CH), 50.5 (CH2), 50.4 (CH2), 34.9 (CH3), 33.3 (CH3); IR (neat) 1745 (s), 1642 (s), 1611 (s).
Methyl 4,5-bis(4-chlorophenyl)-1-(4-methoxybenzyl)-3-methyl-2-thioxoimidazolidine-4-carboxylate 9.
This reaction was carried out under an inert atmosphere of dry argon. A Schlenk tube was charged with imidazolinium iodide 8 (608 mg, 1.0 mmol), KOtBu (118 mg, 1.05 mmol) and S8 (256 mg, 1.0 mmol). THF (30 mL) was added and the reaction mixture was stirred at rt for 2 h. Then, water was added and the mixture was extracted with EtOAc (3×). The organic layers were dried with Na2SO4 and concentrated in vacuo. Purification using flash column chromatography (toluene visualisation on TLC with UV and with iodine) afforded 9a (229 mg, 45%) and 9b (92 mg, 18%) as white solids.
9a (most polar isomer): 1H NMR (250 MHz, CDCl3) δ (ppm) 7.12–7.03 (m, 6H), 6.82 (d, J = 8.6 Hz, 2H), 6.85–6.66 (m, 4H), 5.74 (d, J = 14.7 Hz, 1H), 5.29 (s, 1H), 3.84 (s, 3H), 3.82 (s, 3H), 3.76 (d, J = 14.7 Hz, 1H), 3.26 (s, 3H); 13C NMR (63 MHz, CDCl3) δ (ppm) 170.5 (C), 159.2 (2 × C), 134.6 (C), 134.5 (C), 132.0 (C), 131.3 (C), 129.8 (4 × CH), 128.6 (2 × CH), 128.51 (2 × CH), 128.45 (2 × CH), 127.5 (C), 113.9 (2 × CH), 77.8 (C), 68.1 (CH3), 55.2 (CH3), 53.3 (CH), 49.1 (CH2), 34.2 (CH3); IR (neat) 1751 (s), 1610 (m); HRMS (EI, 70 eV) calculated for C26H24Cl2N2O3S (M+) 514.0885, found 514.0902.
9b (least polar isomer): Mp 120–121 °C; 1H NMR (250 MHz, CDCl3) δ (ppm) 7.36 (d, J = 8.5 Hz, 2H), 2.50 (d, J = 8.7 Hz, 2H), 7.11–7.04 (m, 6H), 6.81 (d, J = 8.6 Hz, 2H), 5.79 (d, J = 14.6 Hz, 1H), 4.73 (s, 1H), 3.80 (s, 3H), 3.62 (d, J = 14.6 Hz, 1H), 3.28 (s, 3H), 3.08 (s, 3H); 13C NMR (63 MHz, CDCl3) δ (ppm) 183.0 (C), 168.1 (C), 159.5 (C), 136.3 (C), 135.4 (C), 135.0 (C), 133.3 (C), 130.2 (4 × CH), 129.1 (2 × CH), 128.1 (4 × CH), 127.2 (C), 114.0 (2 × CH), 77.5 (C), 70.8 (CH3), 55.3 (CH3), 52.2 (CH), 48.7 (CH2), 33.1 (CH3); IR (neat) 1737 (s), 1612 (m); HRMS (EI, 70 eV) calculated for C26H24Cl2N2O3S (M+) 514.0885, found 514.0899.
Methyl 4,5-bis(4-chlorophenyl)-1-(4-methoxybenzyl)-3-methyl-2-oxoimidazolidine-4-carboxylate
10
.
To a cooled (0 °C) solution of imidazolinium iodide 8 (611 mg, 1.0 mmol) in DCM (20 mL), 85% mCPBA (610 mg, 3.0 mmol) was added. The yellow solution abruptly turned red. The reaction mixture was stirred at rt for 18 h, washed twice with saturated Na2CO3 (aq.), dried with Na2SO4 and concentrated in vacuo. Purification using flash column chromatography (c-hexane–EtOAc = 4 : 1) afforded trans-10 (289 mg, 58%) and cis-10 (120 mg, 24%) as white solids.
trans-10 (most polar isomer): Mp 88–90 °C; 1H NMR (250 MHz, CDCl3) δ (ppm) 7.09–6.95 (m, 6H), 6.81–6.68 (m, 6H), 5.03 (s, 1H), 4.96 (d, J = 14.8 Hz, 1H), 3.79 (s, 3H), 3.79 (s, 3H), 3.52 (d, J = 14.8 Hz, 1H), 2.94 (s, 3H); 13C NMR (63 MHz, CDCl3) δ (ppm) 171.2 (C), 160.2 (C), 159.2 (C), 134.3 (C), 134.1 (C), 132.7 (C), 132.1 (C), 130.0 (4 × CH), 128.6 (2 × CH), 128.4 (2 × CH), 128.3 (2 × CH), 128.0 (C), 113.9 (2 × CH), 73.8 (C), 64.3 (CH3), 55.3 (CH3), 53.0 (CH), 45.5 (CH2), 29.6 (CH3); IR (neat) 1738 (s), 1710 (s), 1611 (m); HRMS (EI, 70 eV) calculated for C26H24Cl2N2O4 (M+) 498.1113, found 498.1096.
cis-10 (least polar isomer): Mp 143–145 °C; 1H NMR (200 MHz, CDCl3) δ (ppm) 7.29–6.90 (m, 10H), 6.73 (d, J = 8.7 Hz, 2H), 4.95 (d, J = 14.6 Hz, 1H), 4.39 (s, 1H), 3.72 (s, 3H), 3.40 (d, J = 14.6 Hz, 1H), 3.26 (s, 3H), 2.66 (s, 3H); 13C NMR (63 MHz, CDCl3) δ (ppm) 168.9 (C), 159.5 (C), 159.3 (C), 136.4 (C), 135.0 (C), 134.6 (C), 133.7 (C), 130.1 (2 × CH), 129.6 (2 × CH), 128.8 (4 × CH), 128.4 (2 × CH), 127.7 (C), 114.0 (2 × CH), 73.7 (C), 67.2 (CH3), 55.3 (CH3), 52.0 (CH), 45.2 (CH2), 28.6 (CH3); IR (neat) 1741 (s), 1711 (s), 1611 (m); HRMS (EI, 70 eV) calculated for C26H24Cl2N2O4 (M+) 498.1113, found 498.1107.
General Procedure II for the Cleavage of PMB groups
A 0.25–0.30 M solution of imidazolidinone was refluxed for 1 h. After cooling to rt followed by evaporation of TFA, the crude product was dissolved in EtOAc, washed with saturated NaHCO3 (aq.) solution and brine, dried with Na2SO4 and concentrated in vacuo. Purification was performed using flash column chromatography (c-hexane–EtOAc = 1 : 1, gradient).
trans-Methyl 4,5-bis(4-chlorophenyl)-3-methyl-2-oxoimidazolidine-4-carboxylate 11.
According to General Procedure II, deprotection of imidazolidinone trans-10 (455 mg, 0.88 mmol), followed by column chromatography , afforded 11 (304 mg, 91%) as a white solid. Mp 214–216 °C; 1H NMR (250 MHz, CDCl3) δ (ppm) 7.29–7.06 (m, 4H), 6.93 (d, J = 8.3 Hz, 2H), 6.72 (d, J = 8.4 Hz, 2H), 5.56 (s, 1H), 5.50 (br s, 1H), 3.92 (s, 3H), 2.89 (s, 3H); 13C NMR (63 MHz, CDCl3) δ (ppm) 171.4 (C), 161.1 (C), 135.8 (C), 135.2 (C), 134.4 (C), 134.0 (C), 129.0 (2 × CH), 128.4 (4 × CH), 128.2 (2 × CH), 75.7 (C), 61.5 (CH3), 53.1 (CH), 29.1 (CH3); IR (KBr) 1734 (s), 1716 (s); HRMS (EI, 70 eV) calculated for C18H16Cl2N2O3 (M+) 378.0538, found 378.0526.
cis-Methyl 4,5-bis(4-chlorophenyl)-3-methyl-2-oxoimidazolidine-4-carboxylate.
According to General Procedure II, deprotection of imidazolidinone cis-10 (516 mg, 1.0 mmol), followed by column chromatography , afforded the title compound (396 mg, 99%) as a white solid. 1H NMR (250 MHz, CDCl3) δ (ppm) 7.45 (d, J = 8.8 Hz, 2H), 7.35 (d, J = 8.7 Hz, 2H), 7.26 (d, J = 8.5 Hz, 2H), 7.12 (d, J = 8.5 Hz, 2H), 5.19 (s, 1H), 4.86 (s, 1H), 3.33 (s, 3H), 2.63 (s, 3H); 13C NMR (63 MHz, CDCl3) δ (ppm) 168.6 (C), 160.8 (C), 136.1 (C), 135.8 (C), 135.1 (C), 135.0 (C), 129.1 (2 × CH), 128.8 (2 × CH), 128.75 (2 × CH), 128.72 (2 × CH), 76.0 (C), 65.1 (CH), 52.1 (CH3), 28.2 (CH3); IR (KBr) 1747 (s), 1720 (s), 1494 (m), 1435 (m); HRMS (EI, 70 eV) calculated for C18H16Cl2N2O3 (M+) 378.0538, found 378.0531.
General Procedure III for the thionation of imidazolidinones
Reactions were performed under an inert atmosphere of dry nitrogen at a concentration of 0.020 M of imidazolidinone in dry toluene. A reaction vessel was charged with imidazolidinone (1 equiv.) and Lawesson's reagent (1 equiv.). Toluene was added and the suspension was heated to reflux temperature. The resulting solution was refluxed for 18 h, cooled to room temperature and concentrated in vacuo. The crude product was loaded on to a pre-packed silica column. Impurities were eluted with toluene and then the product was eluted with toluene–EtOAc = 4 : 1, gradient. Visualisation on TLC was performed with UV and with iodine.
trans-Methyl 4,5-bis(4-chlorophenyl)-3-methyl-2-thioxoimidazolidine-4-carboxylate 12.
According to General Procedure III, thionation of imidazolidinone 11 (1.00 g, 2.64 mmol), followed by column chromatography , afforded 12 (2.46 g, 93%) as a white solid. 1H NMR (250 MHz, CDCl3) δ (ppm) 7.06–6.99 (m, 4H), 6.84 (d, J = 8.3 Hz, 2H), 6.60–6.55 (m, 3H), 5.63 (s, 1H), 3.86 (s, 3H), 3.10 (s, 3H); 13C NMR (63 MHz, CDCl3) δ (ppm) 184.4 (C), 170.3 (C), 134.9 (C), 134.5 (C), 133.9 (C), 130.7 (C), 128.8 (2 × CH), 128.6 (2 × CH), 128.4 (4 × CH), 80.0 (C), 65.0 (CH), 53.5 (CH3), 33.5 (CH3); IR (KBr) 3174 (br), 1737 (s), 1596 (w), 1491 (s), 1393 (m), 1256 (s); HRMS (EI, 70 eV) calculated for C18H16Cl2N2O2S (M+) 394.0310, found 394.0303.
cis-Methyl 4,5-bis(4-chlorophenyl)-3-methyl-2-thioxoimidazolidine-4-carboxylate.
According to General Procedure III, thionation of cis-methyl 4,5-bis(4-chlorophenyl)-3-methyl-2-oxoimidazolidine-4-carboxylate (374 mg, 0.98 mmol), followed by column chromatography , afforded the title compound (336 mg, 87%) as a white solid. 1H NMR (250 MHz, CDCl3) δ (ppm) 7.40 (d, J = 8.9 Hz, 2H), 7.34 (d, J = 8.8 Hz, 2H), 7.25 (d, J = 8.3 Hz, 2H), 7.09 (d, J = 8.3 Hz, 2H), 8.95 (s, 1H), 5.04 (s, 1H), 3.28 (s, 3H), 2.92 (s, 3H); 13C NMR (63 MHz, CDCl3) δ (ppm) 183.9 (C), 167.4 (C), 135.7 (C), 135.32 (C), 135.28 (C), 134.8 (C), 129.3 (2 × CH), 128.80 (2 × CH), 128.75 (2 × CH), 128.5 (2 × CH), 80.1 (C), 68.8 (CH), 52.4 (CH3), 32.3 (CH3); IR (KBr) 3162 (br), 1737 (s), 1596 (w), 1491 (s), 1393 (m), 1256 (s); HRMS (EI, 70 eV) calculated for C18H16Cl2N2O2S (M+) 394.0310, found 394.0299.
1-Butyl-4,5-bis(4-chlorophenyl)-3-(4-methoxybenzyl)-4-(methoxycarbonyl)-4,5-dihydro-1H-imidazolium iodide 13.
p-Methoxybenzyl bromide (1.46 g, 7.28 mmol) was added to a solution of imidazoline 5 (2.95 g, 7.28 mmol) and NaI (1.09 g, 7.28 mmol) in acetone (90 mL). The reaction mixture was stirred at rt for 18 h, filtered and concentrated in vacuo to afford 13 (4.75 g, quant.) as a 74 : 26 mixture of diastereomers as a white solid. 1H NMR (400 MHz, CDCl3) δ (ppm) 9.40 (s, 1H), 9.27 (s, 1H), 7.68 (d, J = 8.6 Hz, 2H), 7.51 (d, J = 8.7 Hz, 2H), 7.47–7.42 (m, 2H + 2H), 7.22 (d, J = 8.6 Hz, 2H), 7.16–7.07 (m, 6H + 2H), 6.96 (d, J = 8.5 Hz, 2H), 6.91–6.86 (m, 4H), 6.16 (s, 1H), 5.59 (s, 1H), 5.21 (d, J = 14.5 Hz, 1H), 4.66 (d, J = 14.5 Hz, 1H), 4.52 (d, J = 13.6 Hz, 1H), 4.35 (d, J = 13.7 Hz, 1H), 3.89 (s, 3H), 3.89–3.82 (m, 1H), 3.84 (s, 3H), 3.82 (s, 3H), 3.79–2.65 (m, 1H), 3.38–3.35 (m, 1H), 3.36 (s, 3H), 3.26–3.19 (m, 1H), 1.71–1.57 (m, 2H + 2H), 1.32–1.21 (m, 2H + 2H), 0.90–0.84 (m, 3H + 3H); 13C NMR (101 MHz, CDCl3) δ (ppm) 169.0 (C), 166.0 (C), 160.1 (C), 158.6 (2 × CH), 156.2 (C), 136.9 (C), 136.8 (C), 135.9 (C), 135.8 (C), 133.2 (C), 132.2 (C), 131.7 (2 × CH), 130.3 (2 × CH), 130.1 (2 × CH), 129.93 (C), 129.89 (2 × CH), 129.7 (2 × CH), 129.5 (2 × CH), 129.4 (2 × CH), 129.20 (2 × CH), 129.17 (2 × CH), 129.1 (2 × CH), 128.6 (C), 125.7 (C), 124.0 (C), 114.8 (2 × CH), 114.6 (2 × CH), 80.7 (C), 80.6 (C), 76.9 (CH), 71.4 (CH), 55.4 (CH3), 55.3 (CH3), 54.4 (CH3), 53.0 (CH3), 50.6 (CH2), 50.3 (CH2), 47.6 (CH2), 47.3 (CH2), 29.3 (CH2), 29.0 (CH2), 19.6 (CH2), 19.5 (CH2), 13.5 (CH3), 13.4 (CH3); IR (KBr) 1641 (s), 1513 (m), 1250 (s).
Methyl 1-butyl-4,5-bis(4-chlorophenyl)-3-(4-methoxybenzyl)-2-oxoimidazolidine-4-carboxylate
14
.
To a cooled (0 °C) solution of imidazolinium iodide 13 (265 mg, 0.41 mmol) in DCM (8 mL), 85% mCPBA (247 mg, 1.22 mmol) was added. The yellow solution abruptly turned dark orange. The reaction mixture was stirred at rt for 18 h, while a colour change from dark orange to light pink to dark red was observed. Then, the reaction mixture was washed twice with saturated Na2CO3 (aq.), dried with Na2SO4 and concentrated in vacuo. Purification using flash column chromatography (pentane–EtOAc = 7 : 1, visualisation on TLC with CerMOP [(NH4)6Mo7O24·4H2O (1.1 g L−1), Ce(SO4)2·4H2O (4 g L−1) in H2SO4 (10%)]) afforded trans-14 (109 mg, 49%) and cis-14 (36 mg, 16%) as white solids.
trans-14 (most polar isomer): 1H NMR (250 MHz, CDCl3) δ (ppm) 7.42–7.27 (m, 8H), 7.17–7.06 (m, 8H), 6.88–6.62 (m, 2H + 4H), 6.60 (d, J = 8.7 Hz, 2H), 5.58 (s, 1H), 4.96 (d, J = 15.9 Hz, 1H), 4.81 (s, 1H), 4.35 (d, J = 15.1 Hz, 1H), 4.18–4.11 (m, 1H + 1H), 3.81 (s, 3H), 3.79 (s, 3H), 3.73–3.60 (m, 1H + 1H), 3.38 (s, 3H), 3.23 (s, 3H), 2.75–2.64 (m, 1H + 1H), 1.54–1.22 (m, 4H + 4H), 0.99–0.85 (m, 3H + 3H); 13C NMR (63 MHz, CDCl3) δ (ppm) 170.5 (C), 168.9 (C), 160.7 (C), 160.3 (C), 158.5 (2 × C), 137.3 (C), 134.9 (C), 134.6 (C), 134.3 (C), 134.1 (C), 133.8 (C), 132.7 (C), 132.6 (C), 130.9 (C), 130.3 (C), 129.7 (2 × CH), 128.9 (2 × CH), 128.7 (4 × CH + 4 × CH), 128.2 (2 × CH), 128.2 (2 × CH), 127.9 (2 × CH + 2 × CH), 113.7 (2 × CH), 113.4 (2 × CH), 74.0 (C), 73.8 (C), 69.1 (CH), 64.7 (CH), 55.24 (CH3), 55.20 55.2 (CH3), 52.7 55.2 (CH3), 51.8 55.2 (CH3), 46.8 (CH2), 46.0 (CH2), 42.0 (CH2), 41.8 (CH2), 29.0 (CH2), 28.8 (CH2), 20.0 (CH2), 19.9 (CH2), 13.74 (CH3), 13.67 (CH3); IR (KBr) 1707 (s), 1513 (s), 1244 (s); HRMS (EI, 70 eV) calculated for C29H30Cl2N2O4 (M+) 540.1583, found 540.1587.
cis-14 (least polar isomer): 1H NMR (250 MHz, CDCl3) δ (ppm) 7.42–7.27 (m, 8H), 7.17–7.06 (m, 8H), 6.88–6.62 (m, 2H + 4H), 6.60 (d, J = 8.7 Hz, 2H), 5.58 (s, 1H), 4.96 (d, J = 15.9 Hz, 1H), 4.81 (s, 1H), 4.35 (d, J = 15.1 Hz, 1H), 4.18–4.11 (m, 1H + 1H), 3.81 (s, 3H), 3.79 (s, 3H), 3.73–3.60 (m, 1H + 1H), 3.38 (s, 3H), 3.23 (s, 3H), 2.75–2.64 (m, 1H + 1H), 1.54–1.22 (m, 4H + 4H), 0.99–0.85 (m, 3H + 3H); 13C NMR (63 MHz, CDCl3) δ (ppm) 170.5 (C), 168.9 (C), 160.7 (C), 160.3 (C), 158.5 (2 × C), 137.3 (C), 134.9 (C), 134.6 (C), 134.3 (C), 134.1 (C), 133.8 (C), 132.7 (C), 132.6 (C), 130.9 (C), 130.3 (C), 129.7 (2 × CH), 128.9 (2 × CH), 128.7 (4 × CH + 4 × CH), 128.2 (2 × CH), 128.2 (2 × CH), 127.9 (2 × CH + 2 × CH), 113.7 (2 × CH), 113.4 (2 × CH), 74.0 (C), 73.8 (C), 69.1 (CH), 64.7 (CH), 55.24 (CH3), 55.20 55.2 (CH3), 52.7 55.2 (CH3), 51.8 55.2 (CH3), 46.8 (CH2), 46.0 (CH2), 42.0 (CH2), 41.8 (CH2), 29.0 (CH2), 28.8 (CH2), 20.0 (CH2), 19.9 (CH2), 13.74 (CH3), 13.67 (CH3); IR (KBr) 1707 (s), 1513 (s), 1244 (s); HRMS (EI, 70 eV) calculated for C29H30Cl2N2O4 (M+) 540.1583, found 540.1587.
Methyl 1-butyl-4,5-bis(4-chlorophenyl)-2-oxoimidazolidine-4-carboxylate
15
.
According to General Procedure II, deprotection of imidazolidinone trans-14 (2.15 g, 3.97 mmol), followed by column chromatography , afforded 15 (1.57 g, 94%) as a white solid. 1H NMR (250 MHz, CDCl3) δ (ppm) 7.66 (d, J = 8.8 Hz, 2H), 7.43–7.38 (m, 4H), 7.29 (d, J = 8.5 Hz, 2H), 7.13–7.09 (m, 6H), 6.91 (d, J = 8.5 Hz, 2H), 5.90 (s, 1H), 5.50 (s, 1H), 5.47 (s, 1H), 4.77 (s, 1H), 3.82 (s, 3H), 3.65–3.51 (m, 1H + 1H), 3.35 (s, 3H), 2.60–2.48 (m, 1H + 1H), 1.47–1.35 (m, 2H), 1.33–1.13 (m, 2H + 2H), 1.10–1.07 (m, 2H), 0.90 (t, J = 7.3 Hz, 3H), 0.77 (t, J = 7.1 Hz, 3H); 13C NMR (63 MHz, CDCl3) δ (ppm) 172.5 (C), 169.3 (C), 160.0 (C), 159.4 (C), 139.3 (C), 135.0 (C), 134.7 (C), 134.5 (C), 134.3 (C), 134.2 (C), 133.5 (C), 133.1 (C), 128.9 (2 × CH), 128.9 (2 × CH), 128.49 (2 × CH), 128.46 (2 × CH), 127.3 (4 × CH), 127.2 (4 × CH), 70.3 (CH), 69.4 (C), 68.9 (C), 65.7 (CH), 53.5 (CH3), 52.6 (CH3), 41.0 (CH2), 40.6 (CH2), 29.4 (CH2), 29.3 (CH2), 19.7 (CH2), 19.6 (CH2), 13.6 (CH3), 13.5 (CH3); IR (KBr) 3233 (br), 2956 (m), 1701 (s), 1492 (s), 1231 (s), 1092 (s); HRMS (EI, 70 eV) calculated for C21H22Cl2N2O3 (M+) 420.1007, found 420.0989.
Methyl 1-butyl-4,5-bis(4-chlorophenyl)-2-thioxoimidazolidine-4-carboxylate 16.
According to General Procedure III, thionation of imidazolidinone 15 (250 mg, 0.59 mmol), followed by column chromatography , afforded 16 as a white solid (245 mg, 94%). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.63 (d, J = 8.7 Hz, 2H), 7.43 (d, J = 8.7 Hz, 2H), 7.39 (d, J = 8.6 Hz, 2H), 7.28–7.20 (m, 2H), 7.15–7.10 (m, 6H), 7.04 (d, J = 8.7 Hz, 2H), 6.86 (s, 1H), 6.84 (s, 1H), 5.70 (s, 1H), 4.92 (s, 1H), 4.22–4.14 (m, 1H), 4.09–4.05 (m, 1H), 3.83 (s, 3H), 3.38 (s, 3H), 2.79–2.72 (m, 1H), 2.67–2.63 (m, 1H), 1.57–1.49 (m, 2H), 1.35–1.26 (m, 2H + 2H), 1.09–1.03 (m, 2H), 0.92 (t, J = 7.3 Hz, 3H), 0.77 (t, J = 7.3 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ (ppm) 182.9 (C), 181.8 (C), 171.4 (C), 167.9 (C), 136.2 (C), 135.5 (C), 135.2 (C), 134.7 (2 × C), 133.4 (C), 132.6 (C), 131.9 (C), 129.2 (2 × CH), 128.7 (4 × CH), 128.7 (4 × CH), 127.2 (4 × CH), 126.8 (2 × CH), 74.4 (CH), 72.4 (C), 72.2 (C), 69.7 (CH), 53.8 (CH3), 52.8 (CH3), 45.0 (CH2), 44.3 (CH2), 29.1 (CH2), 28.9 (CH2), 19.7 (CH2), 19.4 (CH2), 13.7 (CH3), 13.5 (CH3); IR (KBr) 3162 (br), 2952 (s), 1729 (s), 1593 (m), 1491 (s), 1231 (s); HRMS (EI, 70 eV) calculated for C21H22Cl2N2O2S (M+) 436.0779, found 436.0777.
General Procedure IV for the microwave-assisted Liebeskind–Srogl reactions
A dry microwave vessel was charged with imidazolidine-2-thione (0.25 mmol), aryl boronic acid (0.38 mmol), Cu(I) thiophene-2-carboxylate (144.7 mg, 0.75 mmol) and Pd(PPh3)4 (8.9 mg, 7.5 µmol). The vessel was flushed with Ar and sealed. Dry DMF was added through the septum and the reaction mixture was irradiated in the microwave at 130 °C for 1 h, unless stated otherwise. After cooling, DMF was removed in vacuo at 50 °C. The crude mixture was diluted with saturated NaHCO3 (aq.) and extracted with DCM. The organic layer was washed twice with saturated NaHCO3 (aq.), dried with Na2SO4 and concentrated in vacuo. Purification was performed with flash column chromatography (c-hexane–EtOAc–Et3N = 5 : 1 : 0.01, gradient).
trans-Methyl 4,5-bis(4-chlorophenyl)-1-methyl-2-phenyl-4,5-dihydro-1H-imidazole-5-carboxylate 17.
According to General Procedure IV, arylation of imidazolidin-2-thione 12 (100 mg, 0.25 mmol) with phenyl boronic acid (46 mg, 0.38 mmol) followed by column chromatography afforded 17 (56 mg, 51%) as a yellow solid together with starting material 12 (2 mg, 2%). 1H NMR (250 MHz, CDCl3) δ (ppm) 7.79–7.68 (m, 2H), 7.57–7.45 (m, 3H), 7.06 (d, J = 8.6 Hz, 2H), 7.03 (s, 4H), 6.79 (d, J = 8.5 Hz, 2H), 5.96 (s, 1H), 3.97 (s, 3H), 2.87 (s, 3H); 13C NMR (63 MHz, CDCl3) δ (ppm) 173.2 (C), 165.5 (C), 137.4 (C), 133.6 (C), 133.0 (C), 132.5 (C), 130.8 (C), 130.8 (CH), 129.5 (2 × CH), 128.7 (2 × CH), 128.5 (2 × CH), 128.4 (2 × CH), 128.1 (2 × CH), 127.6 (2 × CH), 81.1 (C), 76.0 (CH), 52.9 (CH3), 33.0 (CH3); IR (KBr) 1726 (s), 1589 (m), 1487 (m), 1379 (m), 1231 (m); HRMS (EI, 70 eV) calculated for C24H20Cl2N2O2 (M+) 438.0902, found 438.0883.
trans-Methyl 4,5-bis(4-chlorophenyl)-2-(4-methoxyphenyl)-1-methyl-4,5-dihydro-1H-imidazole-5-carboxylate 18.
According to General Procedure IV, arylation of imidazolidin-2-thione 12 (100 mg, 0.25 mmol) with p-methoxyphenyl boronic acid (58 mg, 0.38 mmol) followed by column chromatography afforded 18 (48 mg, 41%) as a yellow solid, together with starting material 12 (5 mg, 5%). 1H NMR (250 MHz, CDCl3) δ (ppm) 7.69 (d, J = 8.4 Hz, 2H), 7.05 (d, J = 8.7 Hz, 2H), 7.01 (br s, 6H), 6.77 (d, J = 8.5 Hz, 2H), 5.92 (br s, 1H), 3.96 (s, 3H), 3.88 (s, 3H), 2.89 (s, 3H); IR (KBr); HRMS (EI, 70 eV) calculated for C25H22Cl2N2O3 (M+) 468.1007, found 468.1002.
Methyl 1-butyl-4,5-bis(4-chlorophenyl)-2-phenyl-4,5-dihydro-1H-imidazole-4-carboxylate
19
.
According to General Procedure IV, arylation of imidazolidin-2-thione 16 (109 mg, 0.25 mmol) with phenyl boronic acid (46 mg, 0.38 mmol) followed by column chromatography afforded 19 (78 mg, 65%) as a yellow solid. 1H NMR (250 MHz, CDCl3) δ (ppm) 7.82 (d, J = 8.7 Hz, 2H), 7.68–7.64 (m, 2H + 2H), 7.50–7.47 (m, 3H + 3H), 7.40–7.33 (m, 2H), 7.09–6.92 (m, 8H + 4H), 5.73 (s, 1H), 4.95 (s, 1H), 3.78 (s, 3H), 3.31–3.21 (m, 1H + 1H), 3.25 (s, 3H), 2.85–2.79 (m, 1H + 1H), 1.37–1.06 (m, 4H + 4H), 0.71 (t, J = 7.2 Hz, 3H), 0.59 (t, J = 7.2 Hz, 3H); 13C NMR (63 MHz, CDCl3) δ (ppm) 174.5 (C), 171.4 (C), 167.7 (C), 167.1 (C), 143.1 (2 × C), 135.9 (C), 134.5 (C), 133.3 (C), 132.6 (C), 132.4 (C), 131.9 (C), 130.0 (2 × C), 131.0 (CH), 130.8 (C), 130.2 (2 × CH), 129.6 (2 × CH), 129.1 (2 × CH), 128.99 (2 × CH), 128.97 (2 × CH + 2 × CH), 128.9 (2 × CH), 128.8 (2 × CH + 2 × CH), 128.7 (2 × CH), 128.6 (2 × CH), 128.2 (2 × CH), 82.8 (C), 82.6 (C), 75.4 (CH), 70.3 (CH), 53.5 (CH3), 52.6 (CH3), 45.9 (CH2), 45.8 (CH2), 30.6 (CH2), 30.1 (CH2), 19.8 (CH2), 19.4 (CH2), 13.9 (CH3), 13.7 (CH3); IR (KBr) 2928 (s), 1726 (s), 1490 (s), 1241 (s); HRMS (EI, 70 eV) calculated for C25H23Cl2N2 (M+ − CO2Me) 421.1238, found 421.1223; the molecular ion could not be detected.
Methyl 1-butyl-4,5-bis(4-chlorophenyl)-2-(4-methoxyphenyl)-4,5-dihydro-1H-imidazole-4-carboxylate
20
.
According to General Procedure IV, arylation of imidazolidin-2-thione 16 (109 mg, 0.25 mmol) with p-methoxyphenyl boronic acid (58 mg, 0.38 mmol) followed by column chromatography afforded 20 (71 mg, 55%) as a yellow solid, together with starting material 16 (14 mg, 13%). 1H NMR (250 MHz, CDCl3) δ (ppm) 7.81 (d, J = 8.5 Hz, 2H), 7.68–7.60 (m, 2H + 2H), 7.39–7.36 (m, 2H), 7.08–6.90 (m, 10H + 6H), 5.70 (s, 1H), 4.90 (s, 1H), 3.87 (s, 3H + 3H), 3.77 (s, 3H), 3.33–3.23 (m, 1H + 1H), 3.23 (s, 3H), 2.88–2.80 (m, 1H + 1H), 1.33–1.01 (m, 4H + 4H), 0.85 (t, J = 6.0 Hz, 3H), 0.72 (t, J = 7.2 Hz, 3H); 13C NMR (63 MHz, CDCl3) δ (ppm) 174.6 (C), 171.6 (C), 167.5 (C), 166.9 (C), 161.8 (C), 161.7 (C), 143.3 (2 × C), 137.5 (C), 136.2 (C), 134.6 (C), 134.0 (C), 133.7 (C), 133.3 (C), 130.7 (2 × CH), 130.5 (2 × CH), 130.2 (2 × CH + 2 × CH), 129.6 (2 × CH), 129.1 (2 × CH), 128.8 (2 × CH), 128.7 (2 × CH), 128.6 (2 × CH), 128.1 (2 × CH), 123.1 (C), 123.0 (C), 114.6 (2 × CH), 114.3 (2 × CH), 82.8 (C), 82.5 (C), 75.5 (CH), 70.3 (CH), 55.8 (CH3 + CH3), 53.5 (CH3), 52.5 (CH3), 46.3 (CH2), 46.2 (CH2), 30.2 (CH2), 30.1 (CH2), 20.1 (CH2), 19.8 (CH2), 14.0 (CH3), 13.8 (CH3); IR (KBr) 2924 (s), 1734 (s), 1611 (s), 1490 (s), 1251 (s); HRMS (EI, 70 eV) calculated for C26H25Cl2N2 (M+ − CO2Me) 451.1344, found 451.1336; the molecular ion could not be detected.
Acknowledgements
Part of this work was performed with financial support from the Dutch Science Foundation (NWO-VICI). Dr M. T. Smoluch (VUA) is kindly acknowledged for the (HR)MS measurements.
References and notes
- For selected examples, see: C. Dardonville and I. Rozas, Med. Res. Rev., 2004, 24, 639 Search PubMed; M. von Rauch, S. Busch and R. Gust, J. Med. Chem., 2005, 48, 466 CrossRef; U. Schäfer, C. Burgdorf, A. Engelhardt, T. Kurz and G. Richardt, J. Pharmacol. Exp. Ther., 2002, 303, 1163 CrossRef CAS; D. Milhaud, L. Fagni, J. Bockaert and M. Lafon-Cazal, Neuropharmacology, 2000, 39, 2244 CrossRef CAS.
- L. T. Vassilev, B. T. Vu, B. Graves, D. Carvajal, F. Podlaski, Z. Filipovic, N. Kong, U. Kammlott, C. Lukacs, C. Klein, N. Fotouhi and E. A. Liu, Science, 2004, 303, 844 CrossRef CAS.
- B. Vogelstein, D. Lane and A. J. Levine, Nature, 2000, 408, 307 CrossRef CAS; L. Römer, C. Klein, A. Dehner, H. Kessler and J. Buchner, Angew. Chem., Int. Ed., 2006, 45, 6440 CrossRef CAS.
- D. C. Fry, S. D. Emerson, S. Palme, B. T. Vu, C.-M. Liu and F. Podlaski, J. Biomol. NMR, 2004, 30, 163 CrossRef CAS.
- V. Sharma and J. J. Tepe, Org. Lett., 2005, 7, 5091 CrossRef CAS; G. H. Merriman, L. Ma, P. Shum, D. McGarry, F. Volz, J. S. Sabol, A. Gross, Z. Zhao, D. Rampe, L. Wang, F. Wirtz-Brugger, B. A. Harris and D. Macdonald, Bioorg. Med. Chem. Lett., 2005, 15, 435 CrossRef CAS; D. C. Braddock, J. M. Redmond, S. A. Hermitage and A. J. P. White, Adv. Synth. Catal., 2006, 348, 911 CrossRef CAS.
- R. S. Bon, C. Hong, M. J. Bouma, R. F. Schmitz, F. J. J. de Kanter, M. Lutz, A. L. Spek and R. V. A. Orru, Org. Lett., 2003, 5, 3759 CrossRef CAS; R. S. Bon, B. van Vliet, N. E. Sprenkels, R. F. Scmitz, F. J. J. de Kanter, C. V. Stevens, M. Swart, F. M. Bickelhaupt, M. B. Groen and R. V. A. Orru, J. Org. Chem., 2005, 70, 3542 CrossRef CAS; N. Elders, R. F. Schmitz, F. J. J. de Kanter, E. Ruijter, M. B. Groen and R. V. A. Orru, J. Org. Chem., 2007, 72, 6135 CrossRef CAS.
- Isocyanoacetate 1 was synthesised from p-chlorophenylglycine according to established procedures in 75% yield over 3 steps. See ref. 6 and the Experimental section for details.
- L. S. Liebeskind and J. Srogl, J. Am. Chem. Soc., 2000, 122, 11260 CrossRef CAS; L. S. Liebeskind and J. Srogl, Org. Lett., 2002, 4, 979 CrossRef CAS.
- C. L. Kusturin, L. S. Liebeskind, H. Rahman, K. Sample, B. Schweitzer, J. Srogl and W. L. Neumann, Org. Lett., 2003, 5, 4349 CrossRef CAS.
- A. Lengar and C. O. Kappe, Org. Lett., 2004, 6, 771 CrossRef CAS.
- D. W. Karkhanis and L. Field, Phosphorus, Sulfur Relat. Elem., 1985, 22, 49 Search PubMed; C. Marshall, M. F. Ward and W. T. A. Harrison, J. Organomet. Chem., 2005, 690, 3970 CrossRef CAS.
- For a review on the applications of this reagent, see: M. P. Cava and M. I. Levinson, Tetrahedron, 1985, 41, 5061 Search PubMed.
- T. Kaiya, S. Aoyama and K. Kohda, Bioorg. Med. Chem. Lett., 1998, 8, 625 CrossRef CAS.
- For the cis-diastereomer, the reactions gave comparable yields (see the Experimental section for details).
|
| This journal is © The Royal Society of Chemistry 2008 |
Click here to see how this site uses Cookies. View our privacy policy here.