A fresh look at natural tropolonoids
Ronald
Bentley
*
Department of Biological Sciences, University of Pittsburgh, Pittsburgh, PA 15260, USA
First published on 22nd November 2007
Abstract
Covering: 1945 to 2007
Tropones and tropolones have been of increasing interest in the past few years; approximately 200 such materials occur in nature. This article reviews the distribution of these materials, the biosynthetic pathways for their production and their biological activities. There are 154 references.
 Ronald Bentley Ronald Bentley | Ronald Bentley worked on penicillin chemistry at Imperial College, London, obtaining his Ph.D. in 1945. After a Commonwealth Fund Fellowship for work on isotope tracers with David Rittenberg (College of Physicians and Surgeons, Columbia University, New York), he set up a mass spectrometer unit at the National Institute of Medical Research, London. Joining the University of Pittsburgh in 1953 he became Professor in 1960, studying the biosynthesis of various secondary metabolites (including pioneer studies on fungal tropolones), ubiquinone and vitamin K. An interest in stereochemistry led to a two-volume text, “Molecular Asymmetry in Biology”, published in 1969–1970. Retired at age 70 (1992) as Professor Emeritus he has continued writing and was an editor for The Oxford Dictionary of Biochemistry and Molecular Biology (1997, revised 2000). He received the Waksman Outstanding Educator Award from the Society for Industrial Microbiology in 2002. He takes pride in more than sixty years of membership of the RSC. |
1 Introduction
Harold Raistrick, F.R.S., 1890–1971, was a brilliant natural product chemist; he and his colleagues isolated more than 200 fungal metabolites and in most cases determined their chemical structures.1 Three of these fungal products, however, defeated him—puberulonic acid, C9H4O7, and puberulic acid, C8H6O6, isolated from Penicillium puberulum and some other fungi in 1932,2 and stipitatic acid, C8H6O5, isolated from Penicillium stipitatum in 1942.3 These materials appeared to resemble aromatic hydroxy acids, but although much information was obtained, especially for stipitatic acid, structures could not be assigned. Perhaps Raistrick's thoughts were elsewhere. In 1932 he was also investigating penicillin and in 1941 his department at The London School of Hygiene and Tropical Medicine had been bombed.In 1945, Michael Dewar made a brilliant leap of imagination and proposed a seven-membered ring structure for stipitatic acid based on 2-hydroxy-2,4,6-cycloheptatrien-1-one.4 This latter compound, 1 R1 = R2 = H, named tropolone, showed certain aromatic properties; Dewar had, in fact, unleashed the new field of non-benzenoid aromatic compounds. In addition, Dewar suggested a tropolone structure for colchicine (see later).5 The impact of Dewar's proposal was rapid and impressive. A review, “The Tropolones ” published in 1951, contained 80 citations;6 only 4 years later, Pauson's comprehensive “Tropones and Tropolones ” required 492 citations.7
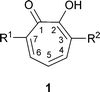
Another theme in the tropolone story concerns the heartwood of Western red cedar. This wood contains materials that are antifungal and are responsible for its durability. Two materials, C10H12O2, had been isolated in 1933 and in 1946 were also found to be present in Swedish-grown Thuja plicata. Extensive investigations by Erdtman's group led to the isolation and structure determination of three, isomeric materials, termed thujaplicins.8 They were clearly shown to be isopropyl tropolones , e.g., α-thujaplicin 1 R1 = H, R2 = isopropyl.
Dewar and Erdtman were working towards the end of World War II, a time when normal scientific communications were often difficult, if not impossible. Such difficulties had impinged on the life of a Japanese chemist, Tetsuo Nozoe born 1902, who died in 1996 just before his 94th birthday.9 In 1926, he had accepted a position in Taipei, Formosa (Taiwan), at that time in Japanese hands and in 1929 he became an Assistant Professor in the Faculty of Science and Agriculture of the newly established Taihoku Imperial University. In early work he studied the components of saponins, wool wax, and essential oils from leaves of taiwanhinoki (Chamaecyparis taiwanensis) and other trees. A dark-red wood pigment, “hinokitin” had been obtained in 1926 from oil of the heartwood of taiwanhinoki. In 1936, Nozoe treated an ether solution of hinokitin with aqueous alkali, obtaining a precipitate of ferric hydroxide and an enolic compound, C10H12O2. He named the latter compound, hinokitiol, and showed that hinokitin was an iron complex of hinokitiol, (C10H11O2)3Fe.
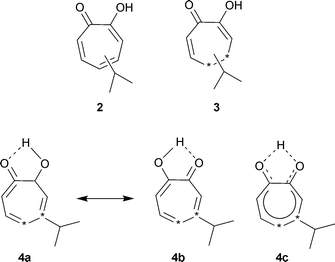
Assigning a structure to hinokitiol proved difficult; one possibility was “the enol form of the seven-membered α-diketone”, 2. Since common knowledge at that time was that “no such compound could exist in nature in a stable form” this structure was temporarily abandoned.10 Further examination of hinokitiol and degradative studies led him to reconsider the seven-membered ring. A tentative tropolone structure, presented in 1940, was actually for a dihydro form, C10H14O23—there had been analytical problems. After the hinokitiol structure had been reconfirmed as C10H12O2, Nozoe obtained a copy of Pauling's, “The Nature of the Chemical Bond” and under its influence considered the seven-membered structure 4a, 4b, 4c as “a new type of aromatic compound stabilized by resonance”. Nozoe states that he presented this “tropolonoid” formula at the Formosa Branch of the Society of the Chemical Industry, in 1941, but that it was received with skepticism.10 Unhappily, there is apparently no published record of this presentation.
With the end of World War II, the Taihoku Imperial University became the National Taiwan University (Republic of China). Nozoe was able to remain there and in 1946 resumed his hinokitiol work, now with a large research group. Substitution reactions convinced him that hinokitiol was indeed an entirely new type of aromatic compound “quite different from the benzene series”. In 1947, his colleague, Katsura, described the isopropyl tropolone structure for hinokitiol at a meeting of the Formosan Medical Association and published it in medical journals. In 1948, Nozoe was repatriated to Japan to become a professor at Tohoku Imperial University in Sendai. There he finally learned of Erdtman's thujaplicin work and of Dewar's 1945 proposal. An account of his work in Formosa, given as lectures in Osaka and Tokyo, was published in Japanese in 1949, and in English translation in 1950.11 Erdtman had suspected the identity of hinokitiol and β-thujaplicin and this identity was directly confirmed by Nozoe in reports of 1949 and 1950, and by Erdtman.10,11
Is there good reason to acknowledge that the first tropolone structure was presented by Nozoe? His early work in Formosa was hindered by the unavailability after 1937 of European chemical journals and texts; by 1941 American materials could not be obtained. His publications in Japanese and in not well-known journals were similarly unavailable to European and American chemists during World War II. Even in 1951, neither his original papers nor abstracts were available to Cook and Loudon for their review.6 His later work from about 1950 slowly received wide attention and acclaim. However, the details of the early Formosa work remained unclear until the publication of his autobiography in 1991.10 This work was originally hand written in Japanese but was issued in translation. Another valuable source of information is the article, “In Memoriam”, written by former students.9 The account given here is largely based on these works.
There is no doubt that Nozoe independently deduced a correct tropolone structure for hinokitiol (β-thujaplicin) and that this was published by Katsura in 1947–1948, well before Nozoe was aware of the work of Dewar and Erdtman. As previously noted, a seven-membered ring structure 3 was published for hinokitiol at a time when the composition was believed to be C10H14O2. Although it is regrettable that there is apparently no written confirmation, Nozoe states that he first presented the correct tropolone structure for hinokitiol at a meeting in Formosa in 1941. Since there is no reason to doubt Nozoe's integrity, it appears that he should be credited as the first to describe a natural tropolone (not then so named) four years prior to Dewar's seminal paper. Nozoe was a highly dedicated chemist, publishing over his long lifetime 402 research papers and 46 other articles (book chapters, reviews, etc.). Had the war not interrupted his research (and note that 1941 was the year of the attack on Pearl Harbor) there is little doubt that he would have published the correct structure for hinokitiol in the usual literature, perhaps as early as 1943. This reappraisal of Nozoe's role in no way detracts from the achievement of Dewar; in fact, Nozoe was reluctant to claim priority in later years. Thus, in an important paper in Nature in 1951 he wrote of “compounds having a tropolone nucleus…as initially proposed by Dewar”.12
Although in 1955 tropolones were described as relatively scarce in nature,7 it can be estimated that more than 200 are now known. The structures of natural tropones and tropolones cover a wide range from tropolone itself to complex multicyclic products and some materials regarded as alkaloids . There is no generally agreed classification system. Tropolonoids are commonly found in fungi and plants; a few have been isolated from bacteria.
2 Natural tropolones and tropones
2.1 Tropolone and hydroxytropolones
Tropolone itself, 1 R1 = R2 = H, has been isolated from Pseudomonas ATCC 31099,13 and from another bacterium, P. plantarii ATCC 43733, which causes seedling blight of rice.147-Hydroxytropolone1 R1 = OH, R2 = H, was isolated from a new actinomycete, Streptomyces neyagawaensis15 and 3,7-dihydroxytropolone1 R1 = R2 = OH from Streptomyces tropolofaciens.16 The compound chamaecin, from the tree Chamaecyparis taiwanensis, was originally thought to be 3- or 7-alkoxy-4-isopropyltropolone but is now known to be 2-hydroxy-4-isopropylbenzaldehyde (macropone).17 However, 3-methoxytropolone1 R1 = H, R2 = OMe, was isolated from the aerobic bacterium, Streptoverticillium hadanonense KY 11449, as an inhibitor of lipoxygenase, MY3-469.182.2 Simple bicyclic tropolones
Cordytropolone 5 was isolated from a fungal insect pathogen, Cordyceps sp. BCC 1681,19 sepedonin 6 from the fungus, Sepedonium chrysospermum,20 and manicol 7, a sesquiterpenoid tropolone from a Guyanan tree, Dalacia guanensis21 (for the related manicoline A see “Tropones ”, Section 2.3). The sepedonin paper20 is of interest as the last, number 117, in Raistrick's classic series “Studies in the Biochemistry of Microorganisms”—some 33 years after the isolation of puberulic acid described in paper 23.2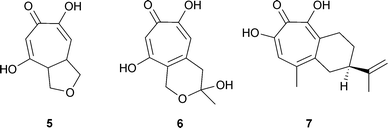
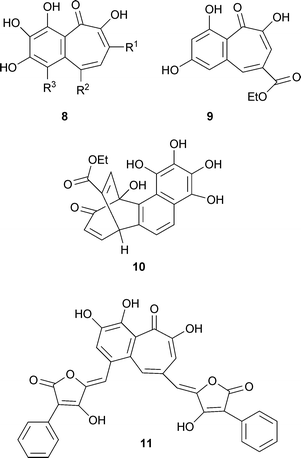
When the oak Quercus sessiliflora is infested with the gall-producing insect Dryophanta taschenbergii (Hymenoptera), it produces “galls”—abnormal swellings or growths. This phenomenon is observed in many other plants in response to the activities of various fungi, insects and mites. The Q. sessiliflora galls contain a red-pigmented diglucoside, of which the aglycone is a material named purpurogallin. This material is also readily obtained synthetically by oxidation of pyrogallol. The structure of purpurogallin remained enigmatic until Barltrop and Nicholson in 1948 proposed correctly that it was a benzotropolone.22 These authors noted the close structural relationships with stipitatic acid and colchicine. This deduction is yet one more instance of the influence of Dewar's proposal. Purpurogallin8 R1 = R2 = R3 = H, occurs widely. Purpurogallin carboxylic acid, 8 R1 = R2 = H, R3 = COOH, a biosynthetic intermediate (see later) is less well known but has recently been reported from “teapigment”. More complex structures that contain a purpurogallin unit are discussed later.
Fomentariol
8 R1 = R3 = –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CH2OH, R2 = H from Fomes fomentarius (a fungus) is a purpurogallin structure modified by two hydroxy-3-propenyl units, one each on the phenyl and tropolone moieties.23 Another structure related to that of purpurogallin is goupiolone A 9 isolated from the aerial parts of Goupia glabra, a plant of the Amazon region of Peru. It is accompanied by a more complex material, goupiolone B 10, apparently derived from an unusual Diels–Alder reaction between a tropolone unit and a benzyne intermediate formed from a naphthalenoid unit with four hydroxyl groups.24 Aurantricholone 11 from the fungus Tricholoma aurantium is another benzotropolone of the purpurogallin type, but having only two hydroxyl groups on the benzene ring.25
CH–CH2OH, R2 = H from Fomes fomentarius (a fungus) is a purpurogallin structure modified by two hydroxy-3-propenyl units, one each on the phenyl and tropolone moieties.23 Another structure related to that of purpurogallin is goupiolone A 9 isolated from the aerial parts of Goupia glabra, a plant of the Amazon region of Peru. It is accompanied by a more complex material, goupiolone B 10, apparently derived from an unusual Diels–Alder reaction between a tropolone unit and a benzyne intermediate formed from a naphthalenoid unit with four hydroxyl groups.24 Aurantricholone 11 from the fungus Tricholoma aurantium is another benzotropolone of the purpurogallin type, but having only two hydroxyl groups on the benzene ring.25
2.3 Tropones
Only a few naturally occurring tropones are known. Two odouriferous dihydrotropones, 12 and 13, were isolated from seaweed (Dictyopteris plagiogramma and D. australis)26 and are apparently metabolites of the cycloheptadiene dictyopterene C′. Nezukone 14 was isolated from wood of Thuja standishii in 1964.27 An aminotropolone, manicoline A 15 was isolated from the same tree as was manicol 7.28Diterpenoid tropones are considered separately.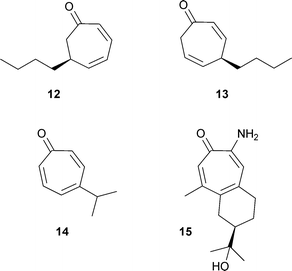
2.4 Fungal hydroxytropolone acids
The fungus producing stipitatic acid was originally named as Penicillium stipitatum.3 It is, in fact, the imperfect, asexual stage (anamorph) of Talaromyces stipitatus and this name will be used here. In addition to stipitatic acid16 R1 = R2 = R3 = H, T. stipitatus also produces stipitatonic acid 16 R1 = R3 = H, R2 = COOH as an anhydride with COOH at C-4,29stipitalide17 R = H,30 and stipitaldehydic acid17 R = OH.31 Although the ethyl ester of stipitatic acid has been reported as a metabolite 32 it is likely to have been an artifact formed by reaction of stipitatonic acid and ethanol.33 T. stipitatus CBS 375.48 also produces non-tropolone antitumor materials, of which one is duclauxin.34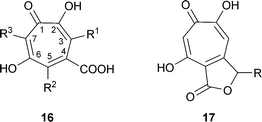
A number of fungi (e.g., P. aurantio-virens, P. cyclopium-viridicatum, P. johannioli, P. puberulum) produce puberulonic acid 16 R1 = COOH as anhydride with COOH at C-4, R2 = H, R3 = OH, and puberulic acid 16 R1 = R2 = H, R3 = OH, but no other tropolone compounds.2 The dicarboxylic acids, stipitatonic acid and puberulonic acid, are actually isolated as the anhydrides .
2.5 Thujaplicins and related compounds
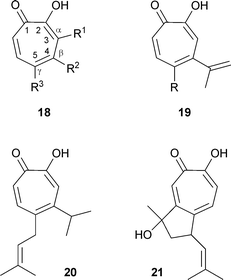
Tropolone compounds of the thujaplicin series occur widely in the family of Cupressaceae (evergreen shrubs and trees);35 a classification of the Order Cupressalles and a listing of metabolites is available.36 The classic names of hinokitiol and thujaplicin derive from the trees taiwanhinoki, Chamaecyparis taiwanensis, and Japanese hinoki, Chamaecyparis obtusa, and from Thuja plicata. While the numbering for the tropolone ring system, shown in 18 is preferred7 the alternative system using Greek letters has also received much use, particularly for many of the thujaplicins from members of the Cupressaceae. In this usage, C-3 is designated as α, C-4 as β and C-5 as γ (see 18). The three commonly found thujaplicins are, therefore, as follows: 18 R1 = isopropyl, R2 = R3 = H, α-thujaplicin; 18 R2 = isopropyl, R1 = R3 = H, β-thujaplicin; 18 R3 = isopropyl, R1 = R2 = H, γ-thujaplicin. In addition to the isopropyl types, the simple 5-ethyltropolone 18 R1 = R2 = H, R3 = Et has been isolated from Calocedrus formosana (Taiwan incense tree, formerly Libocedrus formosana).37 In several materials, there are unsaturated side chains:36 e.g., β-dolabrin19 R = H from Thujopsis dolabrata, procerin19 R = CH2CHC(CH3)2 from Juniperus procera (note: there is also a steroid with this name), nootkatin 20 from Chamaecyparis nootkatensis and chanootin 21 from the same tree (this structure is of interest for the presence of two chiral carbon atoms, although the natural material crystallizes as a racemate).
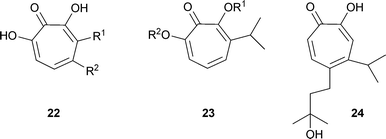
Another structural variation in this series is the presence of further OH groups on the cycloheptatriene ring.36 Examples are 7-hydroxy-3-isopropyltropolone (α-thujaplicinol) 22 R1 = CH(CH3)2, R2 = H from Cupressus pygmaea; 7-hydroxy-4-isopropyltropolone (β-thujaplicinol) 22 R1 = H, R2 = CH(CH3)2; 7-hydroxy-3-isopropenyltropolone (α-dolabrinol) 22 R1 = C(CH3)CH2, R2 = H from C. pygmaea. Related structures with methoxy groups are pygmaein23 R1 = CH3, R2 = H and isopygmaein23 R1 = H, R2 = CH3.38 Compounds with an oxygen atom not directly linked to the cycloheptatriene system are nootkatinol 24 from Chamaecyparis nootkatensis36 and 4-acetyltropolone, a minor component from Thujopsis dolabrata 18 R1 = R3 = H, R2 = COMe.39 β-Thujaplicin-producing cell suspension cultures of Cupressus lusitanica (see later) produce the methyl ether of β-thujaplicin (2-methoxy-6-[methylethyl]cyclohepta-2,4,6-trien-1-one) under certain conditions.40
2.6 Tropones and tropolones containing sulfur
Four naturally occurring tropone derivatives containing two atoms of sulfur have been described: thiotropocin 25 from the bacterium Pseudomonas CB-104 and Caulobacter PK 654;41 troposulfenin 26;42 tropodithietic acid 27 and hydroxytropodithietic acid 28 from marine bacteria of the Roseobacter clade.43 Three of these materials 25–27 are unusual structural isomers and may readily rearrange. More specifically, they can be regarded as tautomers and may possess dynamic behavior.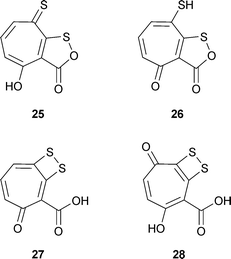
Troposulfenin 26 is only poorly characterized and in any case is a simple prototropic tautomer of thiotropocin 25. It has been implied indirectly that these two materials are identical.41c,41d Moreover, Laatsch has cited unpublished observations of Liang indicating an identity between thiotropocin 25 and tropodithietic acid 27.44 This conclusion is based largely on an X-ray crystal structure for 27 and, also, on a reinterpretation of the 13C NMR data for these two compounds (A. Zeeck, personal communication to RB). A possible source of difference between 25 and 27 is that while the former gave a bromobenzyl derivative the latter did not. However, shortage of material may have prevented the development of appropriate experimental conditions (A. Zeeck, personal communication to RB).
A compound found in Burkholderia cenocepacia (a Gram-negative bacterium formerly Pseudomonas cepacia, ATCC 17759) contains two tropolone rings linked by a single sulfur atom at the 7 and 7′ positions—R–S–R, R = tropolone .45
2.7 Dimeric bicyclic structures, two tropolone units
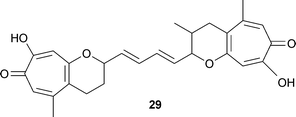
Fusarium moniliforme, a plant pathogenic fungus, forms a number of toxins including the tropolonoid, fusariocin C 29.46 The two tropolone -pyran units are linked by an unsaturated chain of four carbon atoms, –CHCH–CHCH–. It is of interest that these two termini show different methylation patterns; one pyran unit is methylated, the other not.
2.8 Tricyclic structures, one tropolone unit
Malettinins A–C are polyketide metabolites from an unidentified, non-sporulating fungal colonist, Mycelia sterilia, NRRL 29110, of Hypoxylon stromata (found on dead Aspen logs). For malettinin A 30 the structure determined by the usual methods was confirmed by a single crystal X-ray diffraction analysis .47 Malettinin B 31 is a partially reduced analogue of 30 and malettinin C is isomeric at C-9 with 31.48 A further component from this organism is malettinin D, a non-tropolonoid material possibly derived by an oxidative cleavage of the tropolone ring. Chaetospiron 32, a secondary metabolite from the fungus Chaetomium sp. BS 6556, is a dimeric structure containing two units related to maletinnin A.49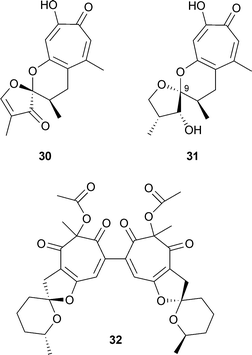
2.9 Tricyclic structures, two tropolone units
The only structure of this type is utahin 33, a ditropolonofuran isolated from the tree Juniperus utahensis.50 Note, however, that chaetospiron 32 is also a ditropolonoid material.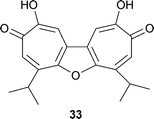
2.10 Tetracyclic structures, one tropolone unit
Two unnamed tropolones 34 R = H and 34 R = OH, were isolated as co-metabolites of xenovulene A in the fungal plant pathogen Acremonium strictum.51 A key feature of these molecules is the fusion of a tropolone unit to a humulene-derived, eleven-membered ring; this feature also appears in other structures (see below). These two tropolones are likely precursors to the xenovulenes, and biosynthesis of the tropolones via 3-methylorsellinic acid (see later) was postulated. More recent molecular and genetic studies have led to the isolation and heterologous expression of a polyketide synthase gene. It appears that the actual precursor to the tropolones and xenovulenes is 3-methyl-orcinaldehyde. A closely related tricyclic structure is that of epolone B 35 isolated from a fungus culture OS-F 69284 (ATCC 74390).52 For epolone A, see below.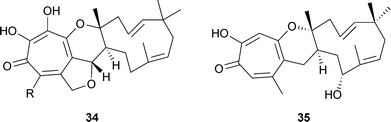
2.11 Pentacyclic structures, one or two tropolone units
Two compounds of this type are pycnidione and eupenifeldin. Pycnidione 36 was isolated from Phoma sp. Strain MF 5726 and, along with epolone B, from fungus OS-F 69284.53 It contains two tropolone units attached to a humulene-derived, eleven-membered ring. Both of these fusions are in the trans sense in 36. The cytotoxic material, eupenifeldin, isolated from the fungus, Eupenicillium brefeldianum, is a stereoisomer in which the linkage of unit 2 in 36 is cis.54 Epolone A 37 is also a pentacyclic structure based on a humulene-derived ring; it has both a phenolic and a tropolone ring.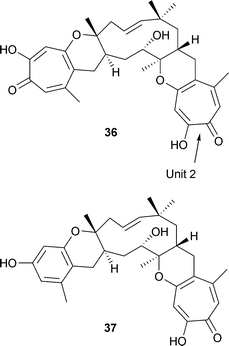
2.12 Hexacyclic structures, one tropone unit
A diterpenoid tropone , hainanolidol 38 was isolated from the plants Cephalotaxus hainanensis and C. fortunei. A further metabolite , harringtonolide (also termed hainanolide) 39 was present in C. harringtonia, a yew;55 unlike haianolidol the latter was a plant growth inhibitor and also had antineoplastic and antiviral activities. Moreover, two structurally related lactones , fortunolide A 40 and fortunolide B 41 were also present in C. fortunei.56 If the published structures (as given here) are correct, the fortunolide skeleton appears to have stereochemistry not congruent with that of hainanolidol.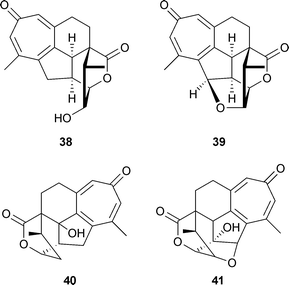
2.13 Nitrogen-containing tropolones
The tropical vines Abuta imene, A. refescens and A. grandifola contain isoquinoline alkaloids with a tropolone unit that have been termed “tropoloisoquinolines”.57 They include grandirubrine42 R1 = R2 = H, R3 = OMe, imerubrine42 R1 = Me, R2 = H, R3 = OMe, isoimerubrine42 R1 = H, R2 = OMe, R3 = OMe, pareirubrine A42 R1 = R3 = H, R2 = OMe, pareirubrine B42 R1 = R2 = R3 = H.58 A further, structurally related material, pareitropone 43 is, as the name implies, a tropone .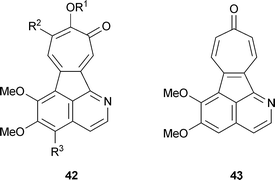
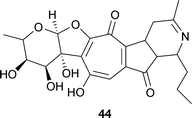
Streptomyces echinoruber sp. nov. produces several pigments, of which the most abundant is rubrulone 44.59 The structure contains one ring containing five carbons and one nitrogen.
2.14 Colchicine and related compounds
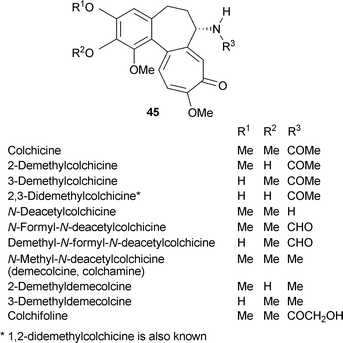
In view of the large number of metabolites, the colchicine group will be treated as a separate classification of nitrogen-containing tropolones . The autumn crocus (meadow saffron, Colchicum autumnale) has been used medicinally since antiquity, often for treatment of gout. This interesting, small, herbaceous perennial typically flowers in autumn after the leaves have disappeared. The major pharmacologically active component, colchicine, was isolated as long ago as 1820, and is now a much studied material. Although the autumn crocus is characteristically linked with the occurrence of colchicine, this alkaloid and related compounds are found in other Colchicum species as well as in many other plants of the Lilliaceae (e.g., Androcymbium, Gloriosa, Merendera, Sandersonia). Colchicine occurs in at least 37 plants.Colchicine, a tricyclic alkaloid , contains a trimethoxyphenyl ring linked to a seven-membered (cycloheptane) ring carrying an acetamide unit, that is itself linked to a tropolone moiety. The correct structure was finally proposed by Dewar in 1945.5 There are many possibilities for variation in this general structural type, and indeed, a large number of colchicine-related materials occur naturally.60 Only a selection of these minor alkaloids can be given here but there are several, more comprehensive treatments.61–63 The chemical synthesis of colchicine has attracted much interest and even today presents a challenge.64 This major topic will not be considered here.
Commonly found variations on the basic colchicine structure 45 R1 = R2 = Me, R3 = COMe, involve methylation and the amide function as listed below:
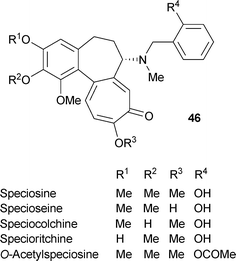
Several compounds containing a benzyl unit on the nitrogen atom are known. A typical structure is that of speciosamine46 R1 = R2 = R3 = Me, R4 = H.
Other examples are as follows:
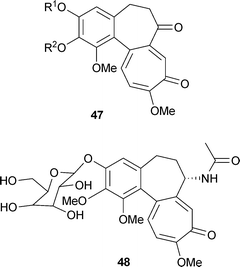
There is a small number of related, non-nitrogenous compounds, for instance in Colchicum ritchii:65colchicone47 R1 = R2 = Me; demethylcolchicone47 R1 = H, R2 = Me; cornigerone47 R1(R2) = –CH2–. In addition to the above compounds, colchicine can occur as the glucoside, colchicoside 48. The common form is 3-O-demethylcolchicine-3-O-β-D-glucopyranoside, but the α anomer has also been reported.66
3 Biosynthesis of fungal tropolones
In the early 1950s, the previously obscure natural product shikimic acid was identified as the precursor for the benzenoid amino acids, phenylalanine, tyrosine, and tryptophan, thus making the very unusual transformation from secondary to primary metabolite . As attention focused on the non-benzenoid aromatic compounds, the question of their biosynthesis became of interest. In those far off days, biosynthetic studies mostly involved the use of isotopic tracers, and these experiments relied almost exclusively on chemical degradations of labelled metabolites to locate the isotope. They were also limited to some extent by the availability of suitably labelled precursor molecules.In the earliest experimental work on tropolone biosynthesis, stipitatic acid16 R1 = R2 = R3 = H was used since it was readily available by growth of Talaromyces stipitatus (then termed Penicillium stipitatum) on simple media. Following its identification, stipitatonic acid 16 R1 = R3 = H, R2 = COOH also became available from this organism.29,67 In 1958, radioactivity from [1-14C]-D-glucose was shown to be readily incorporated into stipitatic acid and chemical degradations of the labelled metabolite indicated that the observed distribution of 14C was not that expected from a shikimate pathway.68 Utilization of acetate was also shown (see later). In similar work, the utilization of [1-14C]-D-glucose was confirmed and the molar specific activity of isolated stipitatic acid was five times higher than that of phenylalanine and tyrosine.69
For chemical degradation, C-9 and C-8 were readily obtained by decarboxylation of stipitatonic and stipitatic acids (Scheme 1). The known conversion of stipitatic acid to malonic and aconitic acids was intensively examined to define optimum conditions, as was the alkaline isomerization, stipitatic acid → 5-hydroxyisophthalic acid.70 Although it was initially assumed that in this isomerization both C-1 and C-2 of the tropolone ring would be extruded, it was shown that, in fact, only C-1 was involved.70–72
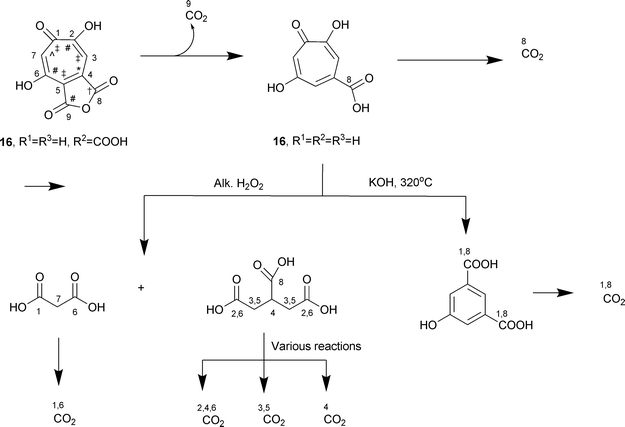 | ||
| Scheme 1 Chemical degradation of stipitatonic and stipitatic acids. For acetate, CH3 = † and COOH = *; for malonate, CH2 = ‡ and COOH = #; one carbon unit from S-adenosylmethionine = ⁁. | ||
Soon after the identification of stipitatic acid as a tropolone , a possible biosynthetic mechanism by a one-carbon addition to a polyhydroxy benzenoid structure, followed by a ring expansion, had been proposed by several authors; early citations are given in reference 73. In fact, substantial utilization of 14C labelled formate and formaldehyde was also observed in the early experiments. In addition, both [1-14C]- and [2-14C]-acetate were well utilized; [14C]-formate labelled only C-7 of stipitatic acid, and [1-14C]-acetate labelled predominantly C-4 and C-6. Utilization of [1-14C]-acetate was confirmed, but location of the isotope was not determined.69 The cyclization of an octulonic acid phosphate, CO2 fixation, as well as a C1 addition to a benzenoid structure were considered as possible biosynthetic mechanisms.69
Since the results pointed to a polyketide + C1 addition, followed by a ring expansion, malonic acid was also examined as a precursor; it, too, was well utilized, as expected for a polyketide process.73 In detailed studies, [1-14C]-acetate contributed four equally labelled carbons, C-2, -4, -6, -9, to stipitatonic acid and only three to stipitatic acid.74 On the other hand, [1,3-14C2]-malonate labelled C-2, -6 and -9 of stipitatonic acid to the extent of 30% each, but C-4 contained substantially less 14C, only 12%. In later work, where C-2 was unambiguously obtained, [1-14C]-acetate was incorporated to the extent of 25% at that position.72a From these results, it became clear that tropolone formation in T. stipitatus could be represented as follows: 1 acetyl-CoA + 3 malonyl-CoA + 1 C-1 unit → nine carbon tropolone + 3 CO2.
The early hypothesis for stipitatic acid formation had postulated the decarboxylation of a tropolone 3,4-dicarboxylic acid. When stipitatonic acid, a 4,5-dicarboxylic acid, was isolated (actually as an anhydride ) in 1959,29 this seemed a more likely possibility. In fact, an enzyme preparation from T. stipitatus, prepared by grinding mycelium with glass beads in phosphate buffer, was obtained75 and shown to decarboxylate stipitatonic acid to stipitatic acid (see later). These preparations also decarboxylated puberulonic acid suggesting that in both series, the dicarboxylic acids are the precursors to the monocarboxylic acids.
Important questions were the nature of the acceptor for the C1 unit and the identity of the transferred moiety. It was likely that the C1 unit was from the methyl group of methionine, actually in its biologically active form, S-adenosylmethionine. Evidence for this supposition was that tropolone formation in T. stipitatus was inhibited by ethionine, leading instead to the formation of the polyketides triacetic acid lactone 49 R1 = H, R2 = Me and tetraacetic acid lactone (6-[2-oxopropyl]-4-hydroxy-2-pyrone) 49 R1 = H, R2 = MeCOCH2, and a small amount of methyl triacetic acid lactone 49 R1 = R2 = Me.76 The latter had also been reported from cultures not treated with ethionine.77 The formation of these classical polyketide structures under ethionine inhibition considerably strengthened the polyketide + C1 pathway, and suggested that C1 addition, probably a methylation, took place prior to aromatization. The role of triacetic acid lactone in fungal metabolism has been discussed in detail.78
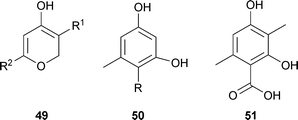
The ethionine-inhibited cultures also contained some orsellinic acid50 R = COOH and orcinol50 R = H, but both of these materials were apparently derived from tetraacetic acid lactone.76 Neither orsellinic acid nor orcylaldehyde were able to function as tropolone precursors in the fungal system. However, 3-methylorsellinic acid 51 was found to be a good precursor when [3-14CH3]- and [1-14COOH]-radiomers were studied as precursors in T. stipitatus. The [3-14CH3]-3-methylorsellinic acid labelled both stipitatonic and stipitatic acids but the carboxyl labelled material labelled only C-9 in stipitatonic acid.79 At this point, the biosynthetic pathway could be summarized as follows: 1 acetyl-CoA + 3 malonyl-CoA + AdoMet → 3-methylorsellinic acid → stipitatonic acid → stipitatic acid + CO2. In the initial condensation, 3 CO2 are lost.
The oxidative mechanism for the ring enlargement of 3-methylorsellinate was envisioned as either the action of a mono-oxygenase or a dioxygenase enzyme (Scheme 2).80 When T. stipitatus was grown in an 18O2 enriched atmosphere, stipitatonic acid and its derivatives gave no evidence for any M + 4 species in high resolution mass spectrometry. An M + 2 peak indicated the presence of a single atom of oxygen. This was the expected result for a mono-oxygenase mechanism both for the enlargement, benzenoid → tropolonoid, and the oxidation, CH3 → COOH (actually as an anhydride ). The arguments relating to possible oxygen exchange reactions are complex and the original text should be consulted. This work did not specifically locate 18O at C-1 of stipitatonic acid as indicated by 52.
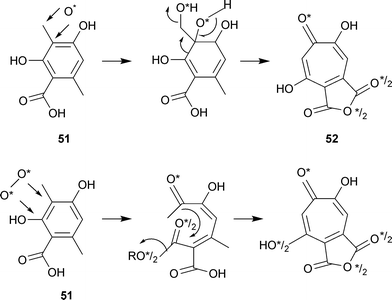 | ||
| Scheme 2 Ring enlargement of 3-methylorsellinic acid. Monooxygenase mechanism, top line. Dioxygenase mechanism, bottom line. | ||
Many years later, the specific incorporation of 18O2 into stipitatic acid was studied in shake cultures of T. stipitatus with the aid of 13C NMR; the 13C NMR spectrum of stipitatic acid having been completely assigned.81 There was an 18O-induced isotope shift for the C-1 carbonyl (0.04 ppm upfield of the 13C16O resonance) but no observable 18O shift associated with the signal at C-6. These results strongly supported the previously suggested monooxygenase enlargement mechanism.
The conversion of the C-6 methyl group, derived from 3-methylorsellinic acid, into the C-9 position of the anhydride ring has been investigated and two likely intermediates have been identified (Scheme 3). Stipitalide 17 R = H had been isolated from shake cultures of T. stipitatus.30 Moreover, [14CHO]-3-methylorcylaldehyde 53 was a more efficient precursor (ten fold) for stipitatonic acid than was [14COOH]-3-methylorsellinic acid 51.31 While no clear reason for this fact was available, it did make possible the identification of a further intermediate, stipitaldehydic acid 17 R = OH, as well as a trace of stipitalide 17 R = H. Labelled stipitaldehydic acid was shown to be further converted to stipitatonic acid. The late stages of stipitatonic acid biosynthesis appear to be as follows: 3-methylorsellinic acid (or 3-methylorcylaldehyde) → stipitalide → stipitaldehydic acid → stipitatonic acid.
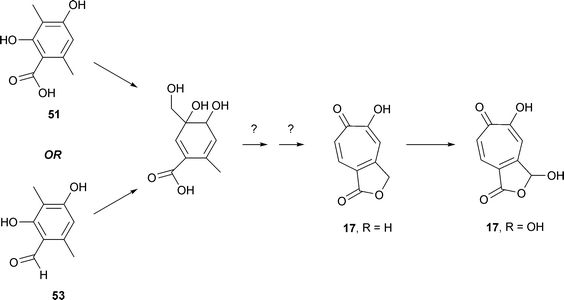 | ||
| Scheme 3 Formation of stipitalide, 17 R = H and stipitaldehydic acid, 17 R = OH. | ||
Environmental factors influencing tropolone formation in growing surface and replacement cultures of T. stipitatus 1006 were described in 195969 and work with fungi growing in continuous culture has also been reported.82
There are two confusing factors in considering the biosynthesis of the puberulonic/puberulic pair. In the first place, if the four contiguous oxygens are treated as OH groups (i. e., ignoring the single CO), both of these compounds have an axis of symmetry. For the dicarboxylic acid anhydride structure of puberulonic acid, the IR spectrum suggests that the solid material is 54a (anhydride of 6,7-dihydroxytropolone-3,4-dicarboxylic acid). However, by rotation of the molecular model about the axis shown in 54a, structure 54b is derived and, because of the tautomerism of CO → C–OH, can be written as the anhydride of 6,7-dihydroxytropolone-4,5-dicarboxylic acid 54c. This is the structure usually used in biosynthetic considerations because of the relationship with stipitatonic acid.
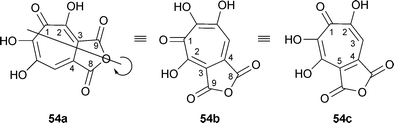
The second difficulty was that an initial study of tropolone biosynthesis in Penicillium aurantio-virens, using labelled acetates and formate, indicated that C-9 of puberulonic acid (numbering as in 54c) was not derived from [1-14C]acetate (as is the case for stipitatonic acid) but from an unidentified one carbon pool.83 However, in a later feeding of [1-14C]acetate to P. aurantio-virens, a distribution pattern of 14C at C-2, -4, -6, and -9, analogous to that found in stipitatonic acid, strongly implied that stipitatonic acid and puberulonic acid 54d are formed from the same set of precursors.84 Moreover, small amounts of stipitatonic acid and methyl triacetic acid lactone 49 R1 = R2 = Me were present in the P. aurantio-virens cultures; however, labelled stipitatonic acid was not converted to puberulonic acid (e.g., by hydroxylation). Surprisingly, neither [14COOH]-3-methylorsellinic acid nor the corresponding [14CHO]orcylaldehyde were utilized for puberulonic acid formation. Furthermore, [14COOH]-3-methylorsellinic acid did not contribute radioactivity to the stipitatonic acid formed by P. aurantio-virens as it does in T. stipitatus. Despite these difficulties, it seems likely that puberulonic/puberulic acid formation follows the pattern of the other fungal tropolones , requiring only the additional hydroxylation at C-7.
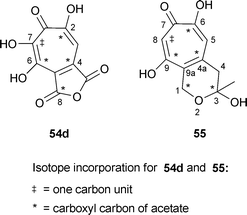
Sepedonin 6, formed by Sepedonium chrysospermum, contains two more carbon atoms than do stipitatonic and puberulonic acids. Using 13C-labelled substrates (acetates, formate) and determination of label positions by proton NMR spectroscopy, as well as experiments with 14C labelled precursors, indicated a labelling pattern 55 generally analogous to that of the other fungal tropolones .85 In particular, C-8 of sepedonin (sepedonin numbering as in 55) was labelled exclusively by formate. In this case, the conventional polyketide acceptor is CH3CO(CH2CO)3CH2CO–S–Enz rather than CH3CO(CH2CO)2CH2CO–S–Enz for the nine carbon fungal tropolones .
Although experimental validation is lacking, it seems likely that the malettinins 30, 31 and chaetospiron 32 are also typical polyketide metabolites.
4 Enzymes for formation of fungal tropolones
4.1 Stipitatonic acid decarboxylase
In 1959, cell free extracts of T. stipitatus NRRL 2104 were shown to decarboxylate stipitatonic acid to stipitatic acid.75 Although pure puberulonic acid was not available, the puberulonic acid in a mixture of puberulonic and puberulic acids was decarboxylated. These preparations were obtained by grinding surface grown mycelia with glass beads and pH 5.8 phosphate buffer using a chilled mortar and pestle. The production of CO2 from the ring-opened dicarboxylic acid form at pH 6.0 was followed manometrically. An acetone powder, stable for 3 months at −20 °C, was also obtained. There was a stoichiometric relationship between the amount of stipitatonic acid removed and the amounts of CO2 and stipitatic acid that were produced. One enzyme unit was defined as that amount catalyzing the formation of 1 µL of CO2 in 10 minutes; initial extracts had a specific activity of 1.5 units mg protein−1. An approximately 20-fold purification was obtained by ammonium sulfate precipitation at 53% saturation followed by calcium phosphate gel treatment. The pH optimum was 6.6. A metal ion requirement, probably for zinc, was shown by the fact that the decarboxylase activity was inhibited by Versene.The enzyme activity was specific for tropolone-4,5-dicarboxylic acids; tropolone 3,4-dicarboxylic acid, and its 5-, 6-, and 7-hydroxy derivatives, were not substrates. These materials, however, showed inhibitory properties, the highest inhibition, 60%, being obtained with tropolone-3,4-dicarboxylic acid and its 6-hydroxy derivative. The highest specific activity of the enzyme was found in the mycelia of 8-day old cultures, considerably before the maximum formation of the tropolone acids. The enzyme has been assigned the EC number, 4.1.1.60.
4.2 Stipitatic acid synthase
An enzyme extract, converting [1,3-14C2]malonyl-CoA, and less efficiently, diethyl [2-14C]malonate, H14COONa and [14CH3]-L-methionine, into stipitatic acid, has been obtained.86 Cultures of T. stipitatus were grown on a modified Czapek-Dox medium containing ammonium ion; 4–5 day old, colorless mycelia, were ground in a blender at pH 7. Following incubations, the crude cell-free extract was deproteinized and extracted with ether. Components in the extract were separated by paper chromatography (rechromatographed three times). Stipitatic acid formation was strongly inhibited by iodoacetamide and sodium monofluoroacetate. Since the isolated product was stipitatic acid, it is probable that the stipitatonic acid decarboxylase was also present. Further details on this system have not been reported.4.3 Methylase activity
An acetone powder preparation of T. stipitatus NRRL 2104 was obtained that converted [14CH3]-S-adenosylmethionine to stipitatic acid.87 In a large scale run, stipitatic acid (obtained by ether extraction) was isolated without addition of carrier material (i.e., the preparation was also a stipitatic acid synthase and contained stipitatonic acid decarboxylase). After five recrystallizations from water, the specific radioactivity remained constant. In several chromatographic systems, there was only a single peak of activity and this was clearly associated with stipitatic acid. There was a broad pH optimum between 6.9 and 7.9. Chemical degradation established that 14C was present only at C-7 of stipitatic acid.Exhaustively dialyzed enzyme preparations produced labelled stipitatic acid from [14CH3]-S-adenosylmethionine without the addition of any organic substrate; potential methyl group acceptors had little or no effect on the system. Radio-activity from labelled acetate was incorporated into the enzymatically active protein , suggesting the presence of an enzyme bound polyketide that could function as methyl group acceptor. The protein was estimated to have a molecular mass of 105![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000.
000.
It is likely that more detailed information on the enzymology of stipitatonic and stipitatic acid biosynthesis will become available as a result of the determination of the genome sequence for T. stipitatus. While not itself a pathogen, this organism is a close relative of Penicillium marneffei, an organism causing invasive disease in immunocompromised humans. The work involves groups headed by W. C. Nierman at TIGR and A. Andrianopoulos of the University of Melbourne (http://www.tigr.org/faculty/Nierman_William/index.shtml; http://www.uninews.unimelb.edu.au/articleid_3206.html). The sequence was said to be complete by May 2007 (http://fungalgenomes.org/blog/). With the genome sequence available, it should be possible to locate genes for polyketide synthases, decarboxylases and methylases and hence to facilitate the identification of enzymes.
5 The role of 3-methylorsellinic acid in fungal metabolism
Although a critical precursor for tropolone formation, 3-methylorsellinic acid has not been identified as a secondary metabolite in tropolone -producing fungi such as T. stipitatus. Perhaps no one has really looked for it. The free acid has been isolated from two strains of Aspergillus terreus—one unidentified88 and the other IFO 6123,89 and from Penicillium spinulosum.90 The ethyl ester of 3-methylorsellinic acid has been isolated from Aspergillus silvaticus.91In recent work, 3-methylorcinaldehyde has been identified as the polyketide -derived intermediate for xenovulene biosynthesis via tropolonoid intermediates (see Section 2.10). For the role of this compound in stipitatonic acid biosynthesis see Section 3.
3-Methylorsellinic acid is also a well-known component of lichen depsides and depsidones.92 Thus, the phenolic group of the depside atranorin is the methyl ester of 3-methylorsellinic acid. Moreover, cells of the lichen Evernia prunastri, immobilized in calcium alginate, produced 3-methylorsellinic acid when a specific oxidase enzyme was inhibited by sodium azide.93
6 Biosynthesis of thujaplicins
The most abundant thujaplicin in nature is the β isomer and essentially all of the biosynthetic information that has been obtained concerns this molecule. Even today, papers from Japanese authors often refer to it by the name used by Nozoe—hinokitiol. There are apparently no papers concerning the biosynthesis of α- or γ-thujaplicin. Work with materials derived from woody tissues is more difficult than that with fungal materials. However, much information has been obtained by use of cell cultures of Cupressus lusitanica (Mexican cypress)94 and to a lesser extent with similar preparations from Thuja plicata.95 There are two major aspects to this work.6.1 Biochemistry of intermediates for thujaplicin formation
The facts that β-thujaplicin has 10 carbon atoms and contains an isopropyl group immediately suggest a terpenoid origin. In early work with callus and suspension cell cultures of C. lusitanica elicited with yeast extract, [U-14C]glucose, [2-14C]mevalonate and [2-14C]malonate were used as precursors.96 Incorporation of radioactivity was observed with the two former materials, whereas that obtained with [2-14C]malonate was very much lower. These samples were not degraded chemically but the malonate result argued against a polyketide origin. Addition of the HMG-CoA reductase inhibitor, compactin, suppressed thujaplicin formation providing some evidence for a mevalonate origin.94,96In further work, [10-14C]geraniol was incorporated into β-thujaplicin by C. lusitanica cell cultures, indicating a biosynthetic route via geranyl pyrophosphate.97 Moreover, this new work indicated a poor utilization of [2-14C]mevalonate; in earlier experiments the purification of the β-thujaplicin may have been incomplete. NMR analysis of β-thujaplicin samples labelled by growth in the presence of [1-13C]-, [2-13C]- and [U-13C]-glucose indicated that the geranyl pyrophosphate was mostly derived by the mevalonate-independent pathway; i.e., by reaction of glyceraldehyde 3-phosphate + pyruvate to form 1-deoxy-D-xylulose phosphate. It is probable that both a mevalonate pathway and a mevalonate-independent pathway may be involved. The acetate/mevalonate pathway is apparently localized in the cytoplasm whereas the mevalonate-independent route is located in plastids; the two pathways are possibly cross-linked.94
Furthermore a new monoterpene , (1S,2S,6S)-(+)-1,6-epoxy-4(8)-p-menthen-2-ol 57, has been isolated from C. lusitanica cultures (Scheme 4); it was accompanied by ten other known monoterpenes , including terpinolene 56 and sabinene 60.98 It seems likely that this new compound is an intermediate in β-thujaplicin biosynthesis.99 The mechanism by which a single methyl group (e.g., in (1S,2S,6S)-(+)-1,6-epoxy-4(8)-p-menthen-2-ol 57) is incorporated into the seven-membered tropolone ring is currently not known. A major competing pathway is believed to be as follows: geranyl pyrophosphate → 4-terpineol 58 → 4-hydroxyphellandric methyl ester 59. The situation is complex and other terpene synthases such as those for sabinene, β-ocimene, and myrcene may also be involved. In view of the large number of monoterpenes produced by the elicited cultures of C. lusitanica, assessing the precise intermediates involved in β-thujaplicin biosynthesis will be difficult and time consuming. It will be of interest to determine whether there is a common pathway for the biosynthesis of all three thujaplicins.
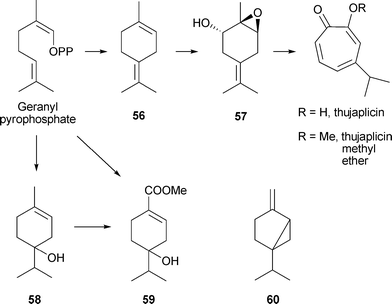 | ||
| Scheme 4 Biosynthesis of β-thujaplicin and related compounds. | ||
6.2 Other considerations in β-thujaplicin production in cell cultures
Thujaplicins and related materials have many physiological activities (see Section 10.2) that may have potential for use in medicine or commerce. In view of these considerations, the conditions for production of cell biomass and secondary metabolites by the C. lusitanica system have been investigated.100 To some extent, there have been overlapping investigations of both physiological and biochemical components (for these, see the preceding section). With respect to inorganic nutrients, optimum conditions are generally different for these two processes. However, strict control of pH and the major inorganic nutrients was not necessary for β-thujaplicin production.One consideration is that there is a feedback regulation of β-thujaplicin production in the cell cultures. When phytoalexin levels reached about 40 mg L−1 in the culture medium, production apparently halted but could be restored by exchange of old medium with new. Under these conditions, a methyl ether of β-thujaplicin was formed; apparently, the excess accumulation of β-thujaplicin was relieved by this metabolic regulation.101
The effects of organic acids (e.g., those of the tricarboxylic acid cycle) on β-thujaplicin production were also investigated. Some small stimulatory effects were noted (e.g., with 1 mM fumaric acid) but there were also inhibitory effects (e.g., 2 mM sodium acetate, 2 mM sodium benzoate).94 However, addition of terpinyl acetate and 2-carene stimulated β-thujaplicin production by 2 to 2.5 fold; addition of other monoterpenes was inhibitory. The metabolism of terpinyl acetate was believed to be as follows: terpinyl acetate → 4-terpineol → thujane → 3-thujone → β-thujaplicin. The action of 2-carene was explained by a different pathway via 3-caren-5-one.
A very active research area concerns the signalling mechanisms indicating the presence of potential pathogens to plant cells and to the activation of appropriate defense mechanisms. In many cases, such responses are initiated by “elicitor” molecules that can be microbial or plant-derived (biotic elicitors) or result from chemical or mechanical stresses (abiotic inducers). This report cannot attempt a detailed account of this extensive topic but will briefly note some of the experimental work dealing with β-thujaplicin production. There is, however, an extensive and recent account of elicitor signal transduction leading to the formation of plant secondary metabolites.102 Also published recently is an account of the molecular regulation of induced terpenoid synthesis in conifers.103
In cell cultures, β-thujaplicin production can be stimulated by a yeast elicitor, methyl jasmonate, and other stresses. The commonly used yeast elicitor preparation contains a 70–80% ethanol-insoluble, polysaccharide component. Elicitor-treated cultures accumulate high amounts of β-thujaplicin at early stages (e.g., 4 days of incubation) and then accumulate other monoterpenes at later stages post-elicitation. Thujaplicin may be the primary defense agent with the other monoterpenes providing a secondary defense.99 The yeast elicitor generally produces an earlier and stronger response than does the octadecanoid plant growth regulator, methyl jasmonate. The content of articles considering many other factors involved in β-thujaplicin production by the C. lusitanica system (e.g., in signalling, biosynthesis and degradation), are noted here for the reader's convenience:
Hydrogen peroxide formation by superoxide anion synthases may mediate elicitor-induced biosynthesis; exogenously applied H2O2 (or use of a generation system) can stimulate biosynthetic activity.104
cAMP stimulates β-thujaplicin biosynthesis, possibly by an involvement of Ca2+ and K+ fluxes. There is cross talk between cAMP treatment and the ethylene signalling pathway.105
In addition to the just-noted role for ethylene signalling, jasmonate signalling is also involved and there is an interaction between these two modes; they are integral parts of the C. lusitanica signalling pathway.106
A further complexity is the role of oxidative stress; under conditions of Fe2+ stress C. lusitanica cells generate significant levels of reactive oxygen species, ROS. The ROS production inhibits cell growth but enhances ethylene and β-thujaplicin formation; as previously noted, H2O2 is a positive signal for β-thujaplicin formation.107
More recently, the interactions (cross talk) between ROS and nitric oxide have been delineated. Nitric oxide is involved in multiple physiological processes and plays a role in the elicitor-induced cell death.108
The metabolic flux of β-thujaplicin formation and that of other monoterpenes in the elicited cultures has been investigated.109
Yeast elicited cultures of T. plicata also show an increased stimulation of thujaplicin formation, but less detail is presently available than for the C. lusitanica system.95
7 Biosynthesis of colchicine
Experiments in the 1960s with Colchicum autumnale had quickly established that colchicine biosynthesis was not a polyketide + C1 process as had been observed for the fungal tropolones . [1-14C]-Acetate was incorporated only into the N-acetyl group and [14CH3]methionine labelled only the O-methyl positions. However, the benzenoid ring was derived from L-phenylalanine (but not from L-tyrosine) as were two atoms of the cycloheptane ring. The non-equivalence of L-phenylalanine and L-tyrosine suggested the formation of a C6–C3 unit via the cinnamic acid pathway, and this supposition was verified by the use of [3-14C]cinnamic acid.109 Moreover, feeding experiments with [3-14C]-L-tyrosine indicated an incorporation of 14C into the tropolone moiety, specifically at C-12.110 It became clear that dopamine, derived from L-tyrosine, and 4-hydroxycinnamaldehyde, derived from L-phenylalanine, could be regarded as the basic units for colchicine biosynthesis. All of this early work has been reviewed111 and an extensive table of various labelled precursors was given in 1973.112 As the elegant work of Battersby and his colleagues has shown, colchicine is essentially a considerably modified phenethylisoquinoline alkaloid . The space available here cannot do justice to all of this work, which includes precise stereochemical detail for loss of certain hydrogen atoms. The experimental details are, however, readily available.113Since the 16 carbon atoms of colchicine derived from a C6–C3 and a C6–C2 unit it was suggested that a dienone 61 with a good leaving group, X, and with the possibility for ring expansion might be involved in the biosynthetic mechanism.114 An important finding was the structure of androcymbine62 R = H, a cometabolite of colchicine in Androcymbium melathiodes. In fact, 3H labelled O-methylandrocymbine, 62 R = CH3, was an excellent precursor for colchicine. In turn, a possible precursor for O-methylandrocymbine was the diphenol 63, a compound termed autumnaline; this too was well incorporated into colchicine and demecolcine. A basic pathway, omitting various hydroxylation and methylation steps, could, therefore, be written to demecolcine 45 R1 = R2 = R3 = Me and hence colchicine 45 R1 = R2 = Me, R3 = COMe (Scheme 5).
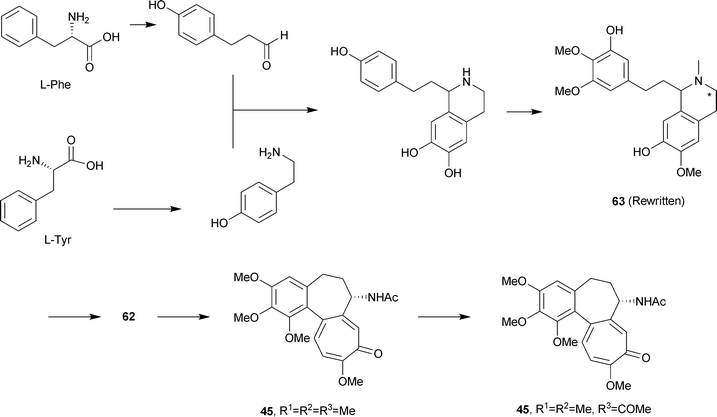 | ||
| Scheme 5 Biosynthesis of colchicine. | ||
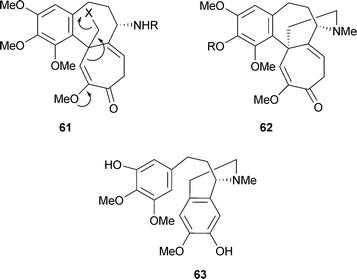
It was established that the N-methyl group of autumnaline and O-methylandrocymbine is retained in demecolcine, but was lost in the formation of colchicine. With appropriately labelled precursor molecules, the following detail was obtained for the O-methylandrocymbine → colchicine portion (Scheme 6): O-methylandrocymbine 62 R = CH3 → N-formyldemecolcine64 R1 = Me, R2 = CHO → demecoline64 R1 = H, R2 = Me → N-formyl-N-deacetylcolchicine64 R1 = H, R2 = CHO → N-deacetylcolchicine64 R1 = R2 = H → colchicine64 R1 = H, R2 = Ac. The N-formyl group of N-formyldemecolcine derives from the C-3 carbon of autumnaline-marked as * in 63. (Of these intermediates, N-formyl-N-deacetylcolchicine, and N-deacetylcolchicine are known as minor alkaloids of C. autumnale and N-formyldemecolcine is present in C. cornigerum).
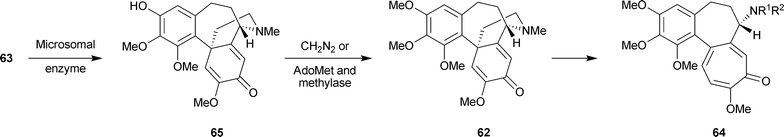 | ||
| Scheme 6 Biosynthesis of colchicine: further details. AdoMet = S-adenosylmethionine. | ||
Further information has come from the use of a microsomal enzyme system present in immature seeds of C. autumnale.115 This system, dependent on NADPH, cytochrome P-450 and O2, catalyzed the intramolecular phenol-oxidative coupling of autumnaline 63 to a new material, termed isoandrocymbine, 65 (Scheme 6). On methylation with diazomethane, 65 was converted to O-methylandrocymbine 62 R = Me. Moreover, isoandrocymbine was converted to androcymbine by a soluble protein extract from C. autumnale seeds in the presence of [14CH3]-S-adenosylmethionine.116 Still more recently, isoandrocymbine 65 and O-methylandrocymbine 62 R = Me were isolated as colchicine cometabolites from C. stevenii, a Jordanian meadow saffron.
8 Biosynthesis of sulfur-containing tropolones
A possible polyketide origin for thiotropocin, obtained from Pseudomonas CB-104, was unlikely since [1-14C]-, [2-14C]-, and [1,2-13C2]acetate did not contribute isotopes to this sulfur-containing metabolite (the question of its identity with tropodithietic acid was discussed earlier).117 Experiments using [U-13C6]glucose indicated a shikimate-derived intermediate, most probably phenylacetic acid66. This would be derived by the well-known pathway: shikimate → chorismate → prephenate → phenylpyruvate → phenylacetate. In fact, [1,2-13C2]phenylacetate contributed two 13C atoms to thiotropocin 25 (Scheme 7). The biosynthetic mechanism requires a ring expansion involving either of the ortho carbons of the benzene ring, and a further oxidation, as well as a sulfur–oxygen exchange. A possible intermediate 67 has been suggested.43 Separate pathways would lead either to the thiotropocin 25 or the tropodithietic acid 27 structure.![Biosynthesis of sulfur-containing tropolonoids. * = 13C from [1,2-13C2]phenylacetate.](/image/article/2008/NP/b711474e/b711474e-s7.gif) | ||
| Scheme 7 Biosynthesis of sulfur-containing tropolonoids. * = 13C from [1,2-13C2]phenylacetate. | ||
9 Benzotropolones. Purpurogallin, theaflavins and related compounds
It will be convenient to give here a general account of a large group of benzotropolones including biosynthetic information where available; a characteristic feature is the formation of the tropolone unit by the action of plant oxidases. Over the past few years, there has been extensive work on the production of materials such as theaflavins and related compounds from catechins. The interest concerns not only chemistry but also the many antibacterial, antiviral, antifungal, antitoxin and antioxidant activities of teas and tea components.118 The green tea polyphenols are also of interest for a broad anticarcinogenic effect.119As indicated earlier, purpurogallin, the simplest of these benzotropolones, is readily obtained from pyrogallol by either chemical or enzymatic oxidation. In vivo, gallic acid68 undergoes decarboxylation to pyrogallol69 followed by a complex enzymatic oxidation (Scheme 8). Purpurogallin is essentially a shikimate-derived product.
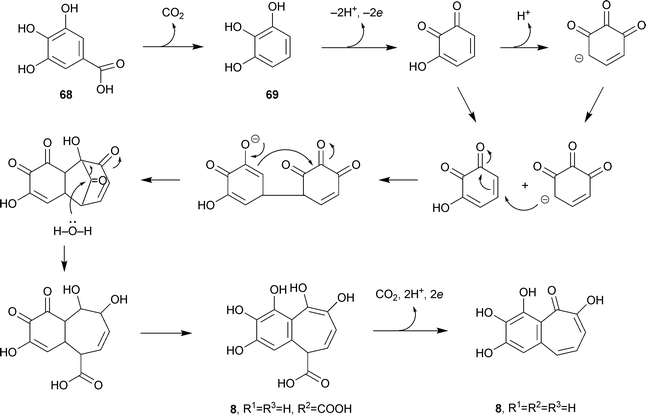 | ||
| Scheme 8 Biosynthesis of purpurogallin. | ||
The leaves of Camellia sinensis are used to produce more than 300 different kinds of tea, with three main types: green tea (non-fermented), oolong tea (semi-fermented) and black tea (fermented). Fermentation is a rather inappropriate term since no new biological components (e.g., yeast) are added in the process. It is an oxidation, brought about by polyphenol oxidases present in the leaves. For green tea, this oxidation is prevented by steaming or drying the fresh leaves at elevated temperatures to inactivate oxidases. Black tea having experienced polyphenol oxidase activity contains various pigments including the orange-red coloured theaflavins and the red or dark brown thearubigins (heterogeneous polymers of flavan-3-ols and flavan-3-ol gallates). The theaflavins contain a benzotropolone nucleus.
Perhaps theaflavins should not be considered as true “natural products”; however, this distinction is not usually enforced. Green tea, closest to the natural leaves, is essentially free of theaflavin components. In a detailed HPLC analysis of 48 green teas, theaflavin components were reported as “not detected” with a single exception where the amount of theaflavin itself was 0.2 ± 0.02 mg g−1.120 Moreover, in this one case, theaflavin gallates were not detected. Typical structures are as follows: theaflavin72 R1 = R2 = H; theaflavin 3-gallate 72 R1 = galloyl, R2 = H; theaflavin 3′-gallate 72 R1 = H, R2 = galloyl; theaflavin 3,3′-digallate 72 R1 = R2 = galloyl (Scheme 9).
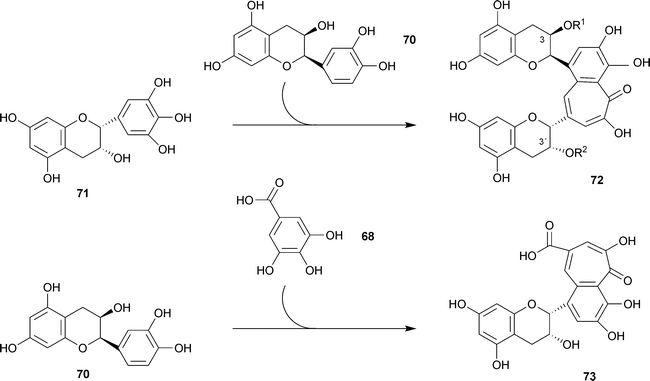 | ||
| Scheme 9 Biosynthesis of theaflavin 72 R1 = R2 = H and epitheaflavic acid 73. | ||
Theaflavins are formed by co-oxidation of catechins during the fermentation process. Thus, for example, theaflavin itself 72 R1 = R2 = H, was formed by incubation of (−)-epicatechin 70 and (−)-epigallocatechin 71 with a crude polyphenol oxidase preparation from bananas (Scheme 9).121 From (−)-epicatechin 70 and gallic acid68, under influence of a polyphenol oxidase, there was obtained epitheaflavic acid 73.122 In other work, 18 benzotropolone derivatives were obtained by reaction of selected pairs of catechins and other materials (e.g., catechol, pyrogallol, gallic acid) with H2O2 in the presence of horseradish peroxidase. For structures, the reader is referred to the original paper.123
Theaflavins can be synthesized from epicatechin and epigallocatechin by many plants, even those not themselves containing catechins. Using homogenates from 62 plants (49 families), 46 were capable of theaflavin synthesis.124 Japanese pear (Pyrus pyrifolia) and unripe loquat (Eriobotrya japonica) homogenates were particularly active.
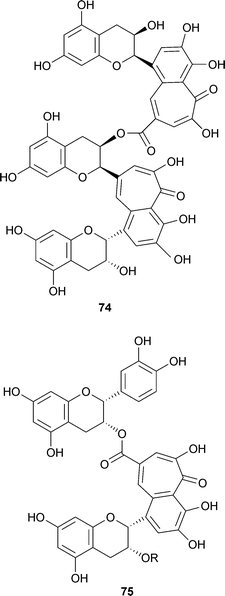
Polymeric compounds with higher molecular masses were obtained by reaction of theaflavins and tea catechins in the presence of horseradish peroxidase and have been identified in black tea itself. To give only one example, reaction of theaflavin 3-gallate 72 R1 = galloyl, R2 = H, with epicatechin gave theadibenzotropolone 74.125 Moreover, a theatribenzotropolone was also obtained. A different structural type of benzotropolone, theaflavate A 75 R = galloyl, was obtained by chemical oxidation of (−)-epicatechin-3-O-gallate with potassium ferricyanide and was also shown to be present in small amounts in black tea.123,126 Theaflavate B 75 R = H lacks the galloyl residue.
10 Biological activities of tropolones
Secondary metabolites frequently exhibit biological properties, such as antibacterial, antifungal and antiviral activities; many of these agents have found widespread applications. Indeed, in 1938, even before the tropolone structure was proposed, β-thujaplicin (then termed hinokitiol) had been shown to have a significant antibacterial activity against a tubercle bacillus and other bacteria.10 Similarly, also prior to structure determination of the fungal metabolites, puberulonic and puberulic acids were shown to be active against Gram-positive bacteria.12710.1 Tropolone
Tropolone itself is bacteriostatic and bactericidal for a wide range of bacterial species.128 It has also been shown to have a strong insecticidal activity against Tyrophagus putrescentiae, Dermatophagoides farinae and Captotermes formosanus, as well as growth-inhibitory effects on some pathogenic fungi, especially Pythium aphanidermatum. It inhibits metalloproteases, with a high level of activity against carboxypeptidase A. Biological activities discovered earlier have been summarized.129Tropolone and also β-thujaplicin analogues have antifungal activity against organisms involved in wood degradation such as white and brown rot fungi. The level of activity is comparable to that of commercially available fungicides.13010.2 Thujaplicin and related compounds
Much attention has focused on β-thujaplicin in Japan and the following medical and commercial uses have been mentioned in Japanese publications where it is almost invariably described as hinokitiol—use as a preservative for flowers, mushrooms and vegetables, plant growth stimulator (inhibits bacteria), antibacterial or fungicidal in food, cosmetics and toothpaste, use as a hair tonic and tick repellent.131,132 Some of the literature on commercial use in Japan (e.g, descriptions in Japanese patents) is not readily available. A recent, comprehensive review of plant tropolonoids including the thujaplicins and related compounds, describes many, varied biological activities.133 Table 1 gives a general indication of the range of the physiological activities of α- and β-thujaplicin.| Compound | Activity | References |
|---|---|---|
| β-Thujaplicin, 18 R1 = R3 = H, R2 = isopropyl | Antimicrobial | 131,132 |
| Antifungal | 130–132 | |
| Insecticidal | 129 | |
| Cytotoxic (human, murine cells) | 137 | |
| Antitumor (due to metal chelation) | 134 | |
| Chlamydia trachomatis inhibition | 135 | |
| α-Thujaplicin, 18 R1 = isopropyl, R2 = R3 = H | Antibacterial | 136b |
| Antifungal | 136b | |
| Plant growth inhibitor | 136a | |
| Insecticidal | 136b | |
| Ascaricidal | 136b | |
| Cytotoxic (murine cells) | 136a,b |
The related compounds, β-dolabrinol, γ-thujaplicin and 4-acetyltropolone (all components of Thujopsis dolabrata) have various antibacterial and antifungal activities, and are cytotoxic in vitro to the murine P 388 lymphocytic leukemia cell line.137 In a separate study of 4-acetyltropolone the following activities were observed: metalloprotease inhibition, plant growth inhibition, cytotoxicity, toxicity to mice.39
It has long been recognized that certain woods from trees of the Order of the Cupressales have a natural durability, and this durability is attributed to tropolone compounds. A present day USA catalogue devoted to products for “eco living with style” notes that hinoki wood and its oils are prized in Japan “for their ability to pamper and heal the skin, kill bacteria, and smooth frazzled nerves”. Moreover, hinoki wood is prized in areas exposed to moisture since “it resists mildew”. Among several hinoki wood items available for sale is a sauna stool for US $98 (Viva Terra catalogue, summer 2007).
As already indicated, the physiological activities of the thujaplicin group of tropolones do include fungicidal and insecticidal activity. Indeed, the commercial use of tropolones as substitutes for toxic wood preservatives (e.g., arsenic compounds) that are already in use, has been seriously considered.36 Since extraction of tropolones from wood, or chemical synthesis, is prohibitively expensive for such use, biotechnological methods have been considered. Cell cultures from Thuja plicata are one possibility95 and the extensive work on the C. lusitanica cell system has already been discussed.
There appears to be little or no use of β-thujaplicin in Europe and the USA. The USA FDA has no documents relating to hinokitiol/β-thujaplicin as of March 2007. Nevertheless, an Internet source indicates that there are 312 USA patents in some way involving hinokitiol. To take one example only, US Patent 7081258 describes its use in a hair growth-promoting composition. Visitors to Russia may be interested to know that, according to Internet sources, the Grand Hotel Europe in St Petersburg has available various cosmetology services including those of “Hinoki Clinical”, described as a “deluxe-category therapeutic cosmetics company” in Japan. The active component in this company's products is hinokitiol, the main qualities of which are said to be as follows: powerful antibacterial action, unique anti-inflammatory activity, deep penetration of skin, normalization and stimulation of cell activity, nourishing activity, treatment of pigmentation marks, improvement of skin tone. The “Spa Club” of the Orient Express also features Hinoki Clinical cosmetics in the “Japanese compartment”.
10.3 Benzotropolones
Several benzotropolones have been investigated for their ability to inhibit the methylation of 2- and 4-hydroxyestradiol. These redox active and carcinogenic metabolites are detoxified by S-adenosylmethionine-dependent methylation with catechol-O-methyltransferase. The most active inhibitors were purpurogallin, purpurogallin carboxylic acid and theaflavin-3,3′-digallate. The inhibition mechanism was of a mixed type. For the purpurogallins, the benzene ring was inserted into the active site of the transferase and for the theaflavin derivative the galloyl ester was inserted. It is apparently not known whether this inhibition is beneficial or deleterious in clinical practice although there are no reports of increased estrogen-related malignancies in humans using selective catechol-O-methyltransferase inhibitors.138Several tropolone types, including purpurogallin, protect cultured cells from oxidative stress-mediated damage, specifically hydrogen peroxide-induced DNA damage and apoptosis. In cultured Jurkat cells (an immortalized line of T lymphocyte cells originally derived from a 14 year old boy with T cell leukemia), tropolone , β-thujaplicin, purpurogallin and trimethylcolchicinic acid were protective; however, colchicine and tetramethyl purpurogallin ester were not. Hydrogen peroxide-induced apoptosis was also inhibited by tropolone . These results were attributed to the formation of a redox-inactive iron complex. Under some conditions (e.g., absence of exogenous hydrogen peroxide) tropolone enhanced iron-mediated DNA damage, possibly by formation of a lipophilic redox-active complex.139
10.4 Hydroxylated tropolones
Synthetic mono- and bistropolones show antitumor activity in vitro (KB cells) and in vivo (P 388 leukemia in mice) possibly by inhibiting intracellular metal-enzymes such as ribonucleotide reductase.140 Of concern here is the action of a small number of natural hydroxylated tropolones and the possibility for the development of novel drugs against HIV infection. Recent research on the cell biology of HIV-1 and other retroviruses has been reviewed.141β-Thujaplicinol 22 R1 = H, R2 = CH(CH3)2 and manicol 7 are potent and selective inhibitors of the HIV-1 ribonuclease H (RNase H) activity of human immunodeficiency virus-type 1 reverse transcriptase. However, β-thujaplicin was inactive. This work emphasized the importance of the 2,7-dihydroxy function in both of these natural tropolones , possibly through a role in metal chelation at the RNase H active site.142
In addition, seven natural tropolones were investigated as inhibitors for the magnesium-dependent strand transfer activity of HIV-1 integrase. The materials were identified by NSC compound numbers as follows (NSC = Nomenclature Standards Committee of the USA FDA): 18806, β-thujaplicinol; 310618, manicol; 43339, nootkatin; 89303, tropolone ; 18804, β-thujaplicin; 18805, β-thujaplicin; 43338, β-thujaplicin. As with the inhibition of the RNase H activity, β-thujaplicinol, and manicol were inhibitors of HIV-1 integrase with Mn2+ as cofactor . The three measurable activities of HIV-1 integrase—3′-processing, strand transfer, disintegration—were studied in detail with β-thujaplicinol; the role of the 7 hydroxy group was again evident. The hydroxy tropolones probably chelate the divalent metal, Mg2+ or Mn2+, at the enzyme's active site. It was also observed that β-thujaplicinol had a weak cytoprotective activity against HIV-1IIIB on lymphoid MY-2 cells.143
In connection with the importance of the 7 hydroxy group, it may be noted that 7-hydroxytropolone 1 R1 = OH, R2 = H, a metabolite of Streptomyces neyagawaensis, is an inhibitor of aminoglycoside-2″-O-adenylyltransferase.15,144 Moreover, 3,7-dihydroxytropolone 1 R1 = R2 = OH, a metabolite of Streptomyces tropolofaciens sp. nov. K611-97, has weak antibacterial activity against a variety of Gram-positive and Gram-negative organisms, weak antifungal activity, but a strong cytotoxicity against murine B16 melanoma cells. It had some antitumor activity against B16 melanoma in mice but was inactive against P388 leukemia.16
10.5 Colchicine
Preparations of the autumn crocus have long been of interest to physicians. The ancient Egyptians probably used it for treatment of gouty arthritis and in the first century AD, Pedanios Dioscorides, a military physician under Nero, recommended it for treatment of a cancer-like disease.145 A full historical account is available.61Colchicum autumnale is native to south, west and central Europe, but is cultivated world-wide as an ornamental plant. Many other plants belonging to the family Colchicaceae contain (−)-colchicine or related metabolites. The true crocus, flowering in spring, is a member of the iris family while the autumn crocus belongs to the lily family. While the spring crocus is not toxic, C. autumnale is definitely a toxic plant. An extensive toxicity description is available.146 A lethal human dose of colchicine is about 10 mg.Despite its toxicity, colchicine has become famous for the treatment of gout—an inherited metabolic disorder, usually in males, involving problems in uric acidmetabolism and progressive chronic arthritis. A classic gout attack is acute in onset and characterized by extreme pain; there can also be marked swelling, warmth, erythema and tenderness of a single joint. The metatarsophalangeal joint of the big toe is often the afflicted member. Colchicine is also used for treatment of Familial Mediterranean Fever, an inherited disorder, frequently but not exclusively found in persons of Mediterranean origin, and characterized by recurrent fever and peritonitis (inflammation of the peritoneal cavity). Colchicine analogues have been tested for anti-cancer activity but toxicity makes their use problematic.147 In medical use, a dose of 1 mg colchicine orally every 2 hours is used to treat an acute gout attack but no more than 7 mg should be taken in a 24 hour period. For chronic disease, the dose is one to three 0.6 mg tablets per day.
Colchicine does not cure gout and the precise mechanism for pain relief is unclear. Elevated blood levels of uric acid lead to crystallization of poorly soluble urates. These foreign materials are then attacked by macrophages and leukocytes, resulting in the release of cytokines and interleukins, thus leading to the inflammatory condition. Colchicine functions by inhibition of the action of the leukocytes. This probably involves the major pharmacological property of colchicine—the ability to bind to tubulin dimers and thus to prevent polymerization to microtubules.147 The microtubules are necessary for several processes including ameboid motility; inhibition of this activity prevents the migration of macrophages and leukocytes and, in turn, the gout-derived inflammation.
As well as gout and Familial Mediterranean Fever, colchicine is approved by the USA FDA for treatment of secondary amyloidosis and scleroderma. In Europe, the European League Against Rheumatism (EULAR) has issued comprehensive recommendations for both diagnosis and treatment of gout.148 Another recent literature review and consensus statement for German, Austrian and Turkish caregivers is available for colchicine use in children and adolescents with Familial Mediterranean Fever.149
In humans, colchicine halts the division and activity of white blood cells; similarly, in plants, the division of plant cells can be arrested or delayed. Injection of colchicine into a plant ovary blocks formation of the mitotic spindle in the dividing, fertilized egg. In consequence, the cell does not divide but the chromosome number in the egg is doubled. This phenomenon provides a useful tool for the study of polyploidy in plants.
Colchicine contains two elements of chirality so four stereoisomeric structures are possible. The carbon at position 7 (carrying the NHAc substituent) is a typical chiral centre. The other stereochemical element is axial chirality (atropisomerism) about the single bond between rings A and C. Specification of axial chirality as aR and aS is described in a standard text.150 In the natural stereoisomer, (−)-colchicine, that binds strongly to tubulin, C-7 has S configuration. Extensive NMR and ORD studies, and X-ray crystallography of an analogue of the thio series, indicate that the axial chirality in (−)-colchicine is aS.147 Hence natural colchicine is (−)-(aS,7S)-colchicine 76. The aR–aS equilibrium, 76 ↔ 77, studied by NMR, indicates an important role for the cycloheptane ring B. The acetamido group at C-7 energetically favours a pseudo-equatorial orientation, and this in turn favours the aS configuration 78. The activation energy for the atropisomerism is 22–24 kcal mol−1. The two remaining stereoisomers are the atropisomers with the C-7 acetamido group in the R configuration—(+)-(aR,7R) and (−)-(aS,7R).
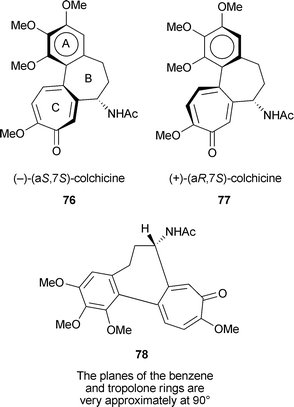
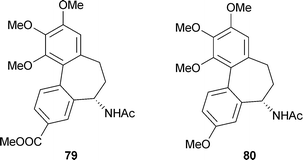
There has been much study of various analogues of (−)-colchicine, including the role of the MeO groups, the C-7 acetamido group, contraction of the tropolone ring to phenyl, and in “iso” compounds reversal of the substituents at C-9 and C-10.62,63,147 One hope is that the design of colchicine analogues may produce compounds with high tubulin-binding activity but lower toxicity; so far however, no practicable anti-cancer agent based on colchicine has been developed. One example is a recent paper describing variations in the B ring of allocolchicinoids (these “allo” compounds have a phenyl ring in place of the usual tropolone ). Thus allocolchicine 79 and N-acetylcolchinol-O-methyl ether (NCME) 80 are known as potent anti-tubulin agents. Although some further, variant synthetic structures inhibited tubulin assembly as well as NCME, none of them revealed growth inhibition of MCF-7 breast cancer cells comparable to that of colchicine itself.151
The binding of colchicine to tubulin is slow and poorly reversible; the exact mechanism by which it stabilizes tubulin against spontaneous decay is unclear. A recent “footprinting” technique shows that colchicine affects mainly the α-subunit of tubulin, involving cysteine residues at positions 295, 305, 315 and 316. This domain is substantially distant from the binding site at the α/β interface. The B ring of colchicine is bound on the α-subunit and the A and C rings are on the β-subunit. An elegant model is available152 as is an X-ray structure showing the colchicine-binding site on tubulin.153
10.6 Eupenifeldin, pycnidione and epolones
As noted earlier (Sections 3.0 and 3.1) these structurally related metabolites are sesquiterpene-tropolones in which the common feature is a humulene-derived, eleven-membered ring. They have a number of interesting physiological activities. Eupenifeldin 36 (cis fusion) is cytotoxic against the HCT-116 cell line and has in vivo antitumor activity against the P388 leukemia model.54 Pycnidione 36 (trans fusion) inhibits stromelysin, an enzyme postulated to cause cartilage degradation.53 A more interesting activity, common to pycnidione and the epolones 35, 37, is that of inducing erythropoietin (Epo) expression.52–54,154 Recombinant Epo, widely used to treat anaemia, is expensive and requires intravenous or subcutaneous administration. These natural, small molecule tropolones are, therefore, of interest as possible alternative treatments for anaemia.11 Acknowledgements
Ann Rogers, Langley Library, University of Pittsburgh is thanked for providing much assistance in obtaining copies of articles. Professors H. Laatsch and A. Zeeck, University of Göttingen, graciously assisted in discussing the structures of the sulfur-containing tropolones and Professor Zeeck informed me of work on chaetospiron. Dr Jian Zhao, Baylor College of Medicine, Houston, TX, is thanked for allowing me to see his comprehensive review,133 titled “Plant Troponoids: Chemistry, Biological Activity, and Biosynthesis” prior to publication. Dr M. G. Banwell, University of Melbourne, Australia, kindly supplied a list of about 90 naturally occurring tropolonoids prepared by one of his students. The NPR reviewer is thanked for very helpful suggestions and information on 3-methylorcinaldehyde synthase.12 References
- R. Bentley and R. Thomas, The Biochemist, 1990, 12, 3–7 Search PubMed.
- J. H. Birkinshaw and H. Raistrick, Biochem. J., 1932, 26, 441–453 CAS.
- J. H. Birkinshaw, A. R. Chambers and H. Raistrick, Biochem. J., 1942, 36, 242–251 CAS.
- M. J. S. Dewar, Nature, 1945, 155, 50–51 CAS.
- M. J. S. Dewar, Nature, 1945, 155, 141–142 CAS.
- J. W. Cook and J. D. Loudon, Q. Rev. Chem. Soc., 1951, 5, 99–130 RSC.
- P. L. Pauson, Chem. Rev., 1955, 55, 9–136 CrossRef CAS.
- (a) H. Erdtman and J. Gripenberg, Nature, 1948, 161, 719 CAS; (b) H. Erdtman and J. Gripenberg, Nature, 1949, 164, 316 CAS.
- T. Asao, S. Itô and I. Murata, Eur. J. Org. Chem., 2004, 899–928 CAS.
- T. Nozoe, Seventy Years in Organic Chemistry, American Chemical Society, Washington, DC, 1991 Search PubMed.
- T. Nozoe, Sci. Rep. Tohoku Univ., Ser. 1: Phys., Chem., Astron., 1950, 34, 199–236 Search PubMed.
- T. Nozoe, Nature, 1951, 167, 1055–1056 CrossRef CAS.
- G. D. Lindberg, J. M. Larkin and H. A. Whaley, J. Nat. Prod., 1980, 43, 592–594 CrossRef CAS.
- K. Azegami, K. Nishiyama and H. Kato, Appl. Environ. Microbiol., 1988, 54, 844–847 CAS.
- N. E. Allen, W. E. Alborn, J. N. Hobbs and H. A. Kirst, Antimicrob. Agents Chemother., 1982, 22, 824–831 CAS.
- K. Sugawara, M. Ohbayashi, K. Shimizu, M. Hatori, H. Kamei, M. Konishi, T. Oki and H. Kawaguchi, J. Antibiot., 1988, 41, 862–868 CAS.
- T.-T. Lin, K.-T. Wang and L.-H. Chang, J. Chin. Chem. Soc., 1963, 10, 139–145.
- S. Kitamura, T. Iida, K. Shirahata and H. Kase, J. Antibiot., 1986, 39, 589–593 CAS.
- P. Seephonkai, M. Isaka, P. Kittakoop, S. Trakulnaleamsai, R. Rattanjot, M. Tanticharoen and Y. Thebtaranonth, J. Antibiot., 2001, 54, 751–752 CAS.
- P. V. Divekar, H. Raistrick, T. A. Dobson and L. C. Vining, Can. J. Chem., 1965, 43, 1835–1848 CrossRef CAS.
- J. Polonsky, J.-C. Beloeil, T. Prangé, C. Pascard, H. Jacquemin, D. M. X. Donnelly and P. T. M. Kenny, Tetrahedron, 1983, 16, 2647–2655 CrossRef.
- J. A. Barltrop and J. S. Nicholson, J. Chem. Soc., 1948, 116–120 RSC.
- N. Arpin, J. Favre-Bonvin and W. Steglich, Phytochemistry, 1974, 13, 1949–1952 CrossRef CAS.
- D. Mesa-Siverio, A. Estévez-Braun, A. G. Ravelo, J. R. Murquia and A. Rodríguez-Afonso, Eur. J. Org. Chem., 2003, 4243–4247 CrossRef CAS.
- D. Klostermeyer, L. Knops, T. Sindlinger, K. Polborn and W. Steglich, Eur. J. Org. Chem., 2000, 603–609 CrossRef CAS.
- R. E. Moore and G. Yost, J. Chem. Soc., Chem. Commun., 1973, 937–938 RSC.
- M. G. Banwell, C. J. Cowden, G. L. Cravatt and C. E. F. Rickard, Aust. J. Chem., 1993, 46, 1941–1954 CAS , and references therein.
- J. Polonsky, T. Varenne, T. Prangé, C. Pascard, H. Jacquemin and A. Fournet, J. Chem. Soc., Chem. Commun., 1981, 731–732 RSC.
- W. Segal, J. Chem. Soc., 1959, 2847–2851 RSC.
- M. Holík and I. Kuhr, J. Chem. Soc., Chem. Commun., 1973, 65–66 RSC.
- R. W. Bryant and R. J. Light, Biochemistry, 1974, 13, 1516–1522 CrossRef CAS.
- P. V. Divekar, P. E. Brenneisen and S. W. Tanenbaum, Biochim. Biophys. Acta, 1961, 50, 588–589 CAS.
- R. Bentley and J. G. Keil, J. Biol. Chem., 1963, 238, 3806–3810 CAS.
- I. Kuhr, J. Fuska, P. Sedmera, M. Podojil, J. Vokun and Z. Vanek, J. Antibiot., 1973, 26, 535–536 CAS.
- E. Zavarin, R. M. Smith and A. B. Anderson, J. Org. Chem., 1959, 24, 1318–1321 CrossRef CAS.
- J.-P. Haluk and C. Roussel, Ann. For. Sci., 2000, 57, 819–829 Search PubMed , and references therein.
- Y.-T. Lin, K.-T. Lin, K.-T. Wang and B. Weinstein, Cell. Mol. Life Sci., 1966, 22, 140–141 CrossRef CAS.
- E. Zavarin, J. Org. Chem., 1962, 27, 3368–3370 CAS.
- Y. Morita, E. Matsumura, H. Tsujibo, M. Yasuda, T. Okabe, Y. Sakagami, Y. Kumeda, N. Ishida and Y. Inamori, Biol. Pharm. Bull., 2002, 25, 981–985 CrossRef CAS.
- J. Yamada, K. Fujita and K. Sakai, Phytochemistry, 2002, 60, 447–450 CrossRef CAS.
- (a) K. Kintaka, H. Ono, S. Tsubotani, S. Harada and H. Okazaki, J. Antibiot., 1984, 37, 1294–1300 CAS; (b) S. Tsubotani, Y. Wada, K. Kamiya, H. Okazaki and S. Harada, Tetrahedron Lett., 1984, 25, 419–422 CrossRef; (c) Y. Kawano, Y. Nagawa, H. Nakanishi, H. Nakajima, M. Matsuo and T. Higashihara, J. Mar. Biotechnol., 1997, 5, 225–229 Search PubMed; (d) Y. Kawano, M. Asada, M. Inoue, K. Nakagomi, S. Oka and T. Higashihara, J. Mar. Biotechnol., 1998, 6, 49–52 Search PubMed.
- (a) B. W. Bycroft, A. D. Roberts and A. A. Higton, Dictionary of Antibiotics and Related Substances, Chapman and Hall, New York, NY, 1988 Search PubMed; (b) K. Tanaka, E. Kondo, K. Matsumoto, M. Hattutori and N. Tsuji, Unexamined patent application, JPO, Kokai, 1984-205994, Shionogi and Co., Ltd., Japan (in Japanese) Search PubMed.
- (a) L. Liang, Ph.D. thesis, University of Göttingen, 2003 Search PubMed; (b) T. Brinkhoff, G. Bach, T. Heidorn, L. Liang, A. Schlingloff and M. Simon, Appl. Environ. Microbiol., 2004, 70, 2560–2565 CrossRef CAS; (c) J. B. Bruhn, K. F. Nielsen, M. Hjelm, M. Hansen, J. Bresciani, S. Schulz and L. Gram, Appl. Environ. Microbiol., 2005, 71, 7263–7270 CrossRef CAS; (d) J. B. Bruhn, L. Gram and R. Belas, Appl. Environ. Microbiol., 2007, 73, 442–450 CAS.
- H. Laatsch, in Frontiers in Marine Biotechnology, ed. P. Proksch and E. G. Müller, Taylor and Francis, 2006, Chapter 6 Search PubMed.
- H. Korth, G. Brüsewitz and G. Pulverer, Zentralbl. Bakteriol. Mikrobiol. Hyg., Abt. 1, Orig. A, 1982, 252, 83–86 Search PubMed.
- T. Itô, T. Arai, Y. Ohashi and Y. Sasada, Agric. Biol. Chem., 1981, 45, 1689–1692 CAS.
- R. H. Angawi, D. C. Swenson, J. B. Gloer and D. T. Wicklow, Tetrahedron Lett., 2003, 44, 7593–7596 CrossRef CAS.
- R. H. Angawi, D. C. Swenson, J. B. Gloer and D. T. Wicklow, J. Nat. Prod., 2005, 68, 212–216 CrossRef CAS.
- J. Bitzer, Ph.D. thesis, University of Göttingen, 2005 Search PubMed.
- B. Karlsson, A.-M. Pilotti and A.-C. Wiehager, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, B32, 3118–3121 CrossRef CAS.
- (a) M. E. Raggatt, T. J. Simpson and M. I. Chicarelli-Robinson, Chem. Commun., 1997, 2245–2246 RSC; (b) A. M. Bailey, R. J. Cox, K. Harley, C. M. Lazarus, T. J. Simpson and E. Skellam, Chem. Commun., 2007, 4053–4055 RSC.
- P. Cai, D. Smiith, B. Cunningham, S. Brown-Shimer, B. Katz, C. Pearce, D. Venables and D. Houck, J. Nat. Prod., 1998, 61, 791–795 CrossRef CAS.
- J. E. Baldwin, A. V. W. Mayweg, K. Neumann and G. P. Pritchard, Org. Lett., 1999, 1, 1933–1935 CrossRef CAS.
- F. Mayerl, Q. Gao, S. Huang, G. E. Klohr, J. A. Matson, D. R. Gustavson, D. M. Pirnik, R. L. Berry, C. Fairchild and W. C. Rose, J. Antibiot., 1993, 46, 1082–1088 CAS.
- (a) J. G. Buta, J. L. Flippen and W. R. Lusby, J. Org. Chem., 1978, 43, 1002–1003 CrossRef CAS; (b) B. Frey, A. P. Wells, D. H. Rogers and L. N. Mander, J. Am. Chem. Soc., 1998, 120, 1914–1915 CrossRef CAS.
- J. Du, M.-H. Chiu and R.-L. Nie, J. Nat. Prod., 1999, 62, 1664–1665 CrossRef CAS.
- J. V. Silverton, C. Kabuto, K. T. Buck and M. P. Cava, J. Am. Chem. Soc., 1977, 99, 6708–6712 CrossRef CAS.
- D. L. Boger and K. Takahashi, J. Am. Chem. Soc., 1995, 117, 12452–12459 CrossRef CAS.
- W. Schüep, J. F. Blount, T. H. Williams and A. Stempel, J. Antibiot., 1978, 31, 1226–1232 CAS.
- K. W. Bentley, The Isoquinoline Alkaloids (Chemistry and Biochemistry of Organic Natural Products), Harwood Academic Press, Amsterdam, 1998, ch. 20, pp. 389–445 Search PubMed.
- O. J. Eigsti and P. Dustin, Colchicine in Agriculture, Medicine, Biology, and Chemistry, Iowa State College Press, Ames, Iowa, 1955 Search PubMed.
- H. G. Capraro and A. Brossi, in The Alkaloids, ed. A. Brossi, Academic Press, New York, NY, 1984, vol. 23, pp. 1–70 Search PubMed.
- O. Boye and A. Brossi, in The Alkaloids, ed. A. Brossi and G. A. Cordell, Academic Press, New York, NY, 1992, vol. 41, pp. 125–178 Search PubMed.
- T. Graening and H. G. Schmalz, Angew. Chem., Int. Ed., 2004, 43, 3230–3256 CrossRef CAS.
- T. H. Al-Tel, M. H. Abu Zarga, S. S. Sabri, A. J. Freyer and M. Shamma, J. Nat. Prod., 1990, 53, 623–629 CAS.
- O. P. Suri, B. D. Gupta, K. A. Suri, A. K. Sharma and N. K. Satti, Nat. Prod. Res., 2001, 154, 217–219 Search PubMed.
- R. Bentley and C. P. Thiessen, J. Biol. Chem., 1963, 238, 1880–1888 CAS.
- R. Bentley, Biochim. Biophys. Acta, 1958, 29, 666–667 CAS.
- S. W. Tanenbaum, E. W. Bassett and N. O. Kaplan, Arch. Biochem. Biophys., 1959, 81, 169–186 CrossRef CAS.
- R. Bentley, J. Biol. Chem., 1963, 238, 1889–1894 CAS.
- W. von E. Doering and D. B. Denney, J. Am. Chem. Soc., 1955, 77, 4619–4622 CrossRef.
- (a) I. G. Andrew and W. Segal, J. Chem. Soc., 1964, 607–613 RSC; (b) W. Segal, J. Chem. Soc., 1964, 613–615 RSC.
- R. Bentley, J. Biol. Chem., 1963, 238, 1895–1902 CAS.
- R. Bentley, Biochem. Biophys. Res. Commun., 1960, 3, 215–219 CrossRef CAS.
- (a) R. Bentley and C. P. Thiessen, Nature, 1959, 184, 552–553 CrossRef CAS; (b) R. Bentley and C. P. Thiessen, J. Biol. Chem., 1963, 238, 3811–3816 CAS.
- (a) R. Bentley, J. A. Ghaphery and J. G. Keil, Arch. Biochem. Biophys., 1965, 111, 80–87 CrossRef CAS; (b) R. Bentley and P. M. Zwitkowits, J. Am. Chem. Soc., 1967, 89, 676–680 CrossRef CAS; (c) R. Bentley and P. M. Zwitkowits, J. Am. Chem. Soc., 1967, 89, 681–685 CrossRef CAS.
- (a) P. E. Brenneisen, T. E. Acker and S. W. Tanenbaum, J. Am. Chem. Soc., 1964, 86, 1264–1265 CrossRef CAS; (b) T. E. Acker, P. E. Brenneisen and S. W. Tanenbaum, J. Am. Chem. Soc., 1966, 88, 834–837 CrossRef CAS.
- (a) R. J. Light, T. M. Harris and C. M. Harris, Biochemistry, 1966, 5, 4037–4043 CrossRef CAS; (b) R. J. Light, J. Agric. Food Chem., 1970, 18, 260–267 CrossRef CAS; (c) S. W. Tanenbaum, S. Nakajima and G. Marx, Biotechnol. Bioeng., 1969, 11, 1135–1156 CrossRef CAS; (d) G. S. Marx and S. W. Tanenbaum, J. Am. Chem. Soc., 1968, 90, 5302–5303 CrossRef CAS.
- A. I. Scott, H. Guilford and E. Lee, J. Am. Chem. Soc., 1971, 93, 3534–3535 CrossRef CAS.
- A. I. Scott and K. J. Wiesner, J. Chem. Soc., Chem. Commun., 1972, 1075–1077 RSC.
- M. C. O'Sullivan and J. M. Schwab, Bioorg. Chem., 1995, 23, 131–143 CrossRef CAS.
- J. D. Linton, R. M. Austin and D. E. Haugh, Biotechnol. Bioeng., 1984, 26, 1464.
- L. D. Ferretti and J. H. Richards, Proc. Natl. Acad. Sci. U. S. A., 1960, 46, 1438–1444 CrossRef CAS.
- A. I. Scott and E. Lee, J. Chem. Soc., Chem. Commun., 1972, 655–656 RSC.
- J. Wright, D. G. Smith, A. J. McInnes and L. C. Vining, Can. J. Biochem., 1969, 47, 945–949 CAS.
- S. W. Tanenbaum and E. W. Bassett, Biochim. Biophys. Acta, 1962, 59, 524–526 CAS.
- G. Gold and R. Bentley, Bioorg. Chem., 1974, 3, 377–385 CrossRef CAS.
- S. Takenaka, N. Ojima and S. Seto, J. Chem. Soc., Chem. Commun., 1972, 391–392 RSC.
- Y. Yamamoto, K.-i. Nishimura and N. Kiriyama, Chem. Pharm. Bull., 1976, 24, 1853–1859 CAS.
- H. Verachtert and L. Hanssens, Ann. Microbiol., 1975, 126A, 143–149 Search PubMed.
- N. Kawahara, K. Nozawa, S. Nakajima, S.-i. Udegawa and K.-i. Kawai, Chem. Pharm. Bull., 1988, 36, 398–400 CAS.
- C. F. Culberson and M. E. Hale, Bryologist, 1973, 76, 77–84 Search PubMed.
- L. X. Filho, E. C. Pereira, C. Vincente and M.-E. Legaz, ARKIVOC, 2004, 6, 5–11.
- (a) J. Zhao, K. Fujita and K. Sakai, Biosci., Biotechnol., Biochem., 2001, 65, 1027–1032 CrossRef CAS , and references therein; (b) J. Zhao, K. Fujita, J. Yamada and K. Sakai, Appl. Microbiol. Biotechnol., 2001, 55, 301–305 CrossRef CAS.
- J.-P. Haluk and C. Roussel-Bousta, Ann. For. Sci., 2003, 60, 271–276 Search PubMed , and references therein.
- K. Sakai, T. Yamaguchi and R. Itose, Mokuzai Gakkaishi, 1997, 43, 696–698 Search PubMed.
- K. Fujita, T. Yamaguchu, R. Itose and K. Sakai, J. Plant Physiol., 2000, 156, 462–467 CAS.
- Y. Matsunaga, K. Fujita, J. Yamada, T. Ashitani and K. Sakai, Nat. Prod. Res., 2003, 17, 441–443 CrossRef CAS.
- J. Zhao, Y. Matsunaga, K. Fujita and K. Sakai, Metab. Eng., 2006, 8, 14–29 CrossRef CAS.
- J. Yamada, K. Fujita and K. Sakai, J. Wood Sci., 2003, 49, 172–175 Search PubMed.
- J. Yamada, K. Fujita and K. Sakai, Phytochemistry, 2002, 60, 447–450 CrossRef CAS.
- J. Zhao, L. C. Davis and R. Verpoorte, Biotechnol. Adv., 2005, 23, 283–333 CrossRef CAS.
- M. A. Phillips, J. Bohlmann and J. Gershenzon, Phytochem. Rev., 2006, 5, 179–189 Search PubMed.
- J. Zhao and K. Sakai, New Phytol., 2003, 159, 719–731 Search PubMed.
- J. Zhao, Y. Guo, K. Fujita and K. Sakai, New Phytol., 2003, 161, 723–733 Search PubMed.
- J. Zhao, S.-H. Zeng, K. Fujita and K. Sakai, J. Exp. Bot., 2004, 55, 1003–1012 CrossRef CAS.
- J. Zhao, K. Fujita and K. Sakai, Biotechnol. Bioeng., 2005, 90, 621–631 CrossRef CAS.
- J. Zhao, K. Fujita and K. Sakai, New Phytol., 2007, 175, 215–219 Search PubMed.
- (a) A. R. Battersby, R. Binks and D. A. Yeowell, Proc. Chem. Soc., 1964, 86 Search PubMed; (b) R. D. Hill and A. M. Unrau, Can. J. Chem., 1965, 43, 709–711 CrossRef CAS.
- A. R. Battersby and R. B. Herbert, Proc. Chem. Soc., 1964, 260 Search PubMed.
- R. B. Herbert, Nat. Prod. Rep., 2001, 18, 50–65 RSC , see references therein and earlier reports in this series.
- E. Leete, in Biosynthesis, Senior Reporter, T. A. Geisman, The Chemical Society, London, 1973, vol. 2, pp. 106–182 Search PubMed.
- P. W. Sheldrake, R. E. Suckling, R. N. Woodhouse, A. T. Murtagh, R. B. Herbert, A. C. Baker, J. Staunton and A. R. Battersby, J. Chem. Soc., Perkin Trans. 1, 1998, 3003–3009 RSC , and references therein.
- A. R. Battersby, R. B. Herbert, E. McDonald, R. Ramage and J. H. Clements, J. Chem. Soc., Perkin Trans. 1, 1972, 1741–1746 RSC.
- A. Nasreen, M. Rueffer and M. H. Zenk, Tetrahedron Lett., 1996, 37, 8161–8164 CrossRef CAS.
- M. S. Al-Mahmoud, F. Q. Alali, K. Tawaha and R. M. Qasaymeh, Nat. Prod. Res., 2006, 20, 153–160 CrossRef CAS.
- D. E. Cane, Z. Wu and J. E. Van Epp, J. Am. Chem. Soc., 1992, 114, 8479–8483 CrossRef CAS.
- (a) M. Friedman, P. R. Henika, C. E. Levin, R. E. Mandrell and N. Kozukue, J. Food Prot., 2006, 69, 354–361 CAS; (b) M. Friedman, Mol. Nutr. Food Res., 2007, 51, 116–134 CrossRef CAS.
- N. Ahmad and H. Mukhtar, Nutr. Revs., 1999, 57, 78–83 Search PubMed.
- M. Friedman, C. E. Levine, S.-H. Choi, E. Kozukue and N. Kozukue, J. Food Sci., 2006, 71, C328–C337 CrossRef CAS.
- T. Tanaka, K. Inoue, Y. Betsumiya, C. Mine and I. Kouno, J. Agric. Food Chem., 2001, 49, 5785–5789 CrossRef CAS.
- J.-W. Jhoo, S. Sang, G.-J. Wei, T. C. Lee, R. T. Rosen and C.-T. Ho, J. Food Lipids, 2004, 11, 89–103 Search PubMed.
- S. Sang, J. D. Lambert, S. Tian, J. Hong, Z. Hou, J.-H. Ryu, R. E. Stark, R. T. Rosen, M.-T. Huang, C. S. Yang and C.-T. Ho, Bioorg. Med. Chem., 2004, 12, 459–467 CrossRef CAS.
- T. Tanaka, C. Mine, K. Inoue, M. Matsuda and I. Kuono, J. Agric. Food Chem., 2002, 50, 2142–2148 CrossRef CAS.
- S. Sang, S. Tian, R. E. Stark, C. S. Yang and C.-T. Ho, Bioorg. Med. Chem., 2004, 12, 3009–3017 CrossRef CAS.
- X. Wan, H. E. Nursten, Y. Cai, A. L. Davis, J. P. G. Wilkins and A. P. Davies, J. Sci. Food Agric., 1997, 74, 401–408 CrossRef CAS.
- A. E. Oxford, H. Raistrick and G. Smith, Chem. Ind., 1942, 61, 485–487 Search PubMed.
- T. J. Trust, Antimicrob. Agents Chemother., 1975, 7, 500–506 CAS.
- Y. Morita, E. Matsumura, T. Okabe, M. Shibata, M. Sugiura, T. Ohe, H. Tsujibo, N. Ishida and Y. Inamori, Biol. Pharm. Bull., 2003, 26, 1487–1490 CrossRef CAS.
- M. Baya, P. Soulounganga, E. Gelhaye and P. Gérardin, Pest Manage. Sci., 2001, 57, 833–838 Search PubMed.
- Y. Arima, Y. Nakai, R. Hayakawa and T. Nishino, J. Antimicrob. Chemother., 2003, 51, 113–122 CrossRef CAS.
- N. Imai, Y. Doi, K. Nabae, S. Tamano, A. Hagiwara, M. Kawabe, T. Ichihara, K. Ogawa and T. Shirai, J. Toxicol. Sci., 2006, 31, 357–370 Search PubMed.
- J. Zhao, Curr. Med. Chem., 2007, 14, 2597–2621 CrossRef CAS.
- S. Liu and H. Yamauchi, Biochem. Biophys. Res. Commun., 2006, 351, 26–32 CrossRef CAS.
- H. Yamano, T. Yamazaki, K. Sato, S. Shiga, T. Hagiwara, K. Ouchi and T. Kishimoto, Antimicrob. Agents Chemother., 2005, 49, 2519–2521 CrossRef CAS.
- (a) Y. Morita, E. Matsumura, H. Tsujibo, M. Yasuda, Y. Sakagami, T. Okabe, N. Ishida and Y. Inamori, Biol. Pharm. Bull., 2001, 24, 607–611 CrossRef CAS; (b) Y. Morita, E. Matsumura, T. Okabe, T. Fukui, M. Shibata, M. Sugiura, T. Ohe, H. Tsujibo, N. Ishida and Y. Inamori, Biol. Pharm. Bull., 2004, 27, 899–902 CrossRef CAS.
- (a) Y. Morita, E. Matsumura, T. Date, H. Tsujibo, M. Yasuda, T. Okabe, N. Ishida and Y. Inamori, Biol. Pharm. Bull., 2001, 24, 299–302 CrossRef; (b) Y. Morita, E. Matsumura, T. Okabe, T. Fukui, T. Ohe, N. Ishida and Y. Inamori, Biol. Pharm. Bull., 2004, 27, 1666–1669 CrossRef CAS.
- J. D. Lambert, D. Chen, C. Y. Wang, N. Ai, S. Sang, C.-T. Ho, W. J. Welsh and C. S. Yang, Bioorg. Med. Chem., 2005, 13, 2501–2507 CrossRef CAS.
- P.-T. Doulias, L. Nousis, B.-Z. Zhu, B. Frei and D. Galaris, Free Radical Res., 2005, 39, 125–135 CrossRef CAS.
- M. Yamato, J. Ando, K. Sakaki, K. Hashigaki, Y. Wataya, S. Tsukagoshi, T. Tashiro and T. Tsuro, J. Med. Chem., 1992, 35, 267–273 CrossRef CAS , and references therein.
- E. O. Freed and A. J. Mouland, Retrovirology, 2006, 3, 77–88 Search PubMed.
- S. R. Budihas, I. Gorshkova, S. Gaidamakov, A. Wamiru, M. K. Bona, M. A. Parniak, R. J. Crouch, J. B. McMahon, J. A. Beutler and S. F. J. Le Grice, Nucleic Acids Res., 2005, 33, 1249–1256 CrossRef CAS.
- E. A. Semenova, A. A. Johnson, C. Marchand, D. A. Davis, R. Yarchoan and Y. Pommier, Mol. Pharmacol., 2006, 69, 1454–1460 CrossRef CAS.
- K. Tomita, Y. Hoshino, Y. Nakakita, S. Umezawa, T. Miyaki, T. Oki and H. Kawaguchi, J. Antibiot., 1989, 42, 317–321 CAS.
- W. H. Blackwell, Poisonous and Medicinal Plants, Prentice Hall, Englewood Cliffs, NJ, 1990 Search PubMed.
- International Programme on Chemical Safety, Poisons Information Monograph 142; http://www.intox.org/databank/document/Plant/colchicum/pim142.htm.
- (a) A. Brossi, H. J. C. Yeh, M. Chrzanowska, J. Wolff, E. Hamel, C. M. Lin, F. Quin, M. Suffness and J. Silverton, Med. Res. Rev., 1988, 8, 77–94 CAS; (b) A. Brossi, J. Med. Chem., 1990, 33, 2311–2318 CrossRef CAS.
- (a) W. Zhang et al., Ann. Rheum. Dis., 2006, 65, 1301–1311 CrossRef; (b) W. Zhang et al., Ann. Rheum. Dis., 2006, 65, 1312–1324 CrossRef.
- T. Kallinich, D. Haffner, T. Niehues, K. Huss, E. Lainka, U. Neudorf, C. Schaefer, S. Stojanov, C. Timmann, R. Keitzer, H. Ozdogan and S. Ozen, Pediatrics, 2007, 119, e474–e483 Search PubMed.
- E. L. Eliel and S. H. Wilen, Stereochemistry of Organic Compounds, John Wiley & Sons Inc, New York, NY, 1994 Search PubMed.
- F. Büttner, S. Bergemann, D. Guénard, R. Gust, G. Seitz and S. Thoret, Bioorg. Med. Chem., 2005, 13, 3497–3511 CrossRef.
- A. R. Chaudhuri, P. Seetharamalu, P. M. Schwarz, F. H. Hausheer and R. F. Ludueña, J. Mol. Biol., 2000, 303, 679–692 CrossRef CAS.
- R. B. G. Ravelli, B. Gigant, P. A. Curmi, I. Jourdain, S. Lachkar, A. Sobel and M. Knossow, Nature, 2004, 428, 198–202 CrossRef CAS.
- R. M. Wanner, P. Spielmann, D. M. Stroka, G. Camenisch, I. Camenisch, A. Scheid, D. R. Houck, C. Bauer, M. Gassmann and R. H. Wenger, Blood, 2000, 96, 1558–1565 CAS.
| This journal is © The Royal Society of Chemistry 2008 |