Marine natural products: synthetic aspects
Jonathan C. Morrisa and Andrew J. Phillips*b
aSchool of Chemistry and Physics, University of Adelaide, Adelaide, Australia 5005
bDepartment of Chemistry and Biochemistry, University of Colorado, Boulder, Colorado 80309, USA. E-mail: Andrew.Phillips@colorado.edu; Fax: +1 303 492 0439; Tel: +1 303 735 2049
First published on 14th January 2008
Abstract
Covering: January to December 2006. Previous review: Nat. Prod. Rep., 2007, 24, 87
An overview of marine natural products synthesis during 2006 is provided. As with earlier installments in this series, the emphasis is on total syntheses of molecules of contemporary interest, new total syntheses, and syntheses that have resulted in structure confirmation or stereochemical assignments.
 Jonathan Morris | Jonathan Morris obtained his B.Sc. (Hons) degree from the University of Western Australia and his Ph.D. degree from The Australian National University in Canberra, Australia. After a postdoctoral appointment with Phil Magnus at the University of Texas at Austin, he took up his first academic position at the University of Canterbury, New Zealand. In 2004, he moved to the University of Adelaide. His research interests focus around the synthesis of biologically active natural products. |
 Andy Phillips | Andy Phillips obtained his B.Sc. (Hons) and Ph.D. degrees from the University of Canterbury in Christchurch, New Zealand. After a postdoctoral appointment with Peter Wipf at the University of Pittsburgh, he joined the faculty at the University of Colorado. His research interests are broadly defined by the development of new methods and strategies for the synthesis of complex natural products. |
1 Introduction
This review is designed to provide an overview of key features of the 2006 literature covering the synthesis of marine natural products and should act as a companion to the Marine Natural Products review published in this journal.1 The emphasis is on total syntheses of molecules of contemporary interest. Tabulated data for other syntheses are also provided. While every effort has been made to be comprehensive within these boundary conditions, we apologize in advance for any oversights.2 Review articles
A number of reviews that cover various aspects of marine natural products synthesis have appeared: ‘Small molecule natural products in the discovery of therapeutic agents: the synthesis connection’,2 ‘New methods of imidazole functionalization—from imidazole to marine alkaloids’,3 ‘Tricyclic marine alkaloids. Synthetic approaches to cylindricines, lepadiformine, and fasicularin’,4 ‘Synthesis of naturally occurring polyynes’,5 ‘Total syntheses of the antibacterial natural product abyssomycin C’,6 ‘Synthesis of naturally derived bioactive compounds of agricultural interest’,7 ‘Total synthesis of spiroketal containing natural products: kinetic vs. thermodynamic approaches’,8 ‘Macrolactonizations in the total synthesis of natural products’,9 ‘Synthetic studies towards the pectenotoxins: a review’,10 ‘Construction of fused polycyclic ethers by strategies involving ring-closing metathesis’,11 ‘Biomimetic synthesis of trans,syn,trans-fused polycyclic ethers’,12 ‘Synthetic studies of marine polycyclic ether, ciguatoxins’,13 ‘Synthesis of marine natural products with bicyclic and/or spirocyclic acetals’,14 ‘Total synthesis of medium-ring ethers from Laurencia red algae’,15 ‘Strategies for the synthesis of manzamine alkaloids’,16 ‘Total synthesis of marine macrolides’,17 and ‘Recent advances in total synthesis of marine polycyclic ethers’.18 Other reviews of relevance are cited in the text.3 Imine polyketide toxins
The azaspiracids continue to attract much attention from synthetic groups. Zhou and Carter have reported a 27-step synthesis of the C1–C26 northern portion of the revised structure of azaspiracid-1 (1, Scheme 1), starting from malic acid.19 The FGHI ring fragment has also proved to be a popular target, with the groups of Forsyth,20 Carter,21 and Sasaki22 all reporting syntheses.![The Nicolaou synthesis of azapiracid-2 and -3. Reagents and conditions: (1) tBuLi (2 eq.), 7 (1 eq.), Et2O, −78 °C; then 8; (2) Dess–Martin periodinane (1.2 eq.), NaHCO3, CH2Cl2, 81% (over 2 steps) for 9 (R1 = Me), 74% (over 2 steps) for 9 (R1 = H); (3) TMSOTf, CH2Cl2, −90 °C, 72% for 10 (R1 = Me), 65% for 10 (R1 = H); (4) 6, (6 eq.), nBuLi/nBu2Mg, THF, 25 °C, then −90 °C, then 4, TBAF, 65% for 11 (R1 = Me), 74% for 11 (R1 = H); (5) DIBAL-H, CH2Cl2, −90 °C; (6) TBAF, THF, 25 °C; (7) AcCl, 2,4,6-collidine, CH2Cl2, −78 °C; (8) PhI(OCOCF3)2, MeCN–pH 7 buffer (4 : 1), 0 °C, 38% (over 4 steps) for 12 (R1 = Me), 32% (over 4 steps) for 12 (R1 = H); (9) 5 (2 eq.), [Pd2(dba)3] (0.3 eq.), AsPh3 (0.3 eq.), LiCl (6 eq.), iPr2NEt (10 eq.), DMF, 25 °C, 80% for 13 (R1 = Me), 69% for 13 (R1 = H); (10) TBAF, THF, 0 °C; (11) NIS (2 eq.), NaHCO3, DMF, 0 °C; 48% for 14 (R1 = Me), 51% for 14 (R1 = H).](/image/article/2008/NP/b701533j/b701533j-s1.gif) | ||
| Scheme 1 The Nicolaou synthesis of azapiracid-2 and -3. Reagents and conditions: (1) tBuLi (2 eq.), 7 (1 eq.), Et2O, −78 °C; then 8; (2) Dess–Martin periodinane (1.2 eq.), NaHCO3, CH2Cl2, 81% (over 2 steps) for 9 (R1 = Me), 74% (over 2 steps) for 9 (R1 = H); (3) TMSOTf, CH2Cl2, −90 °C, 72% for 10 (R1 = Me), 65% for 10 (R1 = H); (4) 6, (6 eq.), nBuLi/nBu2Mg, THF, 25 °C, then −90 °C, then 4, TBAF, 65% for 11 (R1 = Me), 74% for 11 (R1 = H); (5) DIBAL-H, CH2Cl2, −90 °C; (6) TBAF, THF, 25 °C; (7) AcCl, 2,4,6-collidine, CH2Cl2, −78 °C; (8) PhI(OCOCF3)2, MeCN–pH 7 buffer (4 : 1), 0 °C, 38% (over 4 steps) for 12 (R1 = Me), 32% (over 4 steps) for 12 (R1 = H); (9) 5 (2 eq.), [Pd2(dba)3] (0.3 eq.), AsPh3 (0.3 eq.), LiCl (6 eq.), iPr2NEt (10 eq.), DMF, 25 °C, 80% for 13 (R1 = Me), 69% for 13 (R1 = H); (10) TBAF, THF, 0 °C; (11) NIS (2 eq.), NaHCO3, DMF, 0 °C; 48% for 14 (R1 = Me), 51% for 14 (R1 = H). | ||
Full details of the Nicolaou synthesis of azaspiracid-1 (1) have been reported in a series of articles.23 Furthermore, Nicolaou's group has reported the first syntheses of the revised structures of azaspiracid-2 2 and azaspiracid-3 3 (Scheme 1).24 This strategy utilizes a similar retrosynthetic analysis to the azaspiracid-1 synthesis, with the key fragments being pentafluorophenyl ester 4, dithiane 5, and the stannane 6. As azaspiracid-2 and -3 (2 and 3) are very similar, only the synthesis of azaspiracid-2 will be described in the text.
An important difference in the synthesis is the shorter and more efficient preparation of the ABCD ring system 10. This key material was prepared in 3 steps from the readily available iodide 7 and aldehyde 8. Halogen–lithium exchange of 7 was achieved upon treatment with tBuLi, and the resulting lithio compound was added to 8. Oxidation of the resulting alcohol with Dess–Martin periodinane afforded the ketone 9 in 81% yield for the two steps. A cascade reaction was initiated upon reaction of 9 with TMSOTf in CH2Cl2 at −90 °C, which generated the desired tetracyclic alcohol 10 in 72% yield. A seven-step sequence transformed 10 into the pentafluorophenyl ester 4. Reaction of ester 4 with the organometallic species generated when dithiane 5 was treated with nBuLi/nBu2Mg resulted in the formation of ketone 11 in 65% yield. A four-step sequence generated the allylic acetate 12, which was required for the key Stille coupling reaction with stannane 6. The stannane 6 had been previously utilized in the azaspiracid-1 synthesis. Reaction of 12 with stannane 6 under Stille coupling conditions ([Pd2(dba)3], AsPh3, LiCl, iPr2NEt, DMF, 25 °C) resulted in an excellent yield of 13 (80%). Selective removal of the TES group was achieved by reaction of TBAF at 0 °C, and the resulting alcohol was reacted with N-iodosuccinimide to generate the G ring of the azaspiracid 14. Functional group manipulation then transformed 14 into azaspiracid-2 (2). Pleasingly, the spectroscopic data of the synthetic material matched that reported for the natural product, thus confirming the revision of the structure. Nicolaou has also recently published a full account of this synthesis, with the new synthesis also being used to prepare azaspiracid-1 (1).25
With the azaspiracids posing a significant hazard to humans, there is a need for new methods for detecting these toxins, as existing methods are not suitable. Forsyth has been able to generate antibodies that can detect natural azaspiracids.26 The antibodies were raised against a synthetic hapten that represents the common C28–C40 domain of the azaspiracids. This work highlights how synthetic organic chemistry can play a major role in accessing materials that would be otherwise be inaccessible.
The synthesis of pteriatoxins A, B and C (15 and 16, Scheme 2) has been described by the Kishi group using an intramolecular Diels–Alder strategy similar to their earlier synthesis of pinnatoxin A.27 The overall strategy consists of dissection of the molecules into three key building blocks: dithiane 17, iodide 18, and vinyl iodide 19. In the forward sense the first key coupling between 17 and 18 was achieved by deprotonation of the dithiane with tBuLi and alkylation with the iodide. After removal of the TMS group (K2CO3, MeOH), 20 was obtained in 73% yield over the two steps. Removal of the orthoester (PPTS then K2CO3, MeOH, 73%) was followed by oxidative removal of the dithiane with PIFA, which was accompanied by cyclization to the bicycloketal to give 21 in 74% yield. Oxidation of the primary alcohol (Parikh–Doering, 83%) generated aldehyde 22 and was followed by the introduction of the C1–C5 domain by Nozaki–Hiyama–Kishi reaction (22 → 23, 84%). Dess–Martin oxidation (79%) followed by protecting group exchange at C34 and C35 (from TBS to p-methoxybenzoyl—necessary for improved reactivity in the Diels–Alder) gave 25. When 25 was heated to 170 °C, intramolecular Diels–Alder cyclization ensued to give an 88% yield of the cycloadducts, from which 51% of the desired exo product (26) could be isolated. At this juncture the majority of the carbon skeleton was in place, and the final steps commenced with conversion of the diester to the epoxide 27 (K2CO3, then HF/pyr, 74%, followed by TsCl then K2CO3, 78%) and epoxide opening with N-Boc-L-Cys(SH)-OCHPh2. The epoxide opening occurred to give a 2 : 1 ratio of 28 and 29 in 85% yield, although they were not separable at this juncture. The synthesis was then completed by Alloc deprotection (Pd(PPh3)4, AcOH, 72%), cyclization to the imine (2,4,6-triisopropylbenzoic acid, Et3N, xylene, 80 °C) and removal of the diphenylmethyl ester (TFA, CH2 Cl2). Separation and final purification by preparative LC gave pure and (34R,2′R)-pteriatoxin B/C. The same sequence was used to prepare the other C34/C2′ diastereoisomers. After careful analysis and HPLC it was established that natural pteriatoxin A has the 34S,2′R configuration.28 As natural pteriatoxins B and C were isolated as a 1 : 1 mixture, confirmation of the stereochemistry proved to be difficult, but after a suitable HPLC method was established it was found that the natural pteriatoxins B and C matched the synthetic (34R,2′R) and (34S,2′R) stereoisomers respectively.
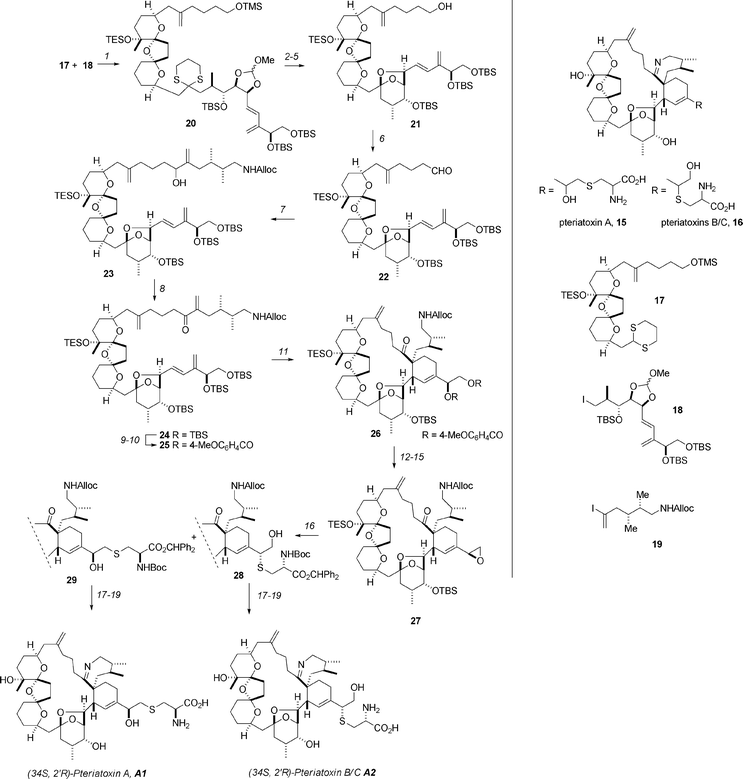 | ||
| Scheme 2 The Kishi synthesis of the pteriatoxins. Reagents and conditions: (1) 17, tBuLi, HMPA; (2) K2CO3, MeOH, 73% over 2 steps; (3) PPTS; (4) K2CO3, MeOH, 73% over 2 steps; (5) PIFA, 74%; (6) SO3·Py, DMSO, 83%; (7) 19, NiCl2, CrCl2, 84%; (8) Dess–Martin periodinane, 79%; (9) HF·Py, Py; (10) 4-MeOC6H4COCl, NEt3; (11) 170 °C, dodecane, 51%; (12) K2CO3; (13) HF·Py, Py, 74% over 2 steps; (14) TsCl; (15) K2CO3, 78% over 2 steps; (16) N-Boc-L-Cys(SH)-OCHPh2 (an inseparable mixture of 28 and 29), 85%; (17) Pd(PPh3)4, AcOH, 72%; (18) 1,3,5-iPr3C6H2CO2H/Et3N salt, 80 °C, xylene, 51%; (19) TFA, CH2Cl2, followed by HPLC separation. PIFA = phenyliodine(III) bis(trifluoroacetate). | ||
4 Polyethers
Sasaki has completed a synthesis of the proposed structure of brevenal 44 and uncovered that the reported spectroscopic data did not match with that obtained for the synthetic material.29 The synthesis employs Sasaki's Suzuki-coupling strategy for the subunit couplings (Scheme 3), the first of which involves the connection of 30 and 31 to give 32. Immediate hydroboration and oxidative workup provided 33 in an impressive 84% for 2 steps. An 8-step sequence of straightforward transformations gave oxathioacetal 35. Protection of the free alcohol with TBSOTf (Et3N, CH2Cl2, 97%) was followed by the oxidation of the sulfide and methylation with Me3Al to give the complete A→E ring-containing domain 37 in 92% yield. The endgame commenced with protecting group manipulation that involved the replacement of the Bn ether with a TBS group (LiDBB, 99% then TBSOTf, Et3N, 98%) and selective removal of the primary TBDPS using AcOH-buffered TBAF. Oxidation of the alcohol to the aldehyde with Dess–Martin periodinane, conversion to the alkyne with the Ohira–Bestmann–Gilbert–Seyferth reagent, and methylation (nBuLi, MeI) gave alkyne 38 in 99% yield for 3 steps. Interchange of the TBS ether for TES ethers, followed by silylcupration–iodination (PhMe2SiLi, CuCN, then NIS) gave iodide 40 in 94% yield over 4 steps and set the stage for introduction of the C1–C3 subunit by Stille coupling. Using Liebskind's conditions, cross-coupling with stannane 45 was readily achieved to give 41 in 63% yield. Silylation of the primary alcohol with TBDPSCl and careful removal of the primary TES ether gave 42. Introduction of the C30–C33 diene could then be achieved by oxidation, olefination with Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2CH2SePh and subsequent selenoxide oxidation–elimination (42 → 43). The removal of the TBDPS group with tris(dimethylamino)sulfonium difluorotrimethylsilicate (TASF, 79%) followed by oxidation with MnO2 gave brevenal's proposed structure. Unfortunately the spectroscopic data for synthetic and isolated material did not match, and ultimately a new structure, the C26-epimer of brevenal (46), was proposed as the correct structure. The newly proposed structure has since been confirmed by synthesis.30
CHCH2CH2SePh and subsequent selenoxide oxidation–elimination (42 → 43). The removal of the TBDPS group with tris(dimethylamino)sulfonium difluorotrimethylsilicate (TASF, 79%) followed by oxidation with MnO2 gave brevenal's proposed structure. Unfortunately the spectroscopic data for synthetic and isolated material did not match, and ultimately a new structure, the C26-epimer of brevenal (46), was proposed as the correct structure. The newly proposed structure has since been confirmed by synthesis.30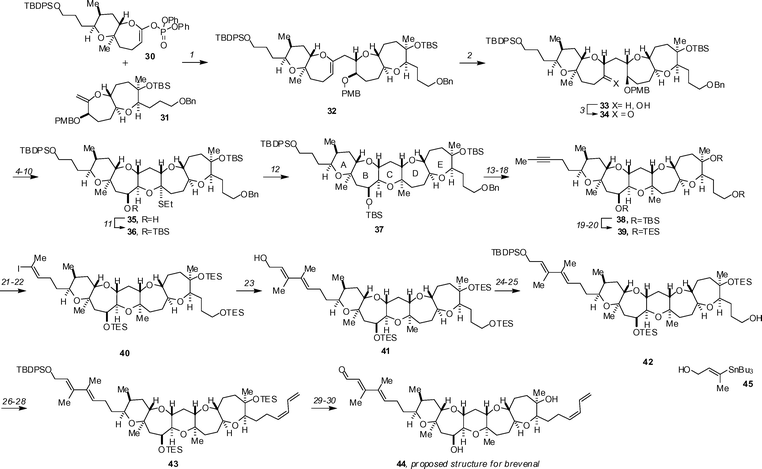 | ||
| Scheme 3 The Sasaki synthesis of the proposed structure of brevenal. Reagents and conditions: (1) 9-BBN, THF, rt; then 3 M aq. Cs2CO3, Pd(PPh3)4, DMF, 50 °C; (2) BH3·SMe2, THF, 0 °C to rt; then aq. NaHCO3, H2O2, 0 °C to rt, 84% (two steps); (3) TPAP, NMO, 4 Å MS, CH2Cl2, 0 °C, 98%; (4) LHMDS, TMSCl, Et3N, THF, −78 °C; (5) OsO4, NMO, THF–H2O, rt, 87% (two steps); (6) DIBALH, THF, −78 °C, 76%; (7) TESOTf, Et3N, CH2Cl2, 0 °C; (8) DDQ, CH2Cl2–pH 7 buffer, rt; (9) TPAP, NMO, 4 Å MS, CH2Cl2, 0 °C, 88% (three steps); (10) EtSH, Zn(OTf)2, THF, rt, 79%; (11) TBSOTf, Et3N, CH2Cl2, 0 °C, 97%; (12) MCPBA, CH2Cl2, −78 °C; then AlMe3, −78 to 0 °C, 92%; (13) LiDBB, THF, −78 °C, 99%; (14) TBSOTf, Et3N, CH2Cl2, 0 °C, 98%; (15) TBAF, AcOH, THF, rt, 78% after three recycles; (16) Dess–Martin periodinane, CH2Cl2, rt; (17) Ohira–Bestmann reagent, K2CO3, MeOH, rt; (18) nBuLi, THF–HMPA, −78 °C; then MeI, rt, 99% (three steps); (19) HF·Py, THF, 0 °C to rt, 96%; (20) TESOTf, Et3N, CH2Cl2, 0 °C, 99%; (21) PhMe2SiLi, CuCN, THF, −78 to 0 °C; (22) NIS, CH3CN–THF, 0 °C to rt, 99% (two steps); (23) 45, Pd2(dba)3, Ph3As, CuTC, 1 : 1 DMSO–THF, rt, 63%; (24) TBDPSCl, imidazole, DMF, 0 °C; (25) PPTS, 4 : 1 CH2Cl2–MeOH, 0 °C, 74% (two steps); (26) SO3·Py, Et3N, DMSO–CH2Cl2, 0 °C; (27) Ph3PCH2CH2CH2SePhBr, nBuLi, THF–HMPA, −78 °C to rt, 97% (two steps); (28) H2O2, NaHCO3, THF, rt, 77%; (29) TASF, DMF–THF, 0 C to rt, 79%; (30) MnO2, CH2Cl2, rt, quant. | ||
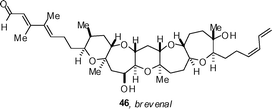
In other papers on brevetoxin-class polyethers, Crimmins has described a synthesis of the GHIJ ring system,31 and an improved route to the A→E ring system.32 Kadota has also provided details on an improved, 37-step, synthesis of the A→G ring fragment of brevetoxin B,33 and Nelson has reported a concise synthesis of a model system for the ABC ring system of hemibrevetoxin B.34
Inoue and Hirama have completed syntheses of ciguatoxin 56 and 51-hydroxyciguatoxin 54, the two most toxic members of the ciguatoxin family (Scheme 4). The syntheses have their foundations in the earlier work from the Tohuku University groups and also exploits newly developed selectivity in a key radical cyclization. The synthesis of 51-hydroxyciguatoxin commences with the coupling of advanced subunits 47 (1 eq.) and 48 (1.3 eq.) by treatment with AgOTf in the presence of di-tert-butylmethylpyridine to give oxa-thioacetal 49 in 70% yield. Removal of the TIPS protecting group (TBAF, 100%) and reaction with pentafluorophenyl propiolate in the presence of PMe3 gave acrylate 50. In an impressive example of the effects of electronics on radical cyclizations, treatment of this compound with Bu3SnH and AIBN in PhMe at 85 °C followed by aqueous workup gave 74% of the desired 7-exo cyclization product 51 (along with 10% of the PFP ester of the 6-exo product). This acid was readily advanced to olefin 52 by methylation (TMSCHN2, 87%), reduction with DIBAL-H and Wittig olefination (98% over 2 steps). Olefin metathesis with 20% Grubbs’ 1st-generation catalyst at 40 °C gave ring closure of the 9-membered ring (52 → 53) in 93% yield. Subsequent oxidative removal of the four (2-naphthyl)methyl ethers (DDQ, 59%) completed the synthesis. In a similar vein, ciguatoxin was assembled by cyclization of oxathioacetal 55. The key radical cyclization in this case provided a 59% yield of the key 7-exo cyclization (55 → 56, Scheme 2). Inoue and Hirama have also reported the synthesis of an A→E ring fragment that employs an acyl radical cyclization for the synthesis of the D ring.35 Other papers on synthetic studies directed toward the ciguatoxins include a synthesis of the FGHI ring domain36 and a synthesis of the H→M ring system.37
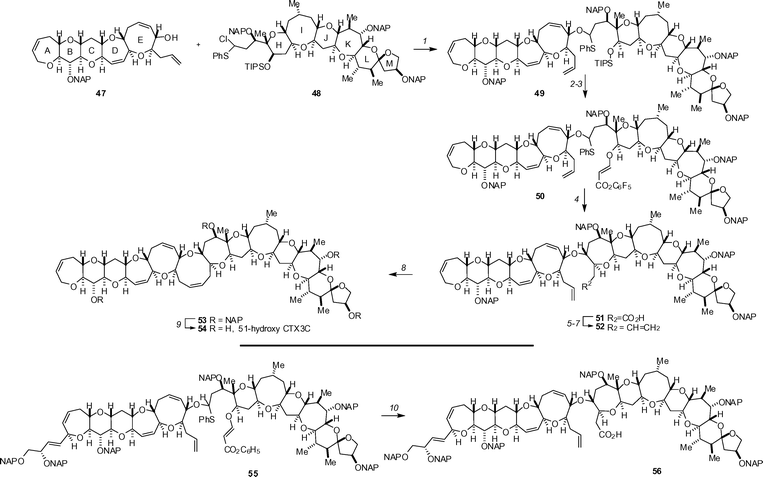 | ||
| Scheme 4 The key reactions of the Inoue–Hirama synthesis of 51-hydroxyciguatoxin (top) and the key radical cyclization en route to ciguatoxin (bottom). Reagents and conditions: (1) 47 (1.0 eq.), 48 (1.3 eq.), AgOTf (2.5 eq.), di-tert-butylmethylpyridine (5.0 eq.), 4 Å MS, CH2Cl2–CCl4, −70 to 0 °C, 70% from 47; (2) TBAF, THF, 35 °C, 100%; (3) pentafluorophenyl propiolate, PMe3, CH2Cl2, rt, 95%; (4) nBu3SnH, AIBN, PhMe, 85 °C, 74%; (5) TMSCHN2, MeOH–PhH, rt, 87%; (6) DIBAL, CH2Cl2, −90 °C; (7) Ph3PCH3Br, tBuOK, THF, 0 °C, 98% (2 steps); (8) Grubbs I catalyst (20 mol%), CH2Cl2, 40 °C, 93%; (9) DDQ (10 eq.), CH2Cl2–H2O (2 : 1), rt, 59%; (10) nBu3SnH, AIBN, PhMe, 85 °C, 59%. | ||
In synthetic studies on yessotoxin, a convergent synthesis of the CDEF ring fragment that employs α-cyanoether chemistry has been reported,38 as has a synthesis of the ABC and IJ ring fragments.39 Full details of the Rainier synthesis of gambierol have been reported.40 Sasaki's synthetic work on gymnocin A has allowed some structure–activity relationships to be elucidated.41
In other papers on polyether synthesis, Jamison has continued his studies of ‘biomimetic’ cyclization of poly-epoxides. The combination of a Me3Si-directing group and Cs2CO3/CsF in MeOH results in control of the cyclization mode to give tetrahydropyrans, and this is exemplified by the synthesis of the THP tetrad 63 (Scheme 5).42
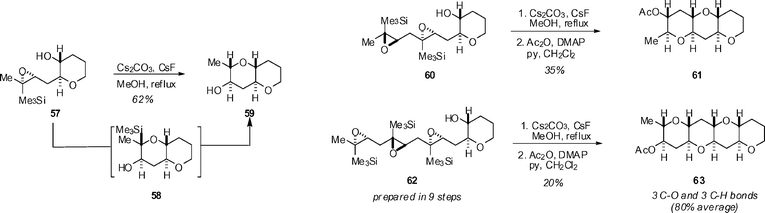 | ||
| Scheme 5 Jamison's Si-directed polyepoxide cyclizations. | ||
5 Guanidinium alkaloids
The synthesis of polycylic guanidinium alkaloids continues to be an active area of research. Gin and co-workers43 have synthesised batzelladine A (73), utilizing their diastereoselective [4 + 2] annulation of vinyl carbodiimides with a chiral N-alkylimine as described in their earlier synthesis44 of batzelladine D (Scheme 6). The mixture of geometrical isomers 64, which was generated in 4 steps from pent-4-yn-1-ol, was reacted with the chiral imine 65 at 23 °C to afford the dihydropyrimidine 66 as a single diastereomer in 89% yield. Cleavage of the pyrrolidine ring of 66 was achieved in a four-step sequence and generated the alkene 67, which underwent cross-metathesis with the TBDPS ether of hept-6-en-1-ol when treated with Grubbs’ second-generation catalyst. Reaction with hydrogen in the presence of palladium hydroxide resulted in the removal of the benzyl and p-methoxybenzyl groups, as well as the hydrogenation of the alkene group, to afford the alcohol 68 in 90% yield. An efficient five-step sequence provided 69, which is an appropriately derivatized bicyclic guanidine core of batzelladine A. This fragment was coupled with the caesium carboxylate of 70, which had previously been utilized in the synthesis of batzelladine D, to form ester 71 in 68% yield. Completion of the synthesis required the formation of the final ring of the tricyclic guanidine core, which was readily achieved by a stereoselective intramolecular iodo-amination, followed by reductive de-iodination. Global deprotection with trifluoroacetic acid completed the total synthesis.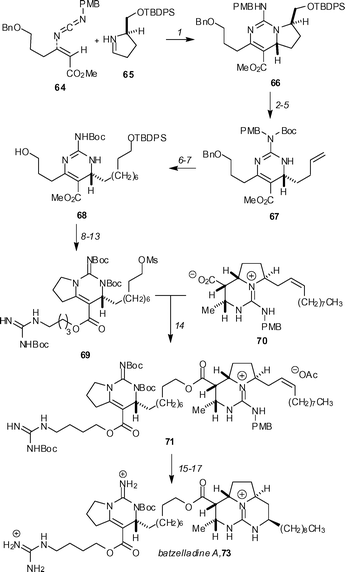 | ||
| Scheme 6 The Gin synthesis of batzelladine A. Reagents and conditions: (1) (ClCH2)2, 23 °C (89%); (2) NaH, Boc2O, DMAP, THF, 23 °C (86%); (3) TBAF, AcOH, THF, 23 °C (91%); (4) I2, PPh3, imidazole, PhMe, 23 °C (97%); (5) tBuLi, THF, −78 °C (93%); (6) CH2CH(CH2)5OTBDPS, Grubbs II, CH2Cl2, 45 °C (73%); (7) 10% Pd(OH)2, AcOH, H2 (1 atm), MeOH, CH2Cl2, 23 °C (90%); (8) DIAD, PPh3, PhMe, 23 °C (93%); (9) NaH, Boc2O, DMAP, THF, 23 °C (85%); (10) EtSLi, HMPA, 23 °C; (11) BOPCl, Et3N, CH2Cl2, 23 °C (55% over two steps); (12) TBAF, THF, 23 °C (92%); (13) MsCl, Et3N, CH2Cl2, 0 °C (91%); (14) Cs2CO3, DMF. 50 °C (68%); (15) I2, Cs2CO3, DME, 23 °C (72%); (16) 10% Pd/C, Et3N, H2 (1 atm), EtOAc (71%); (17) TFA, 0 °C (75%). | ||
Overman's continuing work in the synthesis of the batzelladines has revealed that the proposed structure for batzelladine F 72 is incorrect.45 Cohen and Overman devised a convergent strategy, utilising a fragment-coupling tethered Biginelli reaction, which allowed for the preparation of four potential structures. None of these matched the natural material. Re-investigation of the natural material revealed that the originally proposed connectivity was incorrect.
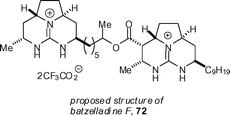
A new structure 74 was proposed, which was confirmed by a 15-step synthesis, again utilizing the fragment-coupling tethered Biginelli methodology (Scheme 7).46 β-Keto ester 77 was prepared in 5 steps and 44% overall yield, starting from aminohexahydropyrrolopyrimidinol 75 and β-keto ester 76. Coupling of 3 equivalents of 77 with readily available guanidine hemiaminal 78 in the presence of 3 equivalents of morpholinium acetate afforded the pentacyclic diguanidine 79 in 59% yield after HPLC separation. The trifluoroacetate counter-ions were exchanged for tetrafluoroborate so as to ensure good yields in the mesylate cyclization step. The synthesis of batzelladine F was completed by mesylation of the alcohol group, followed by heating at 70 °C in the presence of triethylamine to form the final ring. Hydrogenation in the presence of rhodium-on-alumina afforded batzelladine F (74) as its bis-trifluoroacetate salt in 21% yield after HPLC separation. This material was shown to be identical to an authentic sample of the natural product. Overman has also reported concise syntheses of guanidine-containing heterocycles using the Biginelli reaction.47
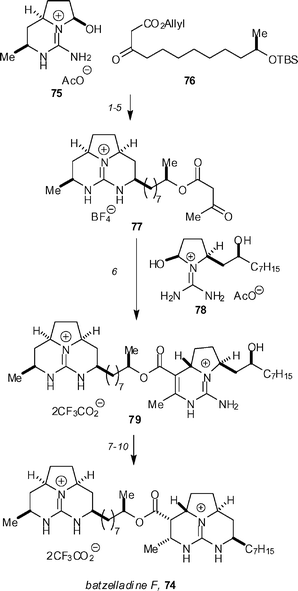 | ||
| Scheme 7 The Overman synthesis of batzelladine F. Reagents and conditions: (1) morpholine, AcOH, Na2SO4, CF3CH2OH, 60 °C (82%); (2) (Ph3P)4Pd, pyrrolidine, THF, MeOH; (3) NaBH4, AcOH; (4) HCl, then aq. NaBF4 (60% for three steps); (5) methyl acetoacetate, DMAP, PhMe, 100 °C (90%); (6) morpholine, AcOH, Na2SO4, CF3CH2OH, 60 °C (59%); (7) aq. NaBF4; (8) MsCl, Et3N, CH2Cl2, 0 °C; (9) Et3N, CHCl3, 70 °C (68% for three steps); (10) H2 (100 psi), Rh/Al2O3, HCO2H, MeOH (21%). | ||
Baran and co-workers have finally realised an enantioselective synthesis of sceptrin 80, having established a procedure for the synthesis of chiral tetrasubstituted cyclobutanes (Scheme 8).48 Enzymatic desymmetrization of meso-diester 81 using pig liver esterase gave a mono-ester 82 in quantative yield and 75% ee. After conversion to the monobenzyl amide 83, irradiation and treatment with sulfuric acid gave the cis,trans,trans-cyclobutane 84 in 50% yield and 75% ee. A single recrystallization resulted in material of >95% ee. This remarkable procedure only proceeded with complete transfer of chirality when a benzylamide group was used. This material was transformed into the all-trans-cyclobutane 85 upon treatment with TsOH and MeOH in toluene at 105 °C. Conversion of this material, using the previously developed synthetic route, resulted in the formation of ent-sceptrin. Access to the natural enantiomer was achieved by preparing the (+)-amide 86 by a straightforward three-step sequence from the same chiral ester 82. Application of the same reaction sequence as before gave cyclobutane 87, which could be converted into (−)-sceptrin 80.
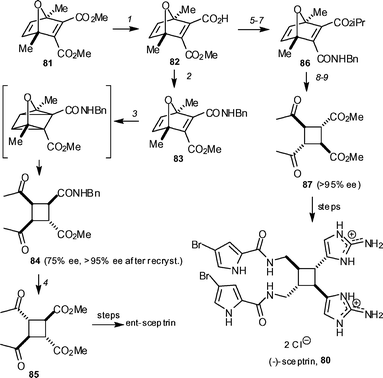 | ||
| Scheme 8 The Baran synthesis of (+)- and (−)-sceptrin 80. Reagents and conditions: (1) pig liver esterase (50 unit mmol−1), acetone–phosphate buffer (pH 8), 23 °C, 1 week (100%, 75% ee); (2) BnNH2, DMT-MM, THF (92%), (3) hν, THF, 72 h, then H2SO4, THF–MeOH (1 : 1) (50%, 75% ee, >95% ee after recrystallization); (4) TsOH, MeOH, PhMe, 105 °C, sealed tube (90%); (5) DMT-MM, iPrOH, DMAP, 55 °C; (6) LiOH, THF–H2O (1 : 1); (7) BnNH2, DMT-MM, THF (80% over three steps); (8) hν, THF, 72 h, then H2SO4, THF–MeOH (1 : 1) (45–50%, 75% ee, >95% ee after recrystallization); (9) TsOH, MeOH, PhMe, 105 °C, sealed tube (50%). DMT-MM = 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride. | ||
(−)-Sceptrin 80 was transformed into (−)-ageliferin 88 upon exposure to microwave irradiation, which triggers a vinylcyclobutane rearrangement. Baran and Houk have reported on the mechanism of this rearrangement, with computational studies indicating that this rearrangement occurs via diradical intermediates that start from the dication form of sceptrin.49 It would appear that this process could be a viable biosynthetic pathway for the ageliferins.
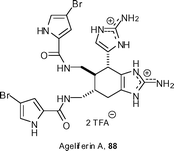
Until Baran's work, it had been speculated that a Diels–Alder cyclodimerization of compounds like the natural product oroidin 89, followed by a double bond migration, would generate the ageliferins. However, Lindel and co-workers found no evidence of such cyclodimerizations, although a Diels–Alder reaction with maleimides was possible to form adducts 90 (Scheme 9).50 However, heating oroidin 89 in a protic solvent, in the absence of a dienophile, resulted in the formation of rac-cyclooroidin 91 as its formate salt, which would suggest that this may be a viable biosynthetic pathway. Minassian and co-workers have confirmed that the absolute configuration of cyclooroidin 91 is S by carrying out a synthesis, starting from (S)-(−)-longamide B.51
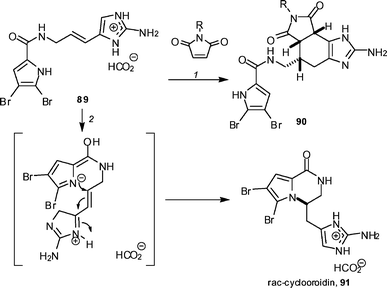 | ||
| Scheme 9 The Lindel synthesis of rac-cyclooroidin. Reagents and conditions: (1) 20 mol% Y(OTf)3, MeOH, 40 °C (R = Ph, 45%; R = H, 54%); (2) H2O–EtOH (4 : 1), 95 °C, sealed tube (93%). | ||
rac-Dibromophakellstatin 92 is a tetracyclic pyrrole-imidazole alkaloid which has promising biological activity. Lindel and co-workers have devised an efficent five-step synthesis, starting from N,O-acetal 93, which can be readily prepared from 4,5-dibromopyrrolyltrichloromethylketone (Scheme 10).52 Removal of the bromo groups and elimination of the alcohol group gave the tricycle 94 in 59% yield. Reaction of 94 with seven equivalents of TsONHCO2Et and calicum oxide gave the desired syn-tetracyclic product 95 in 60% yield. Dibromination with NBS followed by deprotection with 5 equivalents of samarium iodide afforded the target molecule 92 in 78% yield.
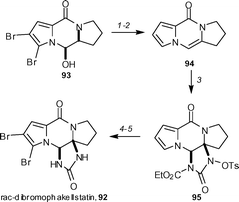 | ||
| Scheme 10 The Lindel synthesis of rac-dibromophakellstatin 92. Reagents and conditions: (1) H2, Pd/C (5 mol%), Et3N, MeOH–CH2Cl2 (1 : 1) (98%); (2) MsCl, DBU, CH2Cl2, 0 −23 °C (60%); (3) TsONHCO2Et, CaO, CH2Cl2, 23 °C (60%); (4) NBS, CH2Cl2, 0 °C (92%); (5) SmI2, THF, 23 °C; then MeOH, 23 °C (85%). | ||
Ageladine A 96 has emerged as a new class of inhibitor of matrix metalloproteinases, which are involved in the regulation of angiogenesis. Meketa and Weinreb reported the first synthesis of this compound, utilizing a 6π-azaelectrocyclization of a vinylimidazole system 97 to generate imidazopyridine 98 (Scheme 11).53 After much experimentation it was found that the chloropyridine 98 could be coupled with N-Boc pyrrole boronic acid and after removal of the protecting groups, pyrrole 99 could be generated in 67% yield. After some functional group manipulation, it was found that bromination of pyrrole 100 did afford ageladine A (96), but it was difficult to control, with mono- and tri-bromination occurring. Ageladine was obtained in 29% yield (based on recovered starting material), with mono- and tribrominated products also being produced.
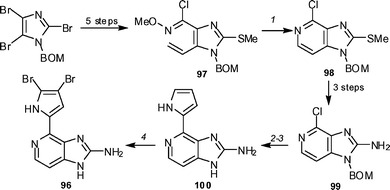 | ||
| Scheme 11 The Meketa and Weinreb synthesis of ageladine A 96. Reagents and conditions: (1) o-xylene, 150 °C (84%); (2) Pd2(dba)3, N-Boc-pyrrole boronic acid, biphenyl-PCy2, K3PO4, 1,4-dioxane, 80 °C, µW; (3) 6 N HCl, EtOH, reflux (67% yield for the two steps); (4) Br2, AcOH, MeOH, 0 °C (17%, 29% brsm). | ||
Shengule and Karuso have reported an alternate synthesis54 of ageladine A 96 based on biomimetic principles originally proposed by Fusetani (Scheme 12).55 The Pictet–Spengler reaction of commercially available 2-aminohistamine 102 with readily available N-Boc-4,5-dibromo-2-formylpyrrole 103 in the presence of scandium triflate afforded tetrahydroageladine A 104 in 44% yield. Ageladine A was obtained in 65% yield by reacting 104 with chloranil in chloroform at reflux. The use of the dibromopyrrole meant that the problematic bromination was avoided.
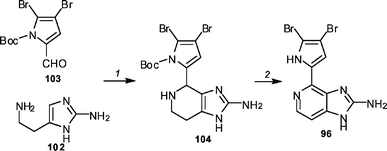 | ||
| Scheme 12 The Shengule and Karuso synthesis of ageladine A 96. Reagents and conditions: (1) Sc(OTf)3, EtOH, 23 °C (44%); (2) chloranil, CHCl3, 80 °C (65%). | ||
Weinreb has reported on his group's attempts to construct the 10-membered ring of chartelline A 105 using an intramolecular aldehyde/β-lactam cyclocondensation strategy.56 Unfortunately, the strain in the macrocyclic system due to the (Z)-vinylogous amide meant that this approach failed. In addition, Weinreb has examined the Cu(I)-catalyzed coupling of lactams with (E)-2-halovinyl iodides to produce the corresponding β-haloenamides, which are a key component of the chartelline alkaloids.57
Baran has taken a different approach to the chartellines, basing his strategy on biosynthetic principles whereby it is postulated that 107, via a bromine-induced ring contraction, could be used to generate the chartelline ring system (Scheme 13).58 Macrocycle 107 was prepared in a seven-step sequence, with the ring being generated by an intramolecular Horner–Wadsworth–Emmons reaction. Bromination of 107 in the presence of calcium carbonate, followed by treatment with N-bromoacetamide, resulted in the formation of the bromoenamide 108 in 36% yield (60% based on recovered starting material). With this key material in hand, the ring contraction process was initiated. This spectacular transformation was carried out in one pot and started with thermolysis at 185 °C to remove the Boc protecting group. Reaction with NBS afforded an unstable tetrabromide intermediate 109, which upon addition of potassium carbonate and 18-crown-6 to the reaction mixture induced the ring contraction to generate 110. An aqueous workup in brine afforded the required β-chlorocarboxyenamide 111 in a remarkable 93% yield for the entire process. To complete the synthesis, the TMSE ester was deprotected and the resulting carboxylic acid thermolysed at 200 °C in o-dichlorobenzene to afford chartelline C 112 in 64% yield.
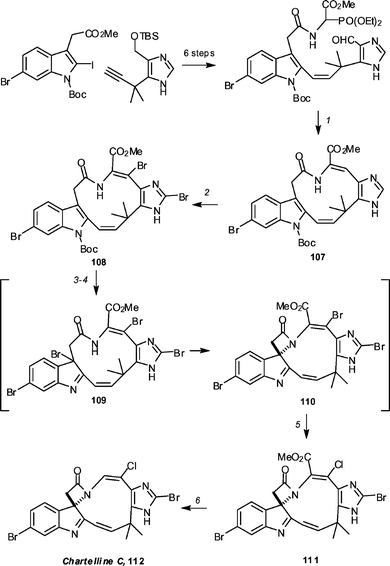 | ||
| Scheme 13 The Baran synthesis of chartelline C 112. Reagents and conditions: (1) LiCl, iPr2NEt, MeCN, 70 °C (56%); (2) Br2, CaCO3, PhH, 20 °C, 6 h, then N-bromoacetamide, PhH, 20 °C (36%, 60% brsm); (3) 185 °C, MeCN, 3 Å MS, NBS, 20 °C (4) 18-crown-6, K2CO3, 20 °C; (5) aq. sat. NaHCO3, then brine (93% for 3 steps); (6) TFA, (CH2Cl)2, 20 °C, then o-C6H4Cl2, 200 °C (64%). | ||
6 Pyrrole alkaloids
Fürstner and co-workers have reported59 syntheses of dictyodendrin C 113 and E 114, which were based on their previously reported synthesis of dictyodendrin B (Scheme 14).60 The synthetic strategy was based around the generation of a common precursor 115. This precursor 115 was generated by cyclization of ketoamide 116 using low-valent titanium and subsequent 6π electrocyclization to form the pyrrolcarbazole ring system. Conversion to dictyodendrin C 113 was achieved in four steps. First, the isopropyl ether was deprotected and then reacted with trichloroethyl chlorosulfuric acid ester to afford an unstable aryl sulfate. Treatment with boron trichloride removed the methoxy groups, and the resulting triphenol was oxidized with aqueous hydrogen peroxide to afford quinone 117 in 57% yield for the two steps. The trichloroethyl ester group was removed using zinc and ammonium formate in methanol, with some over-reduction observed. After removing the excess zinc and stirring the crude material under an oxygen atmosphere, dictyodendrin C 113 was obtained in 76% yield.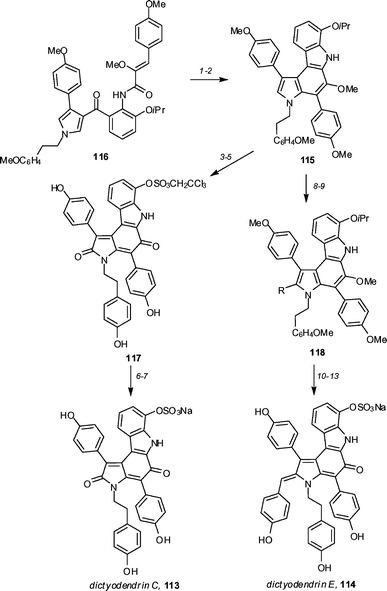 | ||
| Scheme 14 The Fürstner syntheses of dictyodendrins C (113) and E (114). Reagents and conditions: (1) TiCl3/2KC8, DME, Py, reflux (71–93%); (2) hν, MeCN, Pd/C cat., PhNO2 (81%); (3) BCl3, CH2Cl2, 0 °C (75%) (4) Cl3CCH2OSO2Cl, DABCO, CH2Cl2 (92%); (5) (a) BCl3, TBAI, CH2Cl2, 0 °C to rt, (b) aq. H2O2 (30% w/v), MeCN (57%); (6) Zn, HCO2NH4, MeOH; (7) O2 (1 atm), MeOH (76% for two steps); (8) NBS, THF, 0 °C, 69%; (9) p-MeOC6H4CH2MgCl, 9-MeO-9-BBN, Pd(OAc)2 (10 mol%), S-PHOS (20 mol%), DMF–THF (90%); (10) BCl3, CH2Cl2, −20 °C to rt; (11) Cl3CCH2OSO2Cl, DABCO, CH2Cl2 (83% over two steps); (12) BCl3, TBAI, CH2Cl2, 0 °C (85%); (13) (a) Zn, HCO2NH4, MeOH, (b) DDQ, THF (75% for two steps). S-Phos = 2-(2′,6′-dimethoxybiphenyl)dicyclohexylphosphine. | ||
The more complex member of the family, dictyodendrin E 114, was prepared by first brominating precursor 115 to afford bromide 118 (R = Br) in 69% yield (Scheme 14). After some experimentation, it was found that the required p-methoxybenzyl group [118 (R = CH2C6H4OMe)] could be added by coupling 118 (R = Br) with the borate obtained by reaction of p-methoxybenzyl magnesium chloride with 9-methoxy-9-BBN in the presence of 10 mol% Pd(OAc)2 and 20 mol% S-PHOS. Completion of the total synthesis was similar to that reported for dictoydendrin C (113).
The group of Iwao has reported a synthesis of lamellarin α 20-sulfate 119 using a Hinsberg-type pyrrole synthesis and utilizing palladium-catalyzed Suzuki–Miyaura couplings (Scheme 15).61 The symmetrical bistriflate 120 was prepared in five steps, starting from 3-isopropoxy-4-methoxybenzaldehyde. A Suzuki–Miyaura coupling, using boronic acid 106 and 2 mol% of palladium tetrakis(triphenylphosphine), allows for the efficient preparation of mono-arylated pyrrole 121. A second Suzuki–Miyaura coupling, using boronic acid 122 and 8 mol% of palladium tetrakis(triphenylphosphine), generates the diarylated pyrrole 123 in 90% yield. Alkaline hydrolysis, followed by treatment with p-TsOH in dichloromethane, gave the acid, which was decarboxylated by heating in quinoline in the presence of copper(I) oxide, affording 124 in 96% yield. Upon treatment with phenyliodine bis(trifluoroacetate), 124 undergoes an intramolecular oxidative biaryl coupling to generate 125 in 95% yield. This compound can then be utilized to generate lamellarin α 20-sulfate (119) in 5 steps.
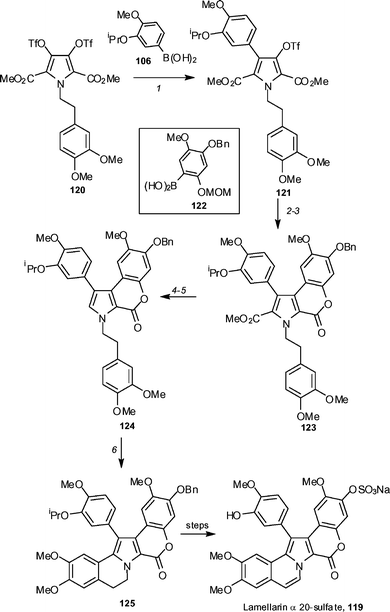 | ||
| Scheme 15 The Iwao synthesis of lamellarin α 20-sulfate 119. Reagents and conditions: (1) 120 (1 eq.), Pd(PPh3)4 (2 mol%), aq. Na2CO3, THF, reflux (80%); (2) 121 (2 eq.), Pd(PPh3)4 (8 mol%), aq. Na2CO3, THF, reflux (90%); (3) conc. HCl, MeOH, reflux (95%); (4) 40% KOH–EtOH (1 : 1), reflux, followed by p-TsOH, CH2Cl2, −40 °C (77%); (5) Cu2O, quinoline, 220 °C (96%); (6) PhI(OTFA)2, BF3·OEt2, CH2Cl2, −40 °C (95%). | ||
Iwao's group has employed a similar strategy to synthesise lamellarins D, L and M.62 Steglich and co-workers have devised a biomimetic synthesis of lamellarins G and K, as well as other related marine alkaloids.63 The pharmaceutical potential of the lamellarins has lead the groups of Ploypradith,64 Banwell,65 and Álvarez66 to report their work on the preparation of a variety of analogs.
7 Amphidinolides
Attention to the amphidinolide family of polyketides as synthetic targets continues unabated, no doubt driven by the impressive cytotoxicity displayed by many members of the class. Zhao and co-workers have described a straightforward strategy for the synthesis of T-class amphidinolides that is potentially quite practical. Their strategy is exemplified by the synthesis of amphidinolde T3 (126, Scheme 16).67 The key subunit coupling involves deprotonation of dithiane 128 using Smith's protocol (tBuLi in THF–HMPA), followed by reaction with aldehyde 127 to give 80% of 129. Although the resulting diastereoisomers (dr = 1.7 : 1) could be separated, it was more convenient to oxidize to ketone 130, and then reduce to the desired alcohol 131 using the Corey–Itsuno reaction. A sequence of 6 steps then advanced this compound to hydroxy acid 132, which was cyclized using Yamaguchi's conditions to yield macrolide 133 in 66% yield. Subsequent oxidative removal of the dithiane in 82% yield using bis(trifluoroacetoxy)iodobenzene completed the synthesis.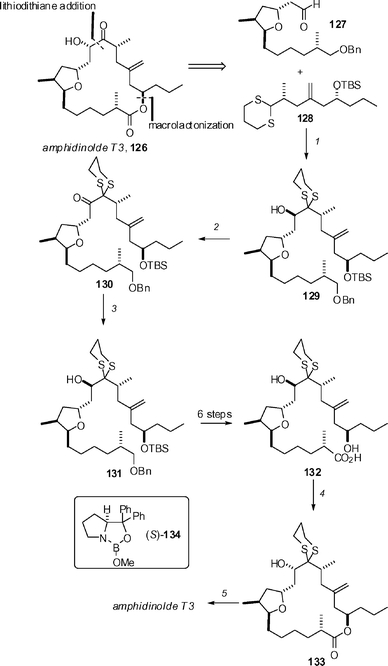 | ||
| Scheme 16 Key steps of Zhao's synthesis of amphidinolide T3. Reagents and conditions: (1) 128, tBuLi, THF–HMPA (9 : 1) −78 °C, then 127, 80% (dr 1.7 : 1); (2) Dess–Martin periodinane, NaHCO3, CH2Cl2, rt, 3 h, 91%; (3) 2 eq. (S)-B-MeO-CBS catalyst 134, Me2S·BH3, 68%; (4) 2,4,6-Cl3C6H2COCl, iPrNEt2, 45 °C, 10 h, then DMAP, toluene, 10 h, 66%; (5) PhI(CF3CO2)2, MeOH–H2O (10 : 1), 15 °C, 15 min, 82%. | ||
The Roush68 and Lee69 groups have independently reported syntheses of amphidinolide E, 135 (Scheme 17). Lee's synthesis is based around the assembly of the tetrahydrofuran ring by radical cyclization onto an alkoxy acrylate, a strategy that the Lee group has employed to good effect in a variety of settings. Known diol 136 was converted to diol 137 by a straightforward 7-step seqence (overall yield 55%). This diol was readily transformed to the key alkoxyacrylate 138 by tosylation of the primary alcohol, conjugate addition of the secondary alcohol onto ethyl propiolate and tosylate → iodide conversion (88% overall). Treatment of 138 with Et3B and (Me3Si)3SiH at −20 °C resulted in the desired cyclization to produce tetrahydrofuran 139 in 92% yield. Hydroboration–oxidation, Dess–Martin oxidation and alkynylation with the Ohira–Bestmann–Gilbert–Seyferth reagent gave alkyne 140 and set the stage for introduction of the trienyl side chain by enyne metathesis. To this end, treatment of 140 with 2-methyl-penta-1,4-diene in the presence of Grubbs’ 2nd-generation catalyst furnished the desired compound 141 in 65% yield and provided a nice example of the power of enyne metathesis in a complex setting. A four-step sequence advanced material to N-phenyltetrazolylsulfone 142, which was readily coupled with aldehyde 143 to give alkene 144 in 74% yield. Removal of the TBDPS ether, oxidation of the resulting primary alcohol to aldehyde and then acid, followed by removal of the TIPS ether, gave hydroxy acid 145. Kita lactonization (145 → 146, 44%) and removal of the protecting groups with aqueous 4 N HCl gave amphidinolide E in 77% yield.
![The Lee (left) and Roush (right) syntheses of amphidinolide E. Reagents and conditions: (1) TsCl, Et3N, CH2Cl2, 0 °C; (2) HCCCO2Et, NMM, CH2Cl2; (3) NaI, acetone, reflux, 88% (3 steps); (4) (TMS)3SiH, Et3B, toluene, −20 °C, 92%; (5) (Sia)2BH, THF, 0 °C; NaBO3·4H2O, H2O; (6) Dess–Martin periodinane, Py, CH2Cl2, 0 °C → rt; (7) Ohira–Bestmann–Gilbert–Seyferth reagent, Cs2CO3, EtOH, 0 °C → rt, 99% (3 steps); (8) CH2CH2, [(H2IMes2)RuCl2(P(c-Hex)3)], CH2Cl2; 15, sealed tube, 40 °C, 65%; (9) LiHMDS, THF, −78 → −40 °C; 5, DMF–DMPU (3 : 1), −78 °C → rt; (10) 15% NaOH–DMPU (1 : 10); (11) IBX, DMSO–THF (1 : 1); (12) NaClO2, NaH2PO4, tBuOH–2-methyl-2-butene–H2O (1 : 1 : 1); (13) TBAF, THF; (14) EtOCCH, (RuCl2(p-cymene))2, toluene, 0 °C → rt; CSA, rt → 50 °C; (15) 4 N HCl, MeOH; (16) BF3·OEt2, CH2Cl2, −78 °C, 4 Å MS, 48% (dr >20 : 1);(17) TBAF·3H2O, DMF, 90 °C; (18) TESOTf, Et3N; (19) DDQ, 76% (3 steps); (20) 2,4,6-trichlorobenzoyl chloride, Et3N, DMAP, THF, 25 °C; (21) CAN, acetone, 0 °C, 94%; (22) Grubbs I catalyst, 73%; (23) Bu3SnAlEt2, CuCN, THF, −30 °C, 58%; (24) NIS, CH2Cl2, −45 °C, 96%; (25) AcOH, H2O, THF, 40 °C, 77%; (26) 155, Pd(PPh3)4, CuCl, THF, 59%.](/image/article/2008/NP/b701533j/b701533j-s17.gif) | ||
Scheme 17 The Lee (left) and Roush (right) syntheses of amphidinolide E. Reagents and conditions: (1) TsCl, Et3N, CH2Cl2, 0 °C; (2) HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCO2Et, NMM, CH2Cl2; (3) NaI, acetone, reflux, 88% (3 steps); (4) (TMS)3SiH, Et3B, toluene, −20 °C, 92%; (5) (Sia)2BH, THF, 0 °C; NaBO3·4H2O, H2O; (6) Dess–Martin periodinane, Py, CH2Cl2, 0 °C → rt; (7) Ohira–Bestmann–Gilbert–Seyferth reagent, Cs2CO3, EtOH, 0 °C → rt, 99% (3 steps); (8) CH2CH2, [(H2IMes2)RuCl2(P(c-Hex)3)], CH2Cl2; 15, sealed tube, 40 °C, 65%; (9) LiHMDS, THF, −78 → −40 °C; 5, DMF–DMPU (3 : 1), −78 °C → rt; (10) 15% NaOH–DMPU (1 : 10); (11) IBX, DMSO–THF (1 : 1); (12) NaClO2, NaH2PO4, tBuOH–2-methyl-2-butene–H2O (1 : 1 : 1); (13) TBAF, THF; (14) EtOCCH, (RuCl2(p-cymene))2, toluene, 0 °C → rt; CSA, rt → 50 °C; (15) 4 N HCl, MeOH; (16) BF3·OEt2, CH2Cl2, −78 °C, 4 Å MS, 48% (dr >20 : 1);(17) TBAF·3H2O, DMF, 90 °C; (18) TESOTf, Et3N; (19) DDQ, 76% (3 steps); (20) 2,4,6-trichlorobenzoyl chloride, Et3N, DMAP, THF, 25 °C; (21) CAN, acetone, 0 °C, 94%; (22) Grubbs I catalyst, 73%; (23) Bu3SnAlEt2, CuCN, THF, −30 °C, 58%; (24) NIS, CH2Cl2, −45 °C, 96%; (25) AcOH, H2O, THF, 40 °C, 77%; (26) 155, Pd(PPh3)4, CuCl, THF, 59%. CCO2Et, NMM, CH2Cl2; (3) NaI, acetone, reflux, 88% (3 steps); (4) (TMS)3SiH, Et3B, toluene, −20 °C, 92%; (5) (Sia)2BH, THF, 0 °C; NaBO3·4H2O, H2O; (6) Dess–Martin periodinane, Py, CH2Cl2, 0 °C → rt; (7) Ohira–Bestmann–Gilbert–Seyferth reagent, Cs2CO3, EtOH, 0 °C → rt, 99% (3 steps); (8) CH2CH2, [(H2IMes2)RuCl2(P(c-Hex)3)], CH2Cl2; 15, sealed tube, 40 °C, 65%; (9) LiHMDS, THF, −78 → −40 °C; 5, DMF–DMPU (3 : 1), −78 °C → rt; (10) 15% NaOH–DMPU (1 : 10); (11) IBX, DMSO–THF (1 : 1); (12) NaClO2, NaH2PO4, tBuOH–2-methyl-2-butene–H2O (1 : 1 : 1); (13) TBAF, THF; (14) EtOCCH, (RuCl2(p-cymene))2, toluene, 0 °C → rt; CSA, rt → 50 °C; (15) 4 N HCl, MeOH; (16) BF3·OEt2, CH2Cl2, −78 °C, 4 Å MS, 48% (dr >20 : 1);(17) TBAF·3H2O, DMF, 90 °C; (18) TESOTf, Et3N; (19) DDQ, 76% (3 steps); (20) 2,4,6-trichlorobenzoyl chloride, Et3N, DMAP, THF, 25 °C; (21) CAN, acetone, 0 °C, 94%; (22) Grubbs I catalyst, 73%; (23) Bu3SnAlEt2, CuCN, THF, −30 °C, 58%; (24) NIS, CH2Cl2, −45 °C, 96%; (25) AcOH, H2O, THF, 40 °C, 77%; (26) 155, Pd(PPh3)4, CuCl, THF, 59%. | ||
The Roush synthesis is orchestrated around an elegantly applied [3 + 2] annulation reaction of allylsilane 147 (available in 9 steps from L-glyceraldehyde pentylidene) and aldehyde 148 (available in 9 steps from isopropylidene dimethyl-D-tartrate). Treatment of 148 with BF3·OEt2 and 2.5 equivalents of 147 provided the 2,5-cis-tetrahydrafuran 149 in 48% yield and with >20 : 1 dr. Excision of the silicon atom was achieved with TBAF·H2O in DMF at 90 °C and was accompanied by removal of the TES ether; however, after reprotection of the secondary alcohol and oxidative removal of the PMB group, alcohol 150 was obtained in 76% yield (over 3 steps). Successful acylation of 150 in advance of ring-closing metathesis required the use of Fe(CO)3-complexed dienoic acid 151 under Yamaguchi conditions, and after oxidative decomplexation of the iron, ester 152 was obtained in 94% yield (two steps). Ring-closing metathesis proceeded smoothly with Grubbs’ 1st-generation catalyst (20 mol%), to provide 153 in 73% yield. Stannylalumination (Bu3SnAlEt2, 58%), iododestannylation (96%) and finally removal of the protecting groups with acetic acid (77%) produced iodide 154. The conversion to amphidinolide E was then achieved by Stille coupling with stannane 155 under Corey and Stoltz's conditions to give the target in 59% yield.
In other work on the amphidinolides, Nicoloau has reported extensive synthetic studies on amphidinolide N 156 and caribenolide I 157 that have ultimately delineated strategies for their synthesis.70 Figadère and Franck have reported related work that is focussed on the C13–C29 fragment of caribenolide I.71 Unfortunately, the stereochemistry of these compounds remains undetermined at this juncture.
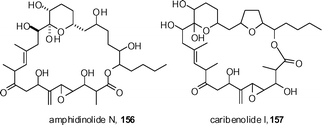
Nelson has described a synthesis of the C7–C20 subunit of amphidinolide B that employs his Al-catalyzed acyl halide aldehyde cycloaddition chemistry.72 Dai has reported a synthesis of the THF domain of amphidinolide X and Y that is based on the ring opening of a vinyl epoxide.73 Kalesse has described a route to a fully functionalized C14–C26 contaning fragment of amphidinolide H2.74 Murga has described a short synthesis of the C19–C26 fragment of amphidinolides G and H from glutamic acid.75 Full details of the Ghosh synthesis of amphidinolide W and Furstner's syntheses of amphidinolides X and Y have been reported.76,77
8 Ecteinascidins and other tetrahydroisoquinolines
Tetrahydroisoquinoline antibiotics such as the ecteinascidins continue to attract attention. This is no doubt a reflection of both the challenges provided to synthesis, and also their clinical potential. This second point is underpinned by the continued progress of ET 743 (Yondelis®, PharmaMar) toward the clinic.78 Zhu has described a total synthesis of ET 743 158 (Scheme 18) which proceeds in 31 steps (longest linear sequence) and that may be amenable to large-scale production.79 The synthesis is based around the assembly of key building blocks (160 → 164) and commences with the Pictet–Spengler reaction of 160 and 161 under catalysis by AcOH in CH2Cl2–trifluorethanol to produce 165 in 84% yield (Scheme 19). A four-step sequence (53% overall) of protecting group manipulations advanced 165 to 166, and 166 was coupled with racemic bromide 162 to provide 68% of the desired compound 167, along with 23% of the epimer 167a. The observed diastereoselectivity can likely be explained by an SN1 mechanism that invokes a quinone methide intermediate such as 162a. Silylation of the primary alcohol with TBSCl (97%), followed by removal of the acetate (K2CO3, MeOH, 94%), yielded alcohol 168. Oxidation with Dess–Martin periodinane and treatment with TMSCN in the presence of ZnCl2, led to a Strecker-type cyclization to give aminonitrile 169 in 74% yield (2 steps). A sequence of ester reduction with LiBH4 (80%), acetylation (Ac2O, pyridine, DMAP, 92%), deprotection of the primary TBS group (aqueous HF, 91%) and Dess–Martin oxidation led to aldehyde 170. Upon exposure to TFA in CH2Cl2, a Pomerantz–Fritsch-type cyclization occurred to produce the full A→E framework, 171, in an impressive 95% yield. Removal of the acetate under standard conditions (K2CO3, MeOH, 96%) and EDCI-mediated coupling with (R)-N-Troc-(S-trimethoxytrityl)cysteine gave 172 in 95% yield. This compound was treated with 1% TFA in trifluorethanol, and a sequence consisting of removal of the trimethoxytrityl group, ionization of the benzylic alcohol (presumably to the quinone methide), and hetero-Michael addition occurred to yield bridged macrocycle 173 in 77% yield after acetylation of the phenol with acetic anhydride. Compound 173 was advanced to ecteinascidin 743 by a six-step sequence that paralleled to a large degree the sequence employed by Corey and Gin80 and Fukuyama81 in their earlier total syntheses. The Zhu group also completed total syntheses of ET 597 and ET 583 by similar strategies,82 and also described studies on the synthesis of highly oxygenated tetrahydroisoquinolines by phenolic Mannich reactions.83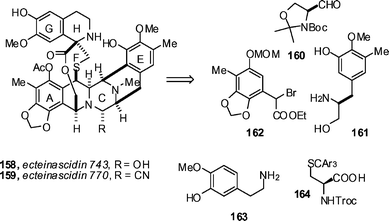 | ||
| Scheme 18 Key building blocks for the Zhu synthesis of ecteinascidin 743. | ||
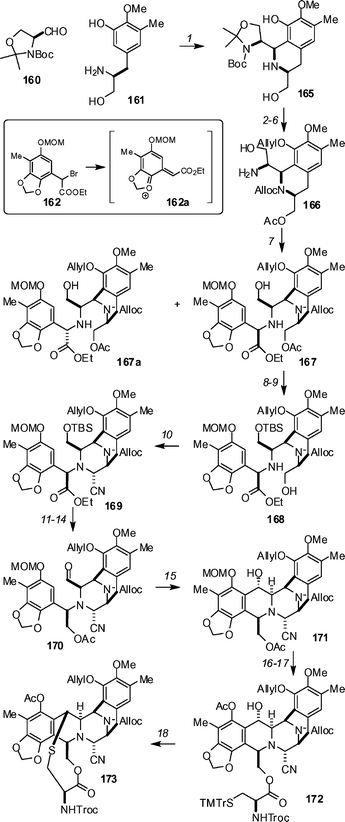 | ||
| Scheme 19 The Zhu synthesis of ecteinascidin 743. Reagents and conditions: (1) AcOH, CH2Cl2–CF3CH2OH (7 : 1), 3 Å MS, rt, 20 h, 84%; (2) 6 N HCl, in MeOH, rt, 95%; (3) AllocCl, NaHCO3, CH2Cl2, rt, 2 h, 88%; (4) AllylBr, Cs2CO3, DMF, rt, 3 h, 86%; (5) Ac2O, Py, DMAP, CH2Cl2, rt, 1 h, 92%; (6) TFA in CH2Cl2, rt, 72%; (7) TEA, MeCN, 0 °C, 91%; (8) TBSCl, imidazole, DMF, rt, 97%; (9) K2CO3, MeOH, rt, 94%; (10) Dess–Martin reagent, rt, then TMSCN, ZnCl2, rt, 78%; (11) LiBH4, MeOH, THF, 0 °C, 80%; (12) Ac2O, Py, DMAP, CH2Cl2, 92%; (13) HF·H2O, MeCN, rt, 91%; (14) Dess–Martin reagent, rt, 93%; (15) TFA, CH2Cl2, rt, 95%; (16) K2CO3, MeOH, rt, 96%; (17) EDCI, DMAP, CH2Cl2, rt, 95%; (18) TFA, TFE, rt, then Ac2O, Py, DMAP, CH2Cl2, 77%. | ||
Danishefsky and co-workers have reported on their synthetic work that has culminated in a formal total synthesis of ET 743 in a series of two papers.84,85 Danishefsky's original plans called for the assembly of the pentacyclic core by a Mannich reaction of a structure such as 174 to give 175 (Scheme 20); however, despite extensive experimentation, this could not be achieved in practice. Thankfully, with only a minimum of changes to the overall synthetic strategy, it was possible to fall back on a related vinylogous Pictet–Spengler cyclization (179 → 180). Subunit coupling commenced with the connection of amine 176 (available in 14 steps from the Borchardt catechol) with acid 177 under the action of BOP-Cl to give amide 178 in 85% yield. Deprotection of the PMB group (buffered aqueous DDQ, 90%), Cu(II)-catalyzed dehydration to the enamide (61%), oxidation to the aldehyde (Dess–Martin periodinane, 94%) and Pd-catalyzed removal of the allyl ether provided phenol 179. After some experimentation, it was determined that difluoroacetic acid (30 equivalents) in benzene was able to mediate the desired intramolecular Pictet–Spengler reaction to yield 180 in 42–58% yield. Refunctionalization of the Δ3,4 olefin required the interchange of the N-methyl group for a Troc group, and this was achieved by a 4-step sequence (180 → 181, 73% overall). Exposure of olefin 181 to dimethyldioxirane followed by 50 equivalents of NaCNBH3 provided the desired alcohol 183 in 78% yield. This alcohol could be advanced to compound 184 by a series of straightforward transformations, and at that point confluence with an intermediate from the Fukuyama synthesis was achieved.
![Danishefsky's formal synthesis of ecteinascidin 743. Reagents and conditions: (1) BOPCl (1.1 eq.), TEA (3 eq.), CH2Cl2, 0 → 25 °C, 24 h, 85%; (2) DDQ (1.6 eq.), CH2Cl2–pH 7 buffer (18 : 1), 25 °C, 30 min, 90%; (3) Cu(OTf)2 (0.2 eq.), benzene, 85 °C, 15 min, 61%; (4) Dess–Martin periodinane (1.5 eq.), CH2Cl2, 25 °C, 5 h, 94%; (5) [(PPh3)2PdCl2] (0.2 eq.), Bu3SnH (1.2 eq.), AcOH (5 eq.), 0 → 25 °C, 1 h, 93%; (6) CHF2CO2H (30 eq.), MgSO4 (4 eq.), benzene, 100 °C, 45 min, 42–58%; (7) TBSOTf (2.5 eq.), TEA (3 eq.), CH2Cl2, 0 °C, 30 min, 100%; (8) TrocCl (20 eq.), TBAI (3 eq.), toluene, 110 °C, 3 h, 92%; (9) TBAF (3 eq.), CH2Cl2, 0 °C, 2 min; (10) MOMCl (3 eq.), Hünig’s base (5 eq.), 30 min, 79%; (11) (a) DMDO (2 eq.), CH2Cl2, 0 → 25 °C, 1.5 h, (b) NaCNBH3 (50 eq.), 10 min, 78%.](/image/article/2008/NP/b701533j/b701533j-s20.gif) | ||
| Scheme 20 Danishefsky's formal synthesis of ecteinascidin 743. Reagents and conditions: (1) BOPCl (1.1 eq.), TEA (3 eq.), CH2Cl2, 0 → 25 °C, 24 h, 85%; (2) DDQ (1.6 eq.), CH2Cl2–pH 7 buffer (18 : 1), 25 °C, 30 min, 90%; (3) Cu(OTf)2 (0.2 eq.), benzene, 85 °C, 15 min, 61%; (4) Dess–Martin periodinane (1.5 eq.), CH2Cl2, 25 °C, 5 h, 94%; (5) [(PPh3)2PdCl2] (0.2 eq.), Bu3SnH (1.2 eq.), AcOH (5 eq.), 0 → 25 °C, 1 h, 93%; (6) CHF2CO2H (30 eq.), MgSO4 (4 eq.), benzene, 100 °C, 45 min, 42–58%; (7) TBSOTf (2.5 eq.), TEA (3 eq.), CH2Cl2, 0 °C, 30 min, 100%; (8) TrocCl (20 eq.), TBAI (3 eq.), toluene, 110 °C, 3 h, 92%; (9) TBAF (3 eq.), CH2Cl2, 0 °C, 2 min; (10) MOMCl (3 eq.), Hünig’s base (5 eq.), 30 min, 79%; (11) (a) DMDO (2 eq.), CH2Cl2, 0 → 25 °C, 1.5 h, (b) NaCNBH3 (50 eq.), 10 min, 78%. | ||
In other synthetic studies on tetrahydroisoquinolines, the Williams group has reported on the utility of radical cyclizations onto glyoxalimines as a method for the synthesis of the AB ring system.86 Chandrasekhar has described the syntheses of building blocks underpinned by Sharpless asymmetric dihydroxylation and aza-Michael reactions,87 and Lemaire has described the synthesis of a number of simplified piperazine-containing alkaloids modeled on the ecteinascidins.88 Several groups have reported studies on the preparation of analogs.89,90
9 Total syntheses of other compounds
Haouamine A 185 (Scheme 21, R = H) was isolated from Aplidium haouarianum and contains several unusual structural features, including a indeno-tetrahydropyridine ring system and an aza-paracyclophane with a bent aromatic ring. Baran and Burns’ retrosynthetic analysis recognized that formation of the highly strained aza-paracyclophane would need to be at the end of the synthesis, and that a suitable intermediate that would allow for such investigations would be the indeno-tetrahydropyridine 186.91 Access to the indeno-tetrahydropyridine was achieved in a remarkable three-step sequence, starting with alkylation of the potassium enolate of ketone 187 with iodide 188. After formation of the oxime 189, a cascade sequence was initiated. First, reaction with 2,4,4,6-tetrabromo-2,5-cyclohexadienone resulted in a 5-exo-trig cyclization to from nitrone 190, which was reduced with sodium borohydride at 50 °C to generate a cis-fused hydroxy pyrrolidine 191. After heating the reaction for 1 h, the piperidine 192 was formed via formation of a hydrooxyazridinium species and subsequent ring expansion. Finally, reduction of the crude material with indium powder afforded the desired material 186 (R = Boc) in 57% overall yield. This procedure allowed for gram quantities of material to be generated. After failing to form the aza-paracyclophane by a variety of methods, Baran and Burns proposed that to achieve this difficult transformation a non-aromatic conformational mimic of the bent aromatic ring would need to be employed. This strategy was realised by utilizing a pyrone–alkyne Diels–Alder reaction to form the macrocycle. The pyrone 193 was generated by carrying out a Stille coupling of the readily available pyrone 194 with the Boc-protected indeno-tetrahydropyridine 186 (R = Boc). Removal of the Boc group and alkylation with 3-butynyl tosylate gave the Diels–Alder precursor 193 (R = Me, R′ = CH2CH2CHCH) in 70% yield. It was found that the Diels–Alder reaction proceeded best if the methoxy groups were replaced with acetates. Microwave irradiation of 193 (R = Ac) in o-dichlorobenzene at 250 °C for 10 h generated the cyclohexadiene adduct 195 (R = Ac), which decarboxylated under the conditions to afford peracetylated (±)-haouamine A 195 (R = Ac). After purification to recover unreacted starting material (30%), the acetate groups were hydrolysed with potassium carbonate and methanol to yield (±)-haouamine A 185 (R = H) in 21% yield. Weinreb has completed a formal synthesis of haouamine A by devising a convergent 13-step synthesis of 186 (R = H).92 The synthesis utilizes an intramolecular nitrone–olefin dipolar cycloaddition.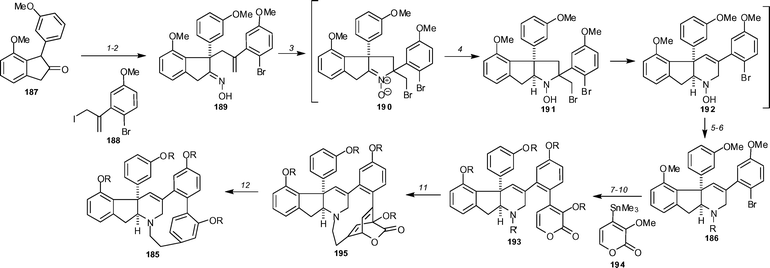 | ||
| Scheme 21 The Baran and Burns synthesis of (±)-haouamine A. Reagents and conditions: (1) KHMDS, THF–DMPU (5 : 1), 0 °C (44%); (2) NH2OH·HCl, NaOAc, EtOH, reflux (75%); (3) 2,4,4,6-tetrabromo-2,5-cyclohexadienone, DCE, 0 °C; (4) NaBH4, EtOH, 50 °C; (5) In powder, EtOH–sat. aq. NH4Cl (2 : 1), reflux (57% from 189); (6) Boc2O, CH2Cl2; (7) 194, Pd(PPh3)4 (0.1 eq.), CuI (0.2 eq.), PhMe, reflux (44% for two steps); (8) CH2Cl2–TFA (10 : 1); (9) 4-tosyloxybutyne, K2CO3, MeCN, reflux (70%); (10) BBr3, CH2Cl2, −78 to 23 °C, Ac2O–Py (1 : 1) (67%); (11) o-C6H4Cl2, 250 °C, 2,6-di-tert-butylmethylphenol; (12) K2CO3, MeOH (21% + 30% 193 (R = Ac, R′ = CH2CH2CHCH)). | ||
Baldwin and co-workers have applied a biomimetic Diels–Alder strategy to synthesise spiculoic acid 196, a recently isolated polyketide (Scheme 22).93 The required Diels–Alder precursor 197 was prepared by reacting aldehyde 198, obtained from the Roche ester in 15 steps, with phosphorane 199. Under the conditions used (toluene, 100 °C), it was found that the Diels–Alder reaction had occurred in situ and the desired bicyclic structure 200 was obtained in 25% yield. A three-step sequence was used to transform 200 into spiculoic acid. The material generated was found to have a specific rotation comparable in magnitude with the natural material, but of the opposite sign.
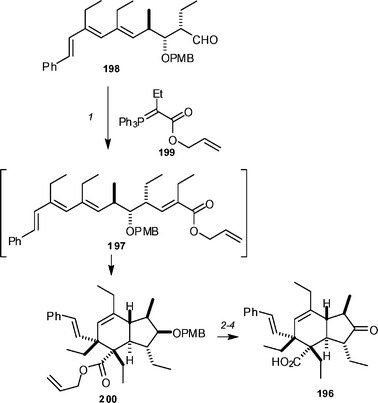 | ||
| Scheme 22 The Baldwin synthesis of spiculoic acid 196. Reagents and conditions: (1) 199, PhMe, 100 °C (25%); (2) DDQ, CH2Cl2, pH 7 buffer; (3) Pd(PPh3)4, morpholine (94% over two steps); (4) IBX, EtOAc, 70 °C (45%). | ||
Prior to the publication of this work, Mehta and Kundu had also reported a Diels–Alder strategy to prepare an analog of spiculoic acid A using what was presumed to be triene 203 as the substrate (Scheme 23).94 However, Kirkham, Lee and Baldwin have found that there were major discrepancies between the proposed structure and the corresponding data.95 They revised the stereochemistry of the Mehta and Kundu triene to that corresponding to 204. After a careful analysis, it was established that Mehta and Kundu had mis-used the Sharpless mnemonic model when carrying out a Sharpless asymmetric epoxidation to form epoxide 202—they had in fact synthesised the diastereomer 204, and thus ultimately carried out their Diels–Alder reaction on triene 205 rather than 203.
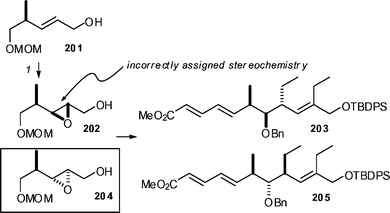 | ||
| Scheme 23 Stereochemical revision of Mehta and Kundu's triene. Reagents and conditions: (1) D-(−)-DET, TBHP, Ti(OiPr)4. | ||
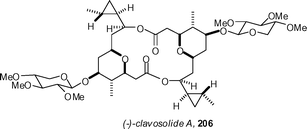
Three total syntheses of the revised structure of clavosolide A 206 have been reported. Lee's synthesis confirmed the proposed relative stereochemistry, but indicated that the absolute configuration needed to be revised.96 The groups of Willis97 and Smith98 have since reported that Lee's revision is incorrect, with both groups obtaining optical rotations matching the sign and magnitude of the natural material. Lee has recently issued a correction indicating that they had made an error when measuring the optical rotation.99
The Kishi group has provided a further example of the determination of stereochemistry using 3JH,H profiles of contiguous polyols. Following their earlier protocol, the 3JH,H profile of the hexaol peracetate region of sagittamide A 207 (Scheme 24) was imaginarily dissected into 3 small stereoclusters. The 3JH,H profile of each of these stereoclusters was compared to 3JH,H profiles for known tetraol acetates to provide a prediction of stereochemistry. A good match in this case was considered to be when the deviation sum between the experimental 3JH,H values and the stereocluster 3JH,H values was less than 3.3 Hz. On the basis of these methods, the relative stereochemistry for sagittamide A was predicted to be as shown. Total synthesis then confirmed the relative stereochemistry and also allowed for the determination of the absolute stereochemistry. It is apparent from the increasing body of data accumulating from the Kishi group that this approach to stereochemical elucidation is a very powerful one.
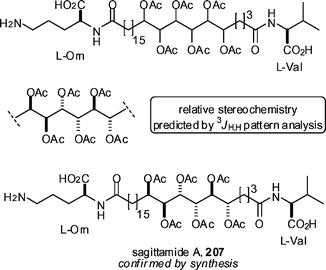 | ||
| Scheme 24 The Kishi group's determination of the stereochemistry of sagittamide A, 207. | ||
In a very nice example of the value of synthesis to structure elucidation/correction, the Burton group has shown that the previously proposed structures for elatenyne 216 and a related enyne 218 are in error (Scheme 25).100 The synthesis of elatenyne involved the conversion of diene 209 to diol 210 by silylation, ozonolysis, and reduction (85% overall). Monosilylation with TESCl gave 70% of the desired compound 211 after one recycle, and the remaining free alcohol was converted to the tosylate 212 (TsCl, DMAP, 93%) and reduced with superhydride to give 213. Removal of the TES group with K2CO3/MeOH (98%) and subsequent oxidation with TPAP (95%) was followed by Peterson olefination (75%), desilylation with p-TsOH/MeOH (75%) and triflation with Tf2O to give 214. Double displacement with tetra(n-butyl)ammonium bromide gave the desired dibromide 215, albeit in a modest 14% yield, although this is no doubt in part due to inductive effects. Removal of the Me3Si group from the alkyne with TBAF gave 216, the originally proposed structure for the natural product. Comparison of the spectroscopic data for the synthetic material with that reported for the natural product showed differences and cast doubt on the proposed structure. A very similar sequence was used to prepare the proposed structure for an enyne (218) from Laurencia majuscula, and it also differed spectroscopically from the reported data. On the basis of the analysis of 13C chemical shifts for the number of compounds, the authors noted the trends in pyrano [3,2-b]pyrans were that the indicated carbon resonates at <76 ppm whereas in 2,2′-bifuranyls these carbons are downfield of 76 ppm. Based on this, and in conjunction with further analysis of the data for the natural products, they propose the gross structures of the compounds to be 217 and 219. Confirmation of this prediction awaits synthesis.
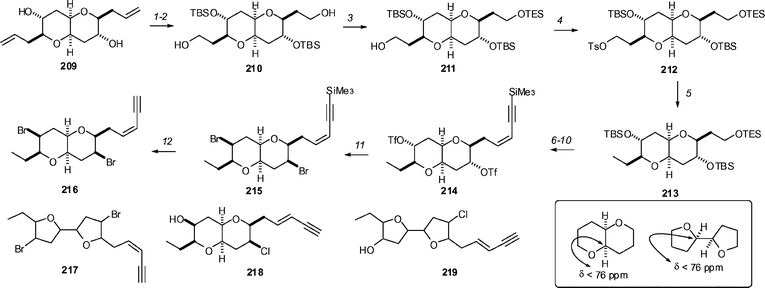 | ||
| Scheme 25 The Burton synthetic studies that produced the proposed structures of elatenyne 216 and the related compound 218. Reagents and conditions: (1) TBSOTf, Et3N, CH2Cl2, 99%; (2) O3/O2, CH2Cl2, MeOH, −78 °C, then Ph3P, −78 °C, 2 h, then NaBH4, −78 °C → rt, 2 h, 82%; (3) Et3SiCl, Et3N, CH2Cl2, 70% after one recycle; (4) TsCl, DMAP, Et3N, CH2Cl2, 93%; (5) LiBHEt3, Et2O, rt, 91%; (6) K2CO3, MeOH, 98%; (7) nPr4NRuO4, NMO, CH2Cl2, 4 Å MS, 95%; (8) Me3SiCCCH2SiMe2tBu, tBuLi, Ti(OiPr)4, THF, −78 °C, add 17, −78 °C → rt, 0.5 h, then (Me3Si)2NK, 75%; (9) TsOH, MeOH, 22 h, 75%; (10) Tf2O, Py, CH2Cl2; (11) nBu4NBr, toluene, reflux, 2 h, 14% from 19; (12) nBu4NF, THF, −5 °C, 98%. | ||
The recently described polyene marinomycins have quickly succumbed to total synthesis by the Nicolaou group.101 The key feature of the synthesis is the sequential use of Suzuki couplings—one intermolecular and one intramolecular—to form the macrocycle (Scheme 26). Alkyne 221 (available in 21 steps from 3-butenol) was hydroborated with catecholborane (in the presence of catalytic Cy2BH, 221 → 224) and coupled with bromide 223 (available in three steps from 221: Mitsunobu with 222, acetate cleavage, and silylation) to give 225 in 63% yield. Mitsunobu reaction with 222 followed by acetate removal and silylation provided 226 (78% over 3 steps), and set the stage for macrocyclization by Suzuki reaction. To this end the alkyne was again hydroborated with catecholborane (in the presence of catalytic Cy2BH) and the intermediate borane was treated with TlOEt and Pd(PPh3)4 in THF–H2O to effect Suzuki coupling. Finally, TBAF removal of the silyl groups gave marinomycin A 220 in 23% yield (over the 3 steps). Marinomycin A was readily isomerized photochemically to marinomycin B (cis Δ8,9, cis Δ8′,9′) and marinomycin C ([trans Δ8,9, cis Δ8′,9′).
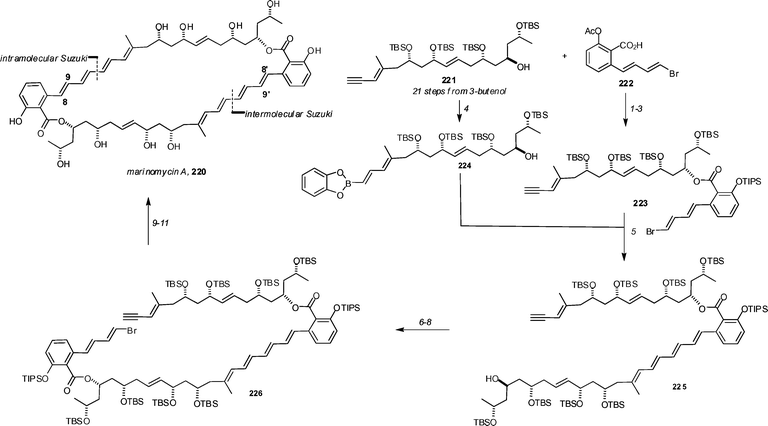 | ||
| Scheme 26 Synthesis of marinomycin A by Nicolaou and co-workers. Reagents and conditions: (1) 221, DEAD, PPh3, 222 (6.0 eq.), THF, 25 °C, 93%; (2) K2CO3, THF–MeOH, 25 °C, 15 min; (3) TIPSOTf, 2,6-lutidine, CH2Cl2, 25 °C, 18 h, 92% over two steps; (4) catecholborane (3.0 eq.), Cy2BH (0.1 M in THF, 0.2 eq.), THF, 25 °C, 1 h; then H2O (5.0 eq.); (5) KOH, 10 mol% Pd(PPh3)4, THF–H2O (4 : 1), 25 °C, 1 h, 63% based on 221; (6) DEAD, PPh3, 222 (6.0 eq.), THF, 25 °C, 1 h; (7) K2CO3, THF–MeOH (1 : 1), 25 °C, 15 min; (8) TIPSOTf, 2,6-lutidine, CH2Cl2, 25 °C, 18 h, 78% over three steps; (9) catecholborane (3.0 eq.), Cy2BH (0.1 M in THF, 0.2 eq.), THF, 25 °C, 1 h; then H2O (5.0 eq.); (10) TlOEt (300 eq.), 100 mol% Pd(PPh3)4, THF–H2O (10 : 1), 25 °C, 4 h; (11) TBAF, THF, 25 °C, 18 h, 23% over three steps. | ||
A large number of other total syntheses of marine natural products were reported in the review period, and papers describing first total syntheses are presented in Table 1. New total syntheses of compounds previously prepared are summarized in Table 2.
| Compound | Reference | Notes |
|---|---|---|
Liphagal  | Marion et al.103 | • Biomimetic total synthesis |
| • Racemic | ||
| • Biological activity: elective inhibitor of phosphatidylinositol-3-kinase α | ||
Viscosamine  | Timm and Köck104 | • 10 Steps from known compound |
| • Bioactivity: unknown | ||
(S)-Plakolide A  | Kanayama et al.105 | • Absolute stereochemistry of naturally occurring material revised to R |
| • 8 Steps | ||
| • Biological activity: nhibitor of iNOS | ||
Putative structure of elisabethadione 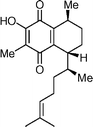 | Davies and Dai106 | • Data doesn't match that reported for natural product. |
| • 8 steps from known compound | ||
| • Non-racemic | ||
| • Biological activity: anti-inflammatory | ||
Plakortone 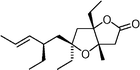 | Akiyama et al.107 | • Absolute configuration established |
| • 12 Steps from crotyl bromide | ||
| • Non-racemic | ||
| • Biological activity: high cytotoxicity on a murine fibrosarcoma cell line | ||
Sporiolide 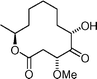 | Chen and Du108 | • 15 Steps from D-xylose |
| • Non-racemic | ||
| • Biological activity: cytotoxicity against L1210 cells, antibacterial activity against Micrococcus luteus | ||
Clavulazine 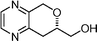 | Kumar et al.109 | • 15 steps from D-mannitol |
| • Non-racemic | ||
| • Biological activity: antibacterial, antimicrobial, herbicidal, plant growth regulator | ||
Gymnasterone 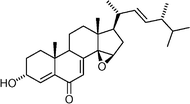 | Li et al.110 | • 26 Steps from cholic acid |
| • Non-racemic | ||
| • Biological activity: cytotoxic | ||
Putative structure of dichomitol 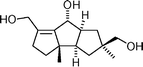 | Mehta and Pallavi111 | • Data doesn't match that reported for natural product. |
| • 22 Steps from known compound | ||
| • Biological activity: unknown | ||
K01-0509B 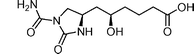 | Tsuchiya et al.112 | • Assignment of absolute configuration |
| • 16 steps from known compound | ||
| • Biological activity: related compounds inhibit Type III secretion | ||
(R)-γ-Indomycinone 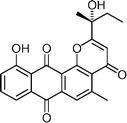 | Tietze et al.113 | • Established that the naturally occurring material has the S configuration |
| • 11 Steps from known compound | ||
| • Non-racemic | ||
| • Biological activity: antibiotic | ||
(R)-Convolutamydine A 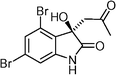 | Luppi et al.114 | • Assignment of absolute configuration |
| • 6 Steps from p-nitroaniline | ||
| • Non-racemic | ||
| • Biological activity: unknown | ||
Convolutamydines B and E 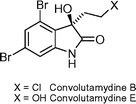 | Nakamura et al.115 | • Assignment of absolute configuration for convolutamydine B |
| • 5 Steps and 8 steps respectively | ||
| • Non-racemic | ||
| • Biological activity: unknown | ||
Agelagalastatin 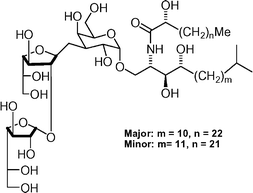 | Lee et al.116 | • Agelagalastatin is a 4 : 1 mixture of structural isomers |
| • Non-racemic | ||
| • Biological activity: significant in vitro inhibitory activities against human cancer cell growth | ||
Clavubicyclone 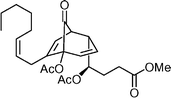 | Ito et al.117 | • 26 Steps from Z-butene-1,4-diol |
| • Racemic | ||
| • Biological activity: unknown | ||
Dysamide 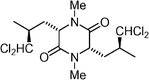 | Durow et al.118 | • 6 steps from (2S,4S)-dichloroleucine |
| • Non-racemic | ||
| • Biological activity: unknown | ||
Antheliolide 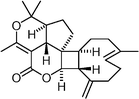 | Mushti et al.119 | • 26 Steps from 2-bromoallyl bromide |
| • An intermediate was resolved to allow for preparation of chiral material | ||
| • Biological activity: unknown | ||
4-Bromopyrrole-2-carboxyarginine and 4-bromopyrrole-2-carboxy-Nε-lysine 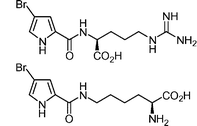 | Grube et al.120 | • Both 3 steps from 4-bromopyrrol-2-yl trichloromethyl ketone |
| • Non-racemic | ||
| • Biological activity: unknown | ||
Callyberyne C 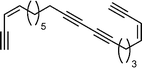 | Lopez et al.121 | • 12 Steps from known compound |
| • Biological activity: potent metamorphosis-inducing action | ||
| • Syntheses of callyberynes A and B also reported | ||
8,12-Iso-iPF3-VI 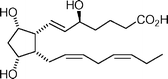 | Jacobo et al.122 | • 20 Steps from known compound |
| • Non-racemic | ||
| • Biological activity: biological marker for Alzheimer's disease and other neurogenerative diseases | ||
Maurenone 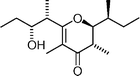 | Crossman and Perkins123 | • 8 Stereoisomers prepared, which allowed relative stereochemistry to be assigned |
| • 9 Steps from known compound | ||
| • Non-racemic | ||
| • Biological activity: unknown | ||
Bromoindole alkaloids from Laurencia brongniartii 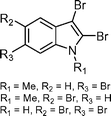 | Suarez-Castillo et al.124 | • 4 Steps from known compound |
| • Biological activity: potential antibacterial | ||
Echinamine A and B 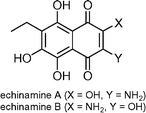 | Pokhilo et al.125 | • 7 Steps from known compound |
| • Biological activity: anti-oxidant | ||
Purealin 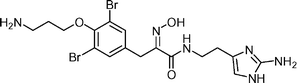 | Zhu et al.126 | • 7 Steps from 4-hydroxybenzaldehyde |
| • Biological activity: inhibits ATPase activity of isolated axonemal dynein and skeletal muscle myosin | ||
Isocladospolide B 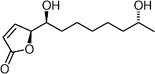 | Trost and Aponick127 | • 11 Steps from acrolein |
| • Non-racemic | ||
| • Biological activity: unknown | ||
(−)-Cladiella-6,11-diene-3-ol 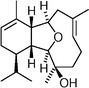 | Kim et al.128 | • 21 Steps from known compounds |
| • Non-racemic | ||
| • Biological activity: potential cytotoxic activity, anti-inflammatory effects, and anti-malarial properties | ||
Chlorodysinosin 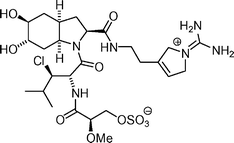 | Hanessian et al.129 | • Structural and stereochemical confirmation |
| • 19 Steps from known compounds | ||
| • Non-racemic | ||
| • Biological activity: potent inhibitory activity against thrombin, trypsin, and factor VIIa | ||
(−)-Panacene 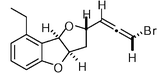 | Boukouvalas et al.130 | • 15 Steps from 2-methoxybenzoic acid |
| • Non-racemic | ||
| • Reassignment of relative and absolute stereochemistry (previously assigned erroneously by synthesis) | ||
| • Biological activity: feeding deterrent for predatory fish | ||
Floresolide 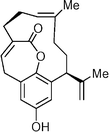 | Nicolaou and Xu131 | • 21 Steps from 2,5-dihydroxybenzaldehyde |
| • Non-racemic | ||
| • Biological activity: cytotoxicity against KB tumor cells | ||
Auripyrone 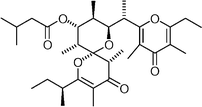 | Lister and Perkins132 | • 16 Steps from known compound |
| • Non-racemic | ||
| • Established absolute stereochemistry | ||
| • Biological activity: moderate cytotoxicity | ||
Azumamides A and E 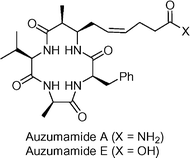 | Izzo et al.133 | • Azumamide E-8 steps from Fmoc-ala |
| • Non-racemic | ||
| • Biological activity: potent inhibitory activity on histone deacetylase (HDAC) enzymes | ||
Luffariellolide 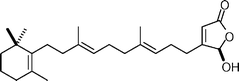 | Boukouvalas et al.134 | • 7 Steps from geranyl bromide |
| • Biological activity: anti-inflammatory |
| Compound | Reference | Compound | Reference |
|---|---|---|---|
| (+)-Lasonolide A | Yoshimura et al.135 | (±)-Debromoflustramine B | Lopez-Alvarado et al.136 |
| Puupehedione and 8-epi-puupehedione | Gansäuer et al.137 | Preclavulone-A methyl ester | Ito et al.138 |
| Panepophenanthrin | Commeira, Moses et al.139 | Mycothiazole | Le Flohic et al.140 |
| SNF4435C, SNF4435D, aureothin, N-acetyl-aureothamine and spectinabilin | Jacobsen et al.141 | Pallescensin | Foot et al.142 |
| (±)-Haemanthidine, (±)-pretazettine, (±)-tazettine, and (±)-crinamine | Zhang et al.143 | (±)-α-Chamigrene, (±)-β-chamigrene and (±)-laurencenone C | Srikrishna et al.144 |
| Aerothionin | Ogamino et al.145 | Membrenone | Yadav et al.146 |
| Lyngbic acid and hermitamides A and B | Virolleaud et al.147 | Cylindricine C | Swidorski et al.148 |
| Adociacetylene | Trost and Weiss149 | Jaspine | Liu et al.150 |
| Lepadiformine | Schar et al.151 | Diisocyanoadociane | Fairweather and Mander152 |
| Dactylolide | Louis et al.153 | Putative structure of pyrostatin | Castellanos et al.154 |
| Apratoxin | Doi et al.155 | Agelasine D | Vik et al.156 |
| Mycalamide A | Kagawa et al.157 | α-Araneosene, dolabellatrienone and palominol | Synder and Corey158 |
| Hybocarpone | Chai et al.159 | Psammaplin A | Godert et al.160 |
| Xestodecalactone A | Yoshino et al.161 | Bengazole A | Bull et al.162 |
| Tetrodotoxin | Urabe et al.163 | Dictyostatin | O’Neil and Phillips164 |
10 Acknowledgements
We would like to thank Professor John Blunt and Professor Murray Munro (University of Canterbury, Christchurch, New Zealand) for a copy of the 2006 version MarinLit database,102 which facilitated data collection for this review. Chris Nasveschuk, Noel Thomsen, and Rhonda Moore are also thanked for their assistance in drawing structures.11 References
- J. W. Blunt, B. R. Copp, W.-P. Hu, M. H. G. Munro, P. T. Northcote and M. R. Prinsep, Nat. Prod. Rep., 20087, 25 10.1039/b701534h (preceding paper in this issue).
- R. M. Wilson and S. J. Danishefsky, J. Org. Chem., 2006, 71, 8329 CrossRef CAS.
- H. Du, Y. He, S. Rasapalli and C. J. Lovely, Synlett, 2006, 965 CAS.
- P. Schar, S. Cren and P. Renaud, Chimia, 2006, 60, 131 CrossRef CAS.
- A. L. K. Shi Shun and R. R. Tykwinski, Angew. Chem., Int. Ed., 2006, 45, 1034 CrossRef.
- R. Peters and D. F. Fischer, Angew. Chem., Int. Ed., 2006, 45, 5736 CrossRef CAS.
- H. Kiyota, Biosci., Biotechnol., Biochem., 2006, 70, 317 CrossRef CAS.
- M. El Sous, D. Ganame, S. Zanatta and M. A. Rizzacasa, ARKIVOC, 2006, 105 CAS.
- A. Parenty, X. Moreau and J. M. Campagne, Chem. Rev., 2006, 106, 911 CrossRef CAS.
- R. Halim and M. A. Brimble, Org. Biomol. Chem., 2006, 4, 4048 RSC.
- J. S. Clark, Chem. Commun., 2006, 3571 RSC.
- J. C. Valentine and F. E. McDonald, Synlett, 2006, 1816 CAS.
- H. Takamura, J. Synth. Org. Chem., Jpn., 2006, 64, 418 CAS.
- H. Kiyota, in Topics in Heterocyclic Chemistry, Springer, Berlin/Heidelberg, 2006, vol. 5, p. 65 Search PubMed.
- K. Fujiwara, in Topics in Heterocyclic Chemistry, Springer, Berlin/Heidelberg, 2006, vol. 5, p. 97 Search PubMed.
- A. Nichida, T. Nagata and M. Nakagawa, in Topics in Heterocyclic Chemistry, Springer, Berlin/Heidelberg, 2006, vol. 5, p. 255 Search PubMed.
- M. Shindo, in Topics in Heterocyclic Chemistry, Springer, Berlin/Heidelberg, 2006, vol. 5, p. 179 Search PubMed.
- M. Sasaki, in Topics in Heterocyclic Chemistry, Springer, Berlin/Heidelberg, 2006, vol. 5, p. 149 Search PubMed.
- X.-T. Zhou and R. G. Carter, Angew. Chem., Int. Ed., 2006, 45, 1787 CrossRef CAS.
- S. Nguyen, J. Xu and C. J. Forsyth, Tetrahedron, 2006, 62, 5338 CrossRef CAS.
- X.-T. Zhou, L. Lu, D. P. Furkert, C. E. Wells and R. G. Carter, Angew. Chem., Int. Ed., 2006, 45, 7622 CrossRef CAS.
- M. Oikawa, T. Uehara, T. Iwayama and M. Sasaki, Org. Lett., 2006, 8, 3943 CrossRef CAS.
- (a) K. C. Nicolaou, D. Y. K. Chen, Y. Li, N. Uesaka, G. Petrovic, T. V. Koftis, F. Bernal, M. O. Frederick, M. Govindasamy, T. Ling, P. M. Pihko, W. Tang and S. Vyskocil, J. Am. Chem. Soc., 2006, 128, 2258 CrossRef CAS; K. C. Nicolaou, T. V. Koftis, S. Vyskocil, G. Petrovic, W. Tang, M. O. Frederick, D. Y. K. Chen, Y. Li, T. Ling and Y. M. A. Yamada, J. Am. Chem. Soc., 2006, 128, 2859 CrossRef CAS; K. C. Nicolaou, P. M. Pihko, F. Bernal, M. O. Frederick, W. Qian, N. Uesaka, N. Diedrichs, J. Hinrichs, T. V. Koftis, E. Loizidou, G. Petrovic, M. Rodriquez, D. Sarlah and N. Zou, J. Am. Chem. Soc., 2006, 128, 2244 CrossRef CAS.
- K. C. Nicolaou, M. O. Frederick, G. Petrovic, K. P. Cole and E. Z. Loizidou, Angew. Chem., Int. Ed., 2006, 45, 2609 CrossRef CAS.
- K. C. Nicolaou, M. O. Frederick, E. Z. Loizidou, G. Petrovic, K. P. Cole, T. V. Koftis and Y. M. A. Yamada, Chem. Asian J., 2006, 1, 245 CrossRef CAS.
- C. J. Forsyth, J. Xu, S. T. Nguyen, I. A. Samdal, L. R. Briggs, T. Rundberget, M. Sandvik and C. O. Miles, J. Am. Chem. Soc., 2006, 128, 15114 CrossRef CAS.
- F. Matsuura, R. Peters, M. Anada, S. S. Harried, J. Hao and Y. Kishi, J. Am. Chem. Soc., 2006, 128, 7463 CrossRef CAS.
- J. Hao, F. Matsuura, Y. Kishi, M. Kita, D. Uemura, N. Asai and T. Iwashita, J. Am. Chem. Soc., 2006, 128, 7742 CrossRef CAS.
- H. Fuwa, M. Ebine and M. Sasaki, J. Am. Chem. Soc., 2006, 128, 9648 CrossRef CAS.
- H. Fuwa, M. Ebine, A. J. Bourdelais, D. G. Baden and M. Sasaki, J. Am. Chem. Soc., 2006, 128, 16989 CrossRef CAS.
- M. T. Crimmins, J. L. Zuccarello, P. A. Cleary and J. D. Parrish, Org. Lett., 2006, 8, 159 CrossRef CAS.
- M. T. Crimmins, P. J. McDougall and J. M. Ellis, Org. Lett., 2006, 8, 4079 CrossRef CAS.
- I. Kadota, H. Nishii, H. Ishioka, H. Takamura and Y. Yamamoto, J. Org. Chem., 2006, 71, 4183 CrossRef.
- C. Crawford, A. Nelson and I. Patel, Org. Lett., 2006, 8, 4231 CrossRef CAS.
- M. Inoue, S. Yamashita, Y. Ishihara and M. Hirama, Org. Lett., 2006, 8, 5805 CrossRef CAS.
- A. Takizawa, K. Fujiwara, E. Doi, A. Murai, H. Kawai and T. Suzuki, Tetrahedron Lett., 2006, 47, 747 CrossRef CAS.
- A. Hamajima and M. Isobe, Org. Lett., 2006, 8, 1205 CrossRef CAS.
- T. Oishi, M. Suzuki, K. Watanabe and M. Murata, Heterocycles, 2006, 69, 91 CrossRef CAS.
- T. Oishi, M. Suzuki, K. Watanabe and M. Murata, Tetrahedron Lett., 2006, 47, 3975 CrossRef CAS.
- U. Majumder, J. M. Cox, H. W. B. Johnson and J. D. Rainier, Chem. Eur. J., 2006, 12, 1736 CrossRef CAS; H. W. B. Johnson, U. Majumder and J. D. Rainier, Chem. Eur. J., 2006, 12, 1747 CrossRef CAS.
- C. Tsukano and M. Sasaki, Tetrahedron Lett., 2006, 47, 6803 CrossRef CAS.
- G. L. Simpson, T. P. Heffron, E. Merino and T. F. Jamison, J. Am. Chem. Soc., 2006, 128, 1056 CrossRef CAS.
- M. A. Arnold, K. A. Day, S. G. Duron and D. Y. Gin, J. Am. Chem. Soc., 2006, 128, 13255 CrossRef CAS.
- M. A. Arnold, S. G. Duron and D. Y. Gin, J. Am. Chem. Soc., 2005, 127, 6924 CrossRef CAS.
- F. Cohen and L. E. Overman, J. Am. Chem. Soc., 2006, 128, 2594 CrossRef CAS.
- F. Cohen and L. E. Overman, J. Am. Chem. Soc., 2006, 128, 2604 CrossRef CAS.
- B. L. Nilsson and L. E. Overman, J. Org. Chem., 2006, 71, 7706 CrossRef CAS.
- P. S. Baran, K. Li, D. P. O'Malley and C. Mitsos, Angew. Chem., Int. Ed., 2006, 45, 249 CrossRef CAS.
- B. H. Northrop, D. P. O'Malley, A. L. Zografos, P. S. Baran and K. N. Houk, Angew. Chem., Int. Ed., 2006, 45, 4126 CrossRef CAS.
- C. Poeverlein, G. Breckle and T. Lindel, Org. Lett., 2006, 8, 819 CrossRef.
- J. Patel, N. Pelloux-Leon, F. Minassian and Y. Vallee, Tetrahedron Lett., 2006, 47, 5561 CrossRef CAS.
- M. Zollinger, P. Mayer and T. Lindel, J. Org. Chem., 2006, 71, 9431 CrossRef.
- M. L. Meketa and S. M. Weinreb, Org. Lett., 2006, 8, 1443 CrossRef CAS.
- S. R. Shengule and P. Karuso, Org. Lett., 2006, 8, 4083 CrossRef CAS.
- M. Fujita, Y. Nakao, S. Matsunaga, M. Seiko, Y. Itoh, J. Yamashita, R. W. M. van Soest and N. Fusetani, J. Am. Chem. Soc., 2003, 125, 15700 CrossRef CAS.
- C. Sun, X. Lin and S. M. Weinreb, J. Org. Chem., 2006, 71, 3159 CrossRef CAS.
- C. Sun, J. E. Camp and S. M. Weinreb, Organic Lett., 2006, 8, 1779 Search PubMed.
- P. S. Baran and R. A. Shenvi, J. Am. Chem. Soc., 2006, 128, 14028 CrossRef CAS.
- A. Furstner, M. M. Domostoj and B. Scheiper, J. Am. Chem. Soc., 2006, 128, 8087 CrossRef.
- A. Fürstner, M. M. Domostoj and B. Scheiper, J. Am. Chem. Soc., 2005, 127, 11620 CrossRef; A. D. Fürstner and M. M. Scheiper, B., J. Am. Chem. Soc., 2005, 127, 11620 CrossRef.
- T. Yamaguchi, T. Fukuda, F. Ishibashi and M. Iwao, Tetrahedron Lett., 2006, 47, 3755 CrossRef CAS.
- N. Fujikawa, T. Ohta, T. Yamaguchi, T. Fukuda, F. Ishibashi and M. Iwao, Tetrahedron, 2006, 62, 594 CrossRef CAS.
- C. Peschko, C. Winklhofer, A. Terpin and W. Steglich, Synthesis, 2006, 3048 CAS.
- P. Ploypradith, T. Petchmanee, P. Sahakitpichan, N. D. Litvinas and S. Ruchirawat, J. Org. Chem., 2006, 71, 9440 CrossRef CAS.
- M. G. Banwell, E. Hamel, D. C. R. Hockless, P. Verdier-Pinard, A. C. Willis and D. J. Wong, Bioorg. Med. Chem., 2006, 14, 4627 CrossRef CAS.
- D. Pla, A. Marchal, C. A. Olsen, A. Francesch, C. Cuevas, F. Albericio and M. Alvarez, J. Med. Chem., 2006, 49, 3257 CrossRef CAS.
- L.-S. Deng, X.-P. Huang and G. Zhao, J. Org. Chem., 2006, 71, 4625 CrossRef CAS.
- P. Va and W. R. Roush, J. Am. Chem. Soc., 2006, 128, 15960 CrossRef CAS.
- C. H. Kim, H. J. An, W. K. Shin, W. Yu, S. K. Woo, S. K. Jung and E. Lee, Angew. Chem., Int. Ed., 2006, 45, 8019 CrossRef CAS.
- K. C. Nicolaou, P. G. Bulger and W. E. Brenzovich, Org. Biomol. Chem., 2006, 4, 2158 RSC; K. C. Nicolaou, W. E. Brenzovich, P. G. Bulger and T. M. Francis, Org. Biomol. Chem., 2006, 4, 2119 RSC.
- G. Jalce, X. Franck, B. Seon-Meniel, R. Hocquemiller and B. Figadere, Tetrahedron Lett., 2006, 47, 5905 CrossRef CAS.
- A. Gopalarathnam and S. G. Nelson, Org. Lett., 2006, 8, 7 CrossRef CAS.
- Y. Chen, J. Jin, J. Wu and W.-M. Dai, Synlett, 2006, 1177 CAS.
- F. P. Liesener, U. Jannsen and M. Kalesse, Synthesis, 2006, 2590 CAS.
- P. Formentin, J. Murga, M. Carda and J. A. Marco, Tetrahedron: Asymmetry, 2006, 17, 2938 CrossRef CAS.
- A. K. Ghosh and G. Gong, J. Org. Chem., 2006, 71, 1085 CrossRef CAS.
- A. Fuerstner, E. Kattnig and O. Lepage, J. Am. Chem. Soc., 2006, 128, 9194 CrossRef.
- J. Jimeno, G. Faircloth, J. M. Fernández Sousa-Faro, P. J. Scheuer and K. Rinehart, Mar. Drugs, 2004, 2, 14 CrossRef CAS.
- J. Chen, X. Chen, M. Bois-Choussy and J. Zhu, J. Am. Chem. Soc., 2006, 128, 87 CrossRef CAS.
- E. J. Corey, D. Y. Gin and R. S. Kania, J. Am. Chem. Soc., 1996, 118, 202 CrossRef CAS.
- A. Endo, A. Yanagisawa, M. Abe, S. Tohma, T. Kan and R. Fukuyama, J. Am. Chem. Soc., 2002, 124, 6552 CrossRef CAS.
- J. Chen, X. Chen, M. Willot and J. Zhu, Angew. Chem., Int. Ed., 2006, 45, 8028 CrossRef CAS.
- X. Chen, J. Chen and J. Zhu, Synthesis, 2006, 4081 CAS.
- C. Chan, S. Zheng, B. Zhou, J. Guo, R. M. Heid, B. J. D. Wright and S. J. Danishefsky, Angew. Chem., Int. Ed., 2006, 45, 1749 CrossRef CAS.
- S. Zheng, C. Chan, T. Furuuchi, B. J. D. Wright, B. Zhou, J. Guo and S. J. Danishefsky, Angew. Chem., Int. Ed., 2006, 45, 1754 CrossRef CAS.
- D. Fishlock and R. M. Williams, Org. Lett., 2006, 8, 3299 CrossRef CAS.
- S. Chandrasekhar, N. R. Reddy and Y. S. Rao, Tetrahedron, 2006, 62, 12098 CrossRef CAS.
- S. Aubry, S. Pellet-Rostaing, B. Fenet and M. Lemaire, Tetrahedron Lett., 2006, 47, 1319 CrossRef CAS.
- P. A. Ceballos, M. Perez, C. Cuevas, A. Francesch, I. Manzanares and A. M. Echavarren, Eur. J. Org. Chem., 2006, 1926 CrossRef CAS.
- Z.-Z. Liu, Y. Wang, Y.-F. Tang, S.-Z. Chen, X.-G. Chen and H.-Y. Li, Bioorg. Med. Chem. Lett., 2006, 16, 1282 CrossRef CAS.
- P. S. Baran and N. Z. Burns, J. Am. Chem. Soc., 2006, 128, 3908 CrossRef CAS.
- J. H. Jeong and S. M. Weinreb, Org. Lett., 2006, 8, 2309 CrossRef CAS.
- J. E. D. Kirkham, V. Lee and J. E. Baldwin, Chem. Commun., 2006, 2863 RSC.
- G. Mehta and U. K. Kundu, Org. Lett., 2005, 7, 5569 CrossRef CAS.
- J. E. D. Kirkham, V. Lee and J. E. Baldwin, Org. Lett., 2006, 8, 5537 CrossRef CAS.
- J. B. Son, S. N. Kim, N. Y. Kim and D. H. Lee, Org. Lett., 2006, 8, 661 CrossRef CAS.
- C. S. Barry, J. D. Elsworth, P. T. Seden, N. Bushby, J. R. Harding, R. W. Alder and C. L. Willis, Org. Lett., 2006, 8, 3319 CrossRef CAS.
- A. B. Smith and V. Simov, Org. Lett., 2006, 8, 3315 CrossRef.
- J. B. Son, S. N. Kim, N. Y. Kim and D. H. Lee, Org. Lett., 2006, 8, 3411 CrossRef CAS.
- H. M. Sheldrake, C. Jamieson and J. W. Burton, Angew. Chem., Int. Ed., 2006, 45, 7199 CrossRef CAS.
- K. C. Nicolaou, A. L. Nold, R. R. Milburn and C. S. Schindler, Angew. Chem., Int. Ed., 2006, 45, 6527 CrossRef CAS.
- MarinLit database, Department of Chemistry, University of Canterbury, Christchurch, New Zealand. http://www.chem.canterbury.ac.nz/marinlit/marinlit.shtml Search PubMed.
- F. Marion, D. E. Williams, B. O. Patrick, I. Hollander, R. Mallon, S. C. Kim, D. M. Roll, L. Feldberg, R. Van Soest and R. J. Andersen, Org. Lett., 2006, 8, 321 CrossRef CAS.
- C. Timm and M. Koeck, Synthesis, 2006, 2580 CAS.
- M. Kanayama, K. Nishiwaki and K. Matsuo, Heterocycles, 2006, 68, 233 CrossRef CAS.
- H. M. L. Davies and X. Dai, Tetrahedron, 2006, 62, 10477 CrossRef CAS.
- M. Akiyama, Y. Isoda, M. Nishimoto, M. Narazaki, H. Oka, A. Kuboki and S. Ohira, Tetrahedron Lett., 2006, 47, 2287 CrossRef CAS.
- Q. Chen and Y. Du, Tetrahedron Lett., 2006, 47, 8489 CrossRef CAS.
- S. P. Kumar, K. Nagaiah and M. S. Chorghade, Tetrahedron Lett., 2006, 47, 7149 CrossRef CAS.
- M. Li, P. Zhou and A. Wu, Tetrahedron Lett., 2006, 47, 3409 CrossRef CAS.
- G. Mehta and K. Pallavi, Tetrahedron Lett., 2006, 47, 8355 CrossRef CAS.
- S. Tsuchiya, T. Sunazuka, T. Hirose, R. Mori, T. Tanaka, M. Iwatsuki and S. Omura, Org. Lett., 2006, 8, 5577 CrossRef CAS.
- L. F. Tietze, R. R. Singidi and K. M. Gericke, Org. Lett., 2006, 8, 5873 CrossRef CAS.
- G. Luppi, M. Monari, R. J. Correa, F. de A. Violante, A. C. Pinto, B. Kaptein, Q. B. Broxterman, S. J. Garden and C. Tomasini, Tetrahedron, 2006, 62, 12017 CrossRef CAS.
- T. Nakamura, S. I. Shirokawa, S. Hosokawa, A. Nakazaki and S. Kobayashi, Org. Lett., 2006, 8, 677 CrossRef CAS.
- Y. J. Lee, B. Y. Lee, H. B. Jeon and K. S. Kim, Org. Lett., 2006, 8, 3971 CrossRef CAS.
- H. Ito, S. Takeguchi, T. Kawagishi and K. Iguchi, Org. Lett., 2006, 8, 4883 CrossRef CAS.
- A. C. Durow, G. C. Long, S. J. O'Connell and C. L. Willis, Org. Lett., 2006, 8, 5401 CrossRef CAS.
- C. S. Mushti, J.-H. Kim and E. J. Corey, J. Am. Chem. Soc., 2006, 128, 14050 CrossRef CAS.
- A. Grube, E. Lichte and M. Koeck, J. Nat. Prod., 2006, 69, 125 CrossRef CAS.
- S. Lopez, F. Fernandez-Trillo, P. Midon, L. Castedo and C. Saa, J. Org. Chem., 2006, 71, 2802 CrossRef CAS.
- S. H. Jacobo, C. T. Chang, G. J. Lee, J. A. Lawson, W. S. Powell, D. Pratico, G. A. FitzGerald and J. Rokach, J. Org. Chem., 2006, 71, 1370 CrossRef CAS.
- J. S. Crossman and M. V. Perkins, J. Org. Chem., 2006, 71, 117 CrossRef CAS.
- O. R. Suarez-Castillo, L. Beiza-Granados, M. Melendez-Rodriguez, A. Alvarez-Hernandez, M. S. Morales-Rios and P. Joseph-Nathan, J. Nat. Prod., 2006, 69, 1596 CrossRef CAS.
- N. D. Pokhilo, M. I. Shuvalova, M. V. Lebedko, G. I. Sopelnyak, A. Y. Yakubovskaya, N. P. Mischenko, S. A. Fedoreyev and V. P. Anufriev, J. Nat. Prod., 2006, 69, 1125 CrossRef CAS.
- G. Zhu, F. Yang, R. Balachandran, P. Hook, R. B. Vallee, D. P. Curran and B. W. Day, J. Med. Chem., 2006, 49, 2063 CrossRef CAS.
- B. M. Trost and A. Aponick, J. Am. Chem. Soc., 2006, 128, 3931 CrossRef CAS.
- H. Kim, H. Lee, J. Kim, S. Kim and D. Kim, J. Am. Chem. Soc., 2006, 128, 15851 CrossRef CAS.
- S. Hanessian, J. R. DelValle, Y. Xue and N. Blomberg, J. Am. Chem. Soc., 2006, 128, 10491 CrossRef CAS.
- J. Boukouvalas, M. Pouliot, J. Robichaud, S. MacNeil and V. Snieckus, Org. Lett., 2006, 8, 3597 CrossRef CAS.
- K. C. Nicolaou and H. Xu, Chem. Commun., 2006, 600 RSC.
- T. Lister and M. V. Perkins, Angew. Chem., Int. Ed., 2006, 45, 2560 CrossRef CAS.
- I. Izzo, N. Maulucci, G. Bifulco and F. De Riccardis, Angew. Chem., Int. Ed., 2006, 45, 7557 CrossRef CAS.
- J. Boukouvalas, J. Robichaud and F. Maltais, Synlett, 2006, 2480 CrossRef CAS.
- T. Yoshimura, F. Yakushiji, S. Kondo, X. Wu, M. Shindo and K. Shishido, Org. Lett., 2006, 8, 475 CrossRef CAS.
- P. Lopez-Alvarado, E. Caballero, C. Avendano and J. C. Menendez, Org. Lett., 2006, 8, 4303 CrossRef CAS.
- A. Gansaeuer, A. Rosales and J. Justicia, Synlett, 2006, 927 CrossRef.
- H. Ito, T. Momose, M. Konishi, E. Yamada, K. Watanabe and K. Iguchi, Tetrahedron, 2006, 62, 10425 CrossRef CAS.
- L. Commeiras, J. E. Moses, R. M. Adlington, J. E. Baldwin, A. R. Cowley, C. M. Baker, B. Albrecht and G. H. Grant, Tetrahedron, 2006, 62, 9892 CrossRef CAS.
- A. Le Flohic, C. Meyer and J. Cossy, Tetrahedron, 2006, 62, 9017 CrossRef CAS.
- M. F. Jacobsen, J. E. Moses, R. M. Adlington and J. E. Baldwin, Tetrahedron, 2006, 62, 1675 CrossRef CAS.
- J. S. Foot, A. T. Phillis, P. P. Sharp, A. C. Willis and M. G. Banwell, Tetrahedron Lett., 2006, 47, 6817 CrossRef CAS.
- F.-M. Zhang, Y.-Q. Tu, J.-D. Liu, X.-H. Fan, L. Shi, X.-D. Hu, S.-H. Wang and Y.-Q. Zhang, Tetrahedron, 2006, 62, 9446 CrossRef CAS.
- A. Srikrishna, B. V. Lakshmi and M. Mathews, Tetrahedron Lett., 2006, 47, 2103 CrossRef CAS.
- T. Ogamino, R. Obata and S. Nishiyama, Tetrahedron Lett., 2006, 47, 727 CrossRef CAS.
- J. S. Yadav, R. Srinivas and K. Sathaiah, Tetrahedron Lett., 2006, 47, 1603 CrossRef CAS.
- M.-A. Virolleaud, C. Menant, B. Fenet and O. Piva, Tetrahedron Lett., 2006, 47, 5127 CrossRef CAS.
- J. J. Swidorski, J. Wang and R. P. Hsung, Org. Lett., 2006, 8, 777 CrossRef CAS.
- B. M. Trost and A. H. Weiss, Org. Lett., 2006, 8, 4461 CrossRef CAS.
- J. Liu, Y. Du, X. Dong, S. Meng, J. Xiao and L. Cheng, Carbohydr. Res., 2006, 341, 2653 CrossRef CAS.
- P. Schar, S. Cren and P. Renaud, Chimia, 2006, 60, 131 CrossRef CAS; P. Schar and P. Renaud, Org. Lett., 2006, 8, 1569 CrossRef.
- K. A. Fairweather and L. N. Mander, Org. Lett., 2006, 8, 3395 CrossRef CAS.
- I. Louis, N. L. Hungerford, E. J. Humphries and M. D. McLeod, Org. Lett., 2006, 8, 1117 CrossRef CAS.
- L. Castellanos, C. Duque, S. Zea, A. Espada, J. Rodriguez and C. Jimenez, Org. Lett., 2006, 8, 4967 CrossRef CAS.
- T. Doi, Y. Numajiri, A. Munakata and T. Takahashi, Org. Lett., 2006, 8, 531 CrossRef CAS.
- A. Vik, E. Hedner, C. Charnock, O. Samuelsen, R. Larsson, L. L. Gundersen and L. Bohlin, J. Nat. Prod., 2006, 69, 381 CrossRef CAS.
- N. Kagawa, M. Ihara and M. Toyota, Org. Lett., 2006, 8, 875 CrossRef CAS.
- S. A. Snyder and E. J. Corey, J. Am. Chem. Soc., 2006, 128, 740 CrossRef CAS.
- C. L. L. Chai, J. A. Elix and F. K. E. Moore, J. Org. Chem., 2006, 71, 992 CrossRef CAS.
- A. M. Godert, N. Angelino, A. Woloszynska-Read, S. R. Morey, S. R. James, A. R. Karpf and J. R. Sufrin, Bioorg. Med. Chem. Lett., 2006, 16, 3330 CrossRef CAS.
- T. Yoshino, F. Ng and S. J. Danishefsky, J. Am. Chem. Soc., 2006, 128, 14185 CrossRef CAS.
- J. A. Bull, E. P. Balskus, R. A. J. Horan, M. Langner and S. V. Ley, Angew. Chem., Int. Ed., 2006, 45, 6714 CrossRef CAS.
- D. Urabe, T. Nishikawa and M. Isobe, Chem. Asian J., 2006, 1, 125 CrossRef CAS.
- G. W. O’Neil and A. J. Phillips, J. Am. Chem. Soc., 2006, 128, 5340 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2008 |