Phenothiazine as a redox-active DNA base substitute: comparison with phenothiazine-modified uridine†
Clemens
Wagner
and
Hans-Achim
Wagenknecht
*
University of Regensburg, Institute for Organic Chemistry, D-93040, Regensburg, Germany. E-mail: achim wagenknecht@chemie.uni-regensburg.de; Fax: +49-941-943-4617; Tel: +49-941-943-4802
First published on 31st October 2007
Abstract
Phenothiazine can be incorporated as a redox-active probe into DNA in two conceptually different ways: the non-nucleosidic DNA base surrogate exhibits similar properties to 10-methylphenothiazine but with no preferential base-pairing properties, whereas the phenothiazine-modified uridine has different optical and electrochemical properties, but exhibits preferred Watson–Crick base pairing with adenine.
Phenothiazine (Pz) is a low-potential reductant (Pz+˙/Pz = 0.7–0.8 vs. SCE) that possesses a spectroscopically well-characterized one-electron oxidized radical (Pz+˙ at 510 nm).1 Thus, Pz represents a promising redox-active probe for DNA. In fact, Kawai, Majima and coworkers applied Pz covalently attached to the 5′-end of oligonucleotides as a charge acceptor for time-resolved measurements of hole transfer in DNA.2 Grinstaff and coworkers incorporated Pz at the 5′-terminal position,3 as a C-nucleoside,4 or attached to the 8-position of guanine.5 Recently, we used 5-(10-methylphenothiazin-3-yl)-2′-deoxyuridine (Pz-dU) as a photoinducable charge donor in order to investigate DNA-mediated electron transfer processes.6 Herein we describe the facile synthesis of a novel Pz DNA base substitute, its synthetic incorporation into oligonucleotides and the comparison of its opto-electronic properties with Pz-modified uridine (Pz-dU).
The 2′-deoxyribofuranoside was replaced by (S)-2-amino-1,3-propanediol as a flexible acyclic linker system (Scheme 1) that provides a high chemical stability during the preparation and allows the chromophore to intercalate perfectly. It has been applied similarly for DNA base substitution by other chromophores, e.g. ethidium,7indole8 and perylene bisimide.9 The Pz derivative 1 can be synthesized as a starting material according to published procedures10 and used subsequently for a nucleophilic substitution with the dimethoxytrityl (DMT)-protected (S)-3-amino-1,2-propanediol (2),7 yielding the Pz derivative 3. For the electrochemical characterization of 4, the DMT group of 3 was cleaved off an analytical sample. After protection of the NH function by a trifluoroacetyl group in 5, the phosphoramidite 6 can be applied for the automated preparation of modified oligonucleotides.
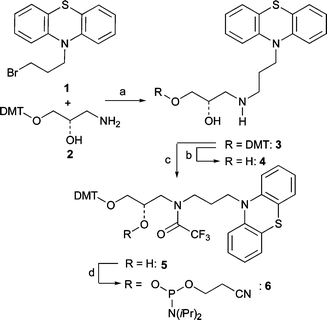 | ||
| Scheme 1 Synthesis of DNA building block 6. Reagents and conditions: a) 2 (2 equiv.), iPr2NEt (4 equiv.), DMF, r.t., 10 d; 54%; b) Cl2CHCOOH, CH2Cl2, r.t., 30 min; 95%; c) Me3SiCl (1 equiv.), pyridine (6 equiv.), CH2Cl2, 0 °C, 3 h; (F3CCO)2O (2 equiv.), 0 °C, 20 min, r.t., 10 min; 1 M (nBu)4NF in THF (1 equiv.), 30 min, r.t.; 95%; d) iPr2NEt (4 equiv.), 2-(cyanoethyl)diisopropylchlorophosphoramidite (2 equiv.), CH2Cl2, r.t., 45 min; 95%. | ||
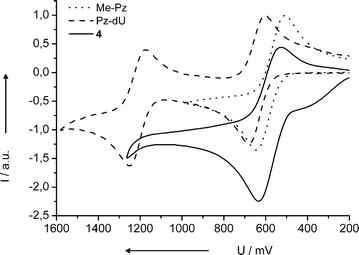
We characterized the nucleoside substitute 4 by optical spectroscopy and electrochemistry methods, each in comparison with the commercially available 10-methylphenothiazine (Me-Pz) and the previously synthesized Pz-dU. Not surprisingly, the absorption and fluorescence properties of 4 are similar to Me-Pz (see Fig. S1† ). In contrast, Pz-dU exhibits an exciplex-type fluorescence due to the strong electronic coupling between the uridine and Pz via a single C–C bond.13,14 The electrochemical potentials were measured by cyclic voltammetry vs.ferrocene (Fc+˙/Fc) and transferred into potentials vs. NHE using a conversion constant of +0.63 V (Fig. 1).11 The Pz derivative 4 as a non-nucleosidic DNA base substitute has a potential of E1/2 = 0.81 V that is identical to that of Me-Pz. The corresponding potential of Pz-dU is shifted by 60 mV to 0.87 V, indicating the small electron-withdrawing effect of the covalently attached uridine moiety. The second potential at 1.44 V can be assigned to the oxidation of the uridine moiety. Comparison with the potential of the structurally similar nucleoside thymine (T+˙/T) at 1.90 V12 shows the strong electron-donating character of the Pz chromophore, which shifts the potential by –0.56 V.
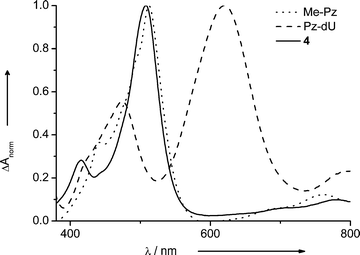
Spectroelectrochemical characterization under oxidizing conditions revealed that the radical cation of 4 absorbs at 508 nm, similar to that of Me-Pz at 512 nm (Fig. 2).1 In contrast, the absorption of the radical cation of Pz-dU is significantly red-shifted to 620 nm. The covalent attachment of the Pz chromophore to uridine changes not only the redox potentials of the Pz moiety slightly, but also the spectro-optical properties significantly.
Using the DNA building block 5, we synthesized a range of duplexes, DNA1a–DNA1e (Scheme 2). Using our previously published synthetic protocol,6 a second set was synthesized, DNA2a–DNA2e, bearing the Pz-modified uridine (Pz-dU). In both DNA sets, the base opposite to Pz modification site was varied, including the abasic site analog S. Representatively for each set, the matched duplexes DNA1a and DNA2a were investigated by absorption (Fig. S2), fluorescence (Fig. S3), and CD spectroscopy (Fig. S4). In these DNA duplexes, the fluorescence of Pz and Pz-dU is quenched significantly. The quantum yields are remarkably low, Φ = 0.1% (DNA1a) and Φ = 0.3% (DNA2a). This result underscores the redox-activity of the Pz chromophore in DNA. As we know from our previous studies, photoexcitation of Pz in DNA initiates very efficient electron hopping via thymines and cytosines as electron carriers.
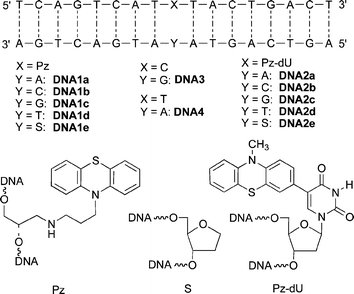 | ||
| Scheme 2 Sequences of DNA1a–DNA1e and DNA2a–DNA2e. | ||
Thermal dehybridization experiments were performed with all duplexes (Table 1). The Tm values of the two duplex sets exhibit a remarkable difference between the non-nucleosidic base surrogate Pz and the modified nucleosides Pz-dU. In the Pz-duplex set DNA1a–DNA1d the Tm values do not reveal any preferential base pairing. In contrast, the correctly matched Pz-dU-modified duplex DNA1a shows a higher Tm value compared to the others (DNA2b–DNA2d), indicating the preferential base pairing with the correct counterbase adenine. Only the presence of the abasic site analog S seems to enhance the hydrophobic interactions of the Pz chromophore inside the DNA, leading to a stabilization of 6 °C.
In conclusion, both Pz modifications presented herein can be used as redox-active probes in DNA for electrochemical analytics or the investigation of charge transfer in DNA. The non-nucleosidic Pz derivative 4 as a DNA base surrogate behaves similarly to the Me-Pz chromophore, but shows no selective base-pairing in DNA, whereas Pz-dU has altered optical and electrochemical properties, but exhibits preferred Watson–Crick base pairing with adenine.
Notes and references
- S. L. Mecklenburg, B. M. Peek, J. R. Schoonover, D. G. McCafferty, C. G. Wall, B. W. Erickson and T. J. Meyer, J. Am. Chem. Soc., 1993, 115, 5479 CrossRef CAS; S. L. Mecklenburg, D. G. McCafferty, J. R. Schoonover, B. M. Peek, B. W. Erickson and T. J. Meyer, Inorg. Chem., 1994, 33, 2974 CrossRef CAS; D. G. McCafferty, D. A. Friesen, E. Danielson, C. G. Wall, M. J. Saderholm, B. W. Erickson and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 8200 CrossRef CAS.
- T. Takada, K. Kawai, M. Fujitsuka and T. Majima, J. Am. Chem. Soc., 2006, 128, 11012 CrossRef CAS; K. Kawai, Y. Osakada, A. Sugimoto, M. Fujitsuka and T. Majima, Chem.–Eur. J., 2007, 13, 2386 CrossRef CAS.
- M. T. Tierny, M. Sykora, S. I. Khan and M. W. Grinstaff, J. Phys. Chem. B, 2000, 104, 7574 CrossRef.
- S. A. N. Hashmi, X. Hu, C. E. Immoos, S. J. Lee and M. W. Grinstaff, Org. Lett., 2002, 4, 4571 CrossRef CAS.
- M. T. Tierney and M. W. Grinstaff, Org. Lett., 2000, 2, 3413 CrossRef CAS.
- C. Wagner and H.-A. Wagenknecht, Chem.–Eur. J., 2005, 11, 1871 CrossRef CAS.
- R. Huber, N. Amann and H.-A. Wagenknecht, J. Org. Chem., 2004, 69, 744 CrossRef CAS; N. Amann, R. Huber and H.-A. Wagenknecht, Angew. Chem., Int. Ed., 2004, 43, 1845 CrossRef CAS.
- C. Wanninger and H.-A. Wagenknecht, Synlett, 2006, 2051 CAS.
- C. Wagner and H.-A. Wagenknecht, Org. Lett., 2006, 8, 4191 CrossRef CAS.
- K. G. Thomas, V. Biju, P. V. Kamat, M. V. George and D. M. Guldi, ChemPhysChem, 2003, 4, 1299–1307 CrossRef CAS.
- V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97–102 CrossRef.
- C. A. M. Seidel, A. Schulz and M. H. M. Sauer, J. Phys. Chem., 1996, 100, 5541 CrossRef CAS.
- E. Mayer, L. Valis, R. Huber, N. Amann and H.-A. Wagenknecht, Synthesis, 2003, 2335 CAS; L. Valis, E. Mayer-Enthart and H.-A. Wagenknecht, Bioorg. Med. Chem. Lett., 2006, 16, 3184 CrossRef CAS; C. Wanninger-Weiß, L. Valis, J. Barbaric and H.-A. Wagenknecht, Bioorg. Med. Chem., 2007 DOI:10.1016/j.bmc.2007.04.064.
- Z. R. Grabowski, K. Rotkiewicz and W. Rettig, Chem. Rev., 2003, 103, 3899 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details; UV/Vis, fluorescence and CD spectra. See DOI: 10.1039/b708904j |
| This journal is © The Royal Society of Chemistry 2008 |