Indolizidine and quinolizidine alkaloids
Joseph P. Michael*
Molecular Sciences Institute, School of Chemistry, University of the Witwatersrand, Wits, 2050, South Africa. E-mail: joseph.michael@wits.ac.za
First published on 13th December 2007
Abstract
Covering: July 2005 to June 2006. Previous review: Nat. Prod. Rep., 2007, 24, 191–222
This review covers the isolation, structure determination, synthesis, chemical transformations and biological activity of indolizidine and quinolizidine alkaloids. Included in the review are the hydroxylated indolizidines lentiginosine, swainsonine, castanospermine and their analogues; alkaloids from animal sources, including arthropods and amphibians; alkaloids from the genera Polygonatum, Prosopis and Poranthera; phenanthroindolizidine and phenanthroquinolizidine alkaloids; Nuphar alkaloids; lupine alkaloids; and alkaloids from marine sources. 130 references are cited.
1 Hydroxylated indolizidine alkaloids
1.1 Lentiginosine
A short synthesis of (+)-lentiginosine1 by Cardona et al. commenced with the highly stereoselective addition of vinylmagnesium bromide to the L-tartrate-derived nitrone21 (Scheme 1). The sole hydroxylamine stereoisomer 3, obtained in 96% yield, was deoxygenated by reduction with zinc in the presence of a catalytic amount of indium, the resulting pyrrolidine4 then being acylated with but-3-enoic acid to give the diene 5 as a mixture of two amide rotamers. Ring-closing metathesis with the first-generation Grubbs catalyst effected the construction of the target's bicyclic core; the product 6 was isolated in a moderate yield of 60%, probably for steric reasons, since the corresponding bis(acetoxy) analogue of 5 underwent a similar ring closure in 89% yield. The synthesis of (+)-1 was completed by reduction of the lactam and alkenegroups followed by cleavage of the tert-butyl ethers with trifluoroacetic acid. The overall yield of this seven-step synthesis was 14% based on nitrone2. The route also afforded the lactam analogues 7 and 8 and 7,8-dehydrolentiginosine9, which were tested for their ability to inhibit a range of 22 commercially available glycosidases. Disappointingly, 7 and 8, apart from showing weak inhibitory activity towards almond β-glucosidase, were inactive; while 9 displayed similar specificity to (+)-lentiginosine itself, although its activity was about an order of magnitude less when tested against the industrially important enzyme amyloglucosidase, which is used to produce glucose from starch.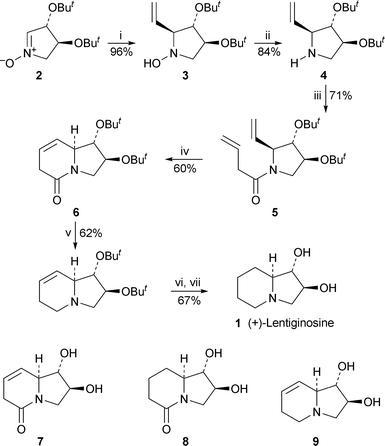 | ||
Scheme 1 Reagents and conditions: i, H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHMgBr (1.2 equiv.), Et2O, 20 °C, 1.75 h; ii, Zn (4 equiv.), In (cat.), aq. NH4Cl, MeOH, reflux, overnight; iii, H2CCHCH2CO2H, HOBt, DCC, CH2Cl2, 20 °C, 20 h; iv, (PCy3)2Cl2RuCHPh (12 mol%), CH2Cl2, reflux, 50 h; v, LiAlH4, THF, reflux, 2 h; vi, H2 (1 atm), 10% Pd/C, MeOH, overnight; vii, TFA, 20 °C, overnight. CHMgBr (1.2 equiv.), Et2O, 20 °C, 1.75 h; ii, Zn (4 equiv.), In (cat.), aq. NH4Cl, MeOH, reflux, overnight; iii, H2CCHCH2CO2H, HOBt, DCC, CH2Cl2, 20 °C, 20 h; iv, (PCy3)2Cl2RuCHPh (12 mol%), CH2Cl2, reflux, 50 h; v, LiAlH4, THF, reflux, 2 h; vi, H2 (1 atm), 10% Pd/C, MeOH, overnight; vii, TFA, 20 °C, overnight. | ||
The unnatural laevorotatory enantiomer of lentiginosine, ent-1, was readily synthesised by Dhavale and co-workers2 from the D-glucose derivative 10 via the known3pyrrolidine intermediate 11 (Scheme 2). Silyl protection of 11 followed by conversion of the ester into an aldehyde and chain extension by Wittig olefination yielded the conjugated ester12 as a mixture of (E)- and (Z)-isomers (4 : 1). Reduction of the alkene under conditions that simultaneously removed the benzyl and silylprotecting groups gave the bicyclic lactam13 in an excellent yield of 94%, after which conventional reduction of the lactam with lithium aluminium hydride completed the synthesis of (−)-lentiginosine, ent-1. A similar approach was used to prepare the 8a-epimer (+)-14 and the pyrrolizidine analogue (+)-15. A different synthesis of (−)-lentiginosine by Génisson and co-workers and employing ring-closing metathesis was previously reported as a communication4 (cf. ref. 5a); a full paper that also contains a synthesis of the pyrrolizidine analogue 15 has now been published.6
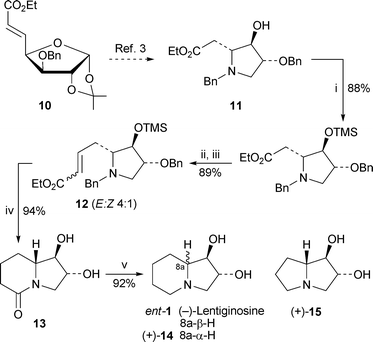 | ||
| Scheme 2 Reagents and conditions: i, (Me3Si)2NH, Me3SiCl (cat.), NH4SCN (cat.), CH2Cl2, 25 °C, 30 min; ii, DIBAL, CH2Cl2. −50 °C, 2.5 h, then aq. NH4Cl; iii, Ph3PCHCO2Et, CH2Cl2, 25 °C, 30 min; iv, HCO2NH4, 10% Pd/C, MeOH, AcOH (cat.), reflux, 1 h; v, LiAlH4, THF, 0 °C, then reflux, 8 h. | ||
1.2 Swainsonine and related compounds
The glycosidase inhibitor (−)-swainsonine 16 has been detected in another two members of the genus Ipomoea (Convolvulaceae), namely I. sericophylla and I. riedelii.7 The lysosomal storage disease contracted by goats grazing on these plants in north-eastern Brazil is undoubtedly due to the presence of the alkaloid, which can eventually result in death caused by vacuolisation of tissue in the brain and other organs. A clinical study of toxicosis in rats by I. carnea has also pinpointed swainsonine as the agent principally responsible for the observed pathological effects.8Guo and O'Doherty have reported a synthesis of both enantiomers of swainsonine by an enantiodivergent route that commenced with addition of 2-furyllithium17 to γ-butyrolactone18 followed by silylation to give the protected hydroxyketone199 (Scheme 3). The key to enantioselectivity was the catalytic transfer hydrogenation of 19 in the presence of the Noyori catalyst (S,S)-20 or its enantiomer. With the (S,S)-catalyst, for example, the (S)-alcohol21 was isolated in 89% yield and an enantiomeric excess (ee) of greater than 96%; comparable results were obtained for the preparation of ent-21 from 19 with the (R,R)-catalyst ent-20. To complete the synthesis of the unnatural (+)-enantiomer of swainsonine, Achmatowicz rearrangement of 21 was effected with N-bromosuccinimide to give the pyranone22 as a mixture of anomers. The corresponding Boc-protected intermediate 23, an 8 : 1 mixture of diastereomers, underwent palladium-induced coupling with benzyl alcohol to give 24 as a single diastereomer. The equatorial allylic alcohol 25, prepared by reduction of 24 with sodium borohydride, was then transformed into the azide26 with retention of configuration by first making a mixed carbonate with methyl chloroformate and then generating a π-allylpalladium intermediate, which was exposed to trimethylsilyl azide to form the product as a single regio- and stereoisomer. Stereoselective cis-dihydroxylation of the late intermediate 27 yielded diol28, exhaustive hydrogenation and hydrogenolysis of which over Pearlman's catalyst on carbon completed the synthesis of (+)-swainsonineent-16, presumably via the bicyclic intermediate 29. A modification of this route entailed dihydroxylation and protection of 26 to give the tetrahydropyran30, which was converted by means of a similar end-game into (+)-swainsoninevia the ketal-protected intermediate 31. In a similar fashion, the natural (−)-enantiomer of swainsonine could be obtained from ent-21 with comparable efficiency. The approach produced both enantiomers of swainsonine in about 17% overall yield from 17.
![Reagents and conditions: i, THF, −78 °C, 5 h, then warm to rt; ii, TBDMSCl, imidazole, DMF, 0 °C, 20 min, then rt, overnight; iii, catalyst (S,S)-20 (5 mol%), HCO2H, NEt3, CH2Cl2, rt, 24 h; iv, NBS, NaHCO3, NaOAc, THF–H2O (4 : 1), 0 °C, 30 min; v, (Boc)2O, DMAP, CH2Cl2, −78 °C, 14 h; vi, BnOH, Pd2(dba)3·CHCl3 (2.5 mol%), PPh3 (5 mol%), CH2Cl2, 0 °C to rt, 2 h; vii, NaBH4, CH2Cl2–MeOH (1 : 1), −78 °C, 4 h; viii, ClCO2Me, DMAP, py, CH2Cl2, rt, 24 h; ix, [Pd(allyl)Cl]2-dppb (1 : 4), Me3SiN3, THF, 50 °C, 24 h; x, Bu4NF, THF, rt, 12 h; xi, MeSO2Cl, NEt3, CH2Cl2, 0 °C, 30 min; xii, OsO4 (1 mol%), NMO, ButOH–Me2CO (1 : 1), 0 °C, 24 h; xiii, H2 (100 psi), Pd(OH)2/C, EtOH, rt, 3 d; xiv, Me2C(OMe)2, p-TsOH, Me2CO, 0 °C, 30 min; xv, H2 (100 psi), Pd(OH)2/C, EtOH–THF (1 : 1), rt, 4 d; xvi, HCl (6 M), THF, rt, overnight, then ion exchange (Dowex 1X8 200 OH−).](/image/article/2008/NP/b612166g/b612166g-s3.gif) | ||
| Scheme 3 Reagents and conditions: i, THF, −78 °C, 5 h, then warm to rt; ii, TBDMSCl, imidazole, DMF, 0 °C, 20 min, then rt, overnight; iii, catalyst (S,S)-20 (5 mol%), HCO2H, NEt3, CH2Cl2, rt, 24 h; iv, NBS, NaHCO3, NaOAc, THF–H2O (4 : 1), 0 °C, 30 min; v, (Boc)2O, DMAP, CH2Cl2, −78 °C, 14 h; vi, BnOH, Pd2(dba)3·CHCl3 (2.5 mol%), PPh3 (5 mol%), CH2Cl2, 0 °C to rt, 2 h; vii, NaBH4, CH2Cl2–MeOH (1 : 1), −78 °C, 4 h; viii, ClCO2Me, DMAP, py, CH2Cl2, rt, 24 h; ix, [Pd(allyl)Cl]2-dppb (1 : 4), Me3SiN3, THF, 50 °C, 24 h; x, Bu4NF, THF, rt, 12 h; xi, MeSO2Cl, NEt3, CH2Cl2, 0 °C, 30 min; xii, OsO4 (1 mol%), NMO, ButOH–Me2CO (1 : 1), 0 °C, 24 h; xiii, H2 (100 psi), Pd(OH)2/C, EtOH, rt, 3 d; xiv, Me2C(OMe)2, p-TsOH, Me2CO, 0 °C, 30 min; xv, H2 (100 psi), Pd(OH)2/C, EtOH–THF (1 : 1), rt, 4 d; xvi, HCl (6 M), THF, rt, overnight, then ion exchange (Dowex 1X8 200 OH−). | ||
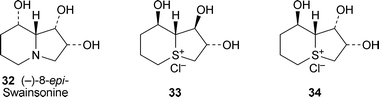
Analogues of swainsonine synthesised during the period under review include (−)-8-epi-swainsonine3210 and its triacetate,11 both by routes involving ring-closing metathesis. New sulfonium ion analogues of swainsonine include the triols (−)-3312 and (−)-34.13
1.3 Castanospermine and related compounds
Diastereoselective addition of vinylmagnesium bromide to the glucose-derived nitrone35 in the presence of trimethylsilyl triflate was the first step in a short synthesis of (+)-castanospermine36 by Dhavale and co-workers14 (Scheme 4). Two separable isomers, 37 and 38, were formed in a ratio of 13 : 87. The major diastereomer was converted in a further four steps into the aldehyde39, a second addition of vinylmagnesium bromide to which produced the two diastereomeric vinyloxazolidinones 40 and 41 in a 1 : 3 ratio. Unfortunately, it was the minor isomer that possessed the requisite stereochemistry for conversion into the target alkaloid. Hydrolysis afforded the amino alcohol42, which was cyclised by an aminomercuration–demercuration sequence to give the pyrrolidine43 in 66% yield after protection of the free hydroxy group as the benzyl ether. Hydrogenolysis of all three benzyl groups followed by re-protection of the amine as the benzyloxycarbonyl (Cbz) derivative yielded 44, which on treatment with aqueous trifluoroacetic acid followed by hydrogenation over palladium on charcoal underwent tandem deprotection and intramolecular reductive amination to complete the synthesis of (+)-castanospermine. In addition, the undesired major epimer41 produced from 39 could also be converted into the alcohol 44 viaketone45, thus permitting alternative access to the target alkaloid. As a bonus, application of the illustrated reaction sequences to the intermediates 41 and 37 led to syntheses of the unnatural alkaloid epimers 1-epi-castanospermine46 and 8a-epi-castanospermine47, respectively. In a further extension, the nitrone35 was treated with allylmagnesium bromide followed by reduction with zinc in acetic acid to produce a separable mixture of amine diastereomers 48.15 Applying the aminomercuration–reductive demercuration sequence then afforded the pyrrolidine diastereomers 49, which could in turn be converted into (+)-1-deoxycastanospermine50 and its 8a-epimer51 as illustrated.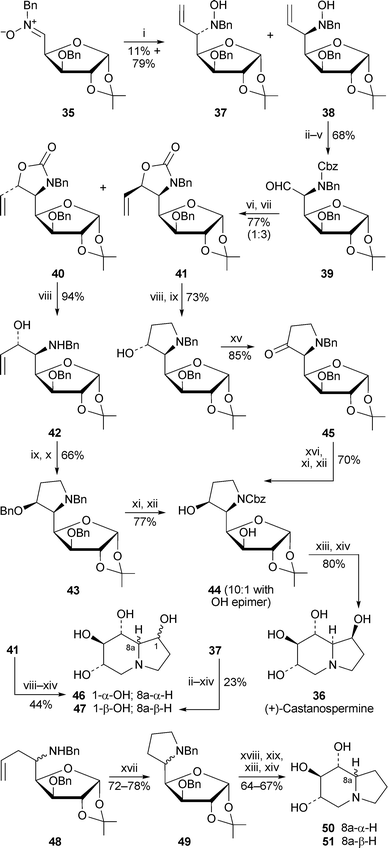 | ||
| Scheme 4 Reagents and conditions: i, Me3SiOTf, THF, −10 °C, 10 min, then H2CCHMgBr, −78 °C, 1 h; ii, Zn, Cu(OAc)2, AcOH, 80 °C, 1 h; iii, ClCO2Bn (CbzCl), NaHCO3, MeOH–H2O (4 : 1), 0 °C, 3 h; iv, K2OsO4·2H2O, NMO, Me2CO–H2O (8 : 1), rt, 12 h; v, NaIO4, Me2CO–H2O (9 : 1), 0 °C, 6 h; vi, H2CCHMgBr, THF, −50 °C, 1 h; vii, aq. KOH (40%), MeOH, 90 °C, 10 min; viii, aq. KOH (40%), MeOH, 90 °C, 48 h; ix, Hg(OAc)2, THF–H2O (1 : 1), rt, 3 h, then NaBH4; x, NaH, BnBr, Bu4NI, THF, 0 °C to rt, 6 h; xi, HCO2NH4, 10% Pd/C, MeOH, 80 °C, 1 h; xii, CbzCl, NaHCO3, MeOH–H2O (9 : 1), 0 °C, 3 h; xiii, TFA–H2O (3 : 2), 25 °C, 2.5 h; xiv, H2 (80 psi), 10% Pd/C, MeOH, rt, 12 h; xv, Swern oxidation, −78 °C to rt; xvi, NaBH4, MeOH–H2O (9 : 1), −50 °C, 1 h; xvii, Hg(OAc)2 (2.5 equiv.), THF–H2O (1 : 1), rt, 18 h, then NaBH4; xviii, H2 (80 psi), 10% Pd/C, MeOH, rt, 16 h; xix, CbzCl, NaHCO3, EtOH, 25 °C, 2 h. | ||
Mariano and co-workers recently demonstrated that the tandem ring opening-ring closing metathesis of the N-allyl derivative 52 could be catalysed by the Grubbs second-generation catalyst to give the tetrahydropyridine53 (R = H) as a single regioisomer16 (cf. ref. 5b). It was shown by X-ray crystallography that the free alcohol 54 exists in the diaxial conformation in order to relieve A1,3 strain between the N-acetyl and 6-allyl substituents. They have now exploited the axial orientation of the hydroxy group to direct selective epoxidation of the endocyclic alkene to produce the epoxide55, which was a pivotal intermediate in a concise new synthesis of (+)-castanospermine3617 (Scheme 5). trans-Diaxial ring opening of the epoxide under mild basic conditions yielded the triol56 which, after protection as the tetrabenzyl derivative 57, was hydroborated to give the primary alcohol58 in rather poor yield (31%), the N-acetyl substituent being fortuitously removed in the process. Cyclisation under Mitsunobu conditions followed by hydrogenolysis of the benzylprotecting groups completed the synthesis of (+)-36.
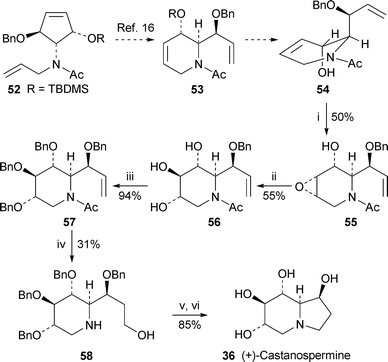 | ||
| Scheme 5 Reagents and conditions: i, VO(acac)2, ButOOH, CH2Cl2, 25 °C, 2.5 h; ii, C6H5CO2Na, H2O, 130 °C, 12 h; iii, NaH, BnBr, DMF, 0 °C, 2 h; iv, BH3·THF, THF, 0 °C, 3 h, then aq. NaOH (3 M), aq. H2O2 (30%), 25 °C, 3 h; v, DEAD, PPh3, THF, 25 °C, 12 h; vi, H2 (1 atm), PdCl2, MeOH–EtOAc (1 : 1), 25 °C, 4 h. | ||
Castanospermine continues to inspire the design and synthesis of novel polyhydroxylated analogues, some of which have been tested as glycosidase inhibitors. The bicyclic carbamates and thiocarbamates 59–68, all possessing an axial hydroxy substituent in the pseudoanomeric position at C-5 (as shown in 59′ and 60′) were designed as reducing analogues of the alkaloid,18 since it has previously been shown that the enhanced configurational stability of similar compounds can lead to dramatic improvements in α-anomeric selectivity when tested against glycosidases.19 In the event, compounds 59 and 60, which have the same glucose-like orientations of hydroxy groups as in castanospermine itself, were potent competitive inhibitors of yeast α-glucosidase (Ki 40 and 2.2 µM, respectively). However, inverting the configurations at C-6 or C-8 destroyed inhibitory activity; only the 7-epi analogue 64 retained weak activity (Ki 540 µM). However, compounds 65 and 66, which have the D-galacto configuration, inhibited coffee bean α-galactosidase (Ki 137 and 259 µM, respectively), while 65 was also weakly effective against bovine liver cytosolic β-galactosidase (Ki 267 µM). The results show the critical role that the hydroxylation profiles play in the specific inhibition of glycosidases.
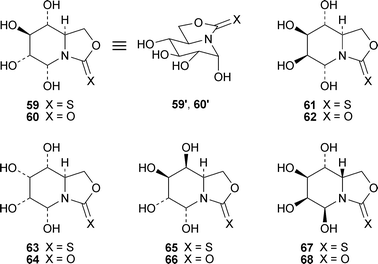
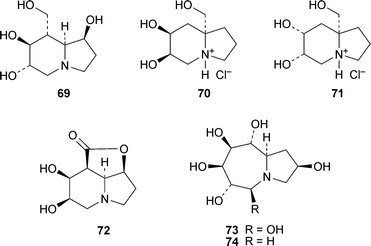
8-Homocastanospermine69, on the face of it a closer structural analogue of the parent alkaloid than any of those described above, showed only trace inhibition of α-glucosidase activity, and none at all of β-glucosidase.20 The bridgehead hydroxymethyl salts 70 and 71 were weak inhibitors of α-glucosidases (IC50 0.57 and 0.59 mM, respectively); and 70 also inhibited β-galactosidase (IC50 0.56 mM).21 The lactone72, also a likely precursor of hydroxymethyl variants of castanospermine, was synthesised by methodology involving nitronecycloadditions,22 but was not developed further. Finally, the interesting hexahydroxylated perhydroazaazulene73, a ring homologue of castanospermine, showed 38% and 65% inhibition of bovine liver β-galactosidase and Rhizopus mould amyloglucosidase, respectively, at a concentration of 1 mM.23 The figures for the corresponding pentahydroxy compound 74 were 36% and 62% respectively; and its inhibition of Aspergillus nigeramyloglucosidase reached 56%.
1.4 Uniflorine-A
The structure 75 for the putative pentahydroxyindolizidinealkaloiduniflorine-A, first reported in 2000,24 was disproved by Pyne and co-workers,25 who showed that the spectroscopic properties of synthetic 75 differed from those reported for the natural product (cf. ref. 5c). They suggested that the reported J1,8a coupling constant of 4.5 Hz is more consistent with a syn relationship between 1-H and 8a-H, implying the same 1β-OH stereochemistry as in castanospermine. Mariano's team has now reported the synthesis of the two alternative 2-hydroxycastanospermines 76 and 77 (Scheme 6).17 Stereocontrolled catalytic dihydroxylation of the same intermediate 57 that featured in their synthesis of (+)-castanospermine (vide supra; Scheme 5) produced 78, which was deacetylated and cyclised under Mitsunobu conditions to produce the protected indolizidine79. Hydrogenolytic removal of the benzyl groups produced the first target (+)-76, a compound that had previously been synthesised by Bell et al.26 Isomer (+)-77 was obtained by Mitsunobu inversion of the secondary alcohol of 79 followed by cleavage of the benzyl groups. Once again, however, the physical and spectroscopic properties of these two products and their hydrochloride salts failed to match those reported for uniflorine-A, leaving the question of the alkaloid's stereostructure (and perhaps even its gross structure) unresolved.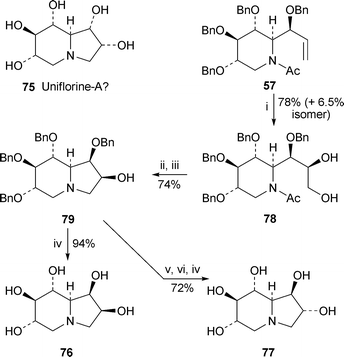 | ||
| Scheme 6 Reagents and conditions: i, OsO4 (cat.), NMO, Me2CO, 25 °C, 4 h; ii, aq. HCl (6 M), THF, 70 °C, 4 h; iii, DEAD, PPh3, py, molecular sieves, 0 °C, 4 h; iv, H2 (1 atm), PdCl2, MeOH–EtOAc (1 : 1), 25 °C, 4 h; v, C6H5CO2H, DEAD, PPh3, THF, 25 °C, 12 h; vi, NaOMe, MeOH, 25 °C, 12 h. | ||
2 Alkaloids from arthropods and amphibians
2.1 Occurrence
The 1999 survey by Daly and co-workers of alkaloids isolated from, or detected in, amphibian skin listed over 500 compounds, not all of them fully characterised.27 This invaluable resource has now been superseded by an even more comprehensive compilation in which a whopping 800-plus alkaloids—many of which still require full structural elucidation and disclosure in the primary literature—are itemised.28 While it is obviously impossible to present a detailed account of all the relevant classes of metabolites, let alone the individual compounds, in this review, several features are worth noting. The most populous class of alkaloids has turned out to be the 5,8-disubstituted indolizidines (e.g., indolizidine 203A 80), of which about 80 (including stereoisomers) have been detected. Many of the proposed structures are tentative, and, like those of most of the other novel metabolites, are based on mass spectral and FTIR spectroscopic data only. Dehydro-5,8-disubstituted indolizidines, hitherto unrecognised, form a major new class of alkaloids, with approximately 30 representatives, among them the alkaloids coded as 245F and 245H (81 and 82, respectively), which were converted into recognisable saturated analogues by catalytichydrogenation. Also emerging as a more widely represented group than had previously been suspected are the 5,6,8-trisubstituted indolizidines (e.g., indolizidine 223A 83), now comprising about 70 members. Other classes with growing representation are the 1,4-disubstituted quinolizidines (e.g.quinolizidine 233A 84), with about 20 members; and the lehmizidines (e.g.lehmizidine 275A 85), of which nine examples have been identified. Less secure are a number of other dehydro-izidines and ring-hydroxylated izidines; while the well-known pumiliotoxins, allopumiliotoxins and homopumiliotoxins have been augmented by an increasing range of deoxy and desmethyl analogues (e.g., 8-deoxypumiliotoxin 193H 86 and 9-desmethylhomopumiliotoxin 209H 87). It now appears indisputable that all of the izidine alkaloids found in amphibian skins have a dietary source; the pumiliotoxins are sequestered from ants and perhaps mites (vide infra), ants are the likely source of other izidines, and coccinelline-like tricyclic alkaloids come from beetles.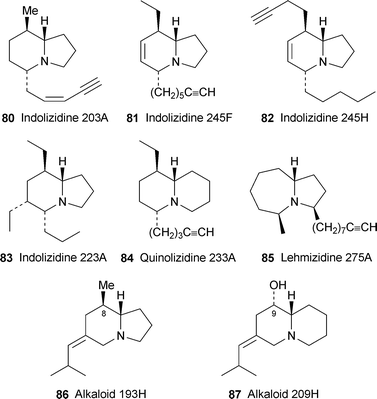
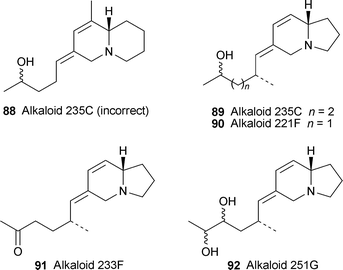
The putative dehydrohomopumiliotoxin structure 88 for alkaloid 235C was recently disproved by Stevenson and co-workers29 (cf. ref. 5d), who showed that the mass spectrometric data for their synthetic product were different from those of the natural product. Daly's team has recently managed to obtain larger quantities of the alkaloid from skin extracts of the Madagascan swamp-dwelling frog Mantella crocea.30 On the basis of 1H NMR spectra, the structure of alkaloid 235C (shown by GC–MS to be a pair of apparent diastereomers) has now been revised to 89, an 8-desmethyldehydropumiliotoxin. From coupling constants supported by molecular modelling, the indolizidine ring system appears to possess an unusual cis-ring fusion. In the light of these results, the dehydrohomopumiliotoxin structures initially proposed for the related alkaloids 221F, 233F and 251G should now be revised to 90, 91 and 92, respectively.
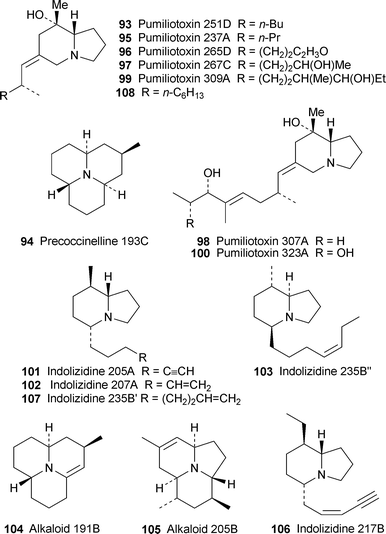
Several typical frog alkaloids have been detected in two species of mite belonging to the suborder Orobatida (cohort Brachypylina)—the first report of such compounds in animals occupying such a low trophic level.31GC–MS and GC–FTIR analysis of hexane extracts from adults (but not nymphs) of the species Scheloribates azumaensis contained three major alkaloidal components: pumiliotoxin 251D93, precoccinelline 193C 94 and another coccinelline-type alkaloid. A second but unidentified Scheloribates species yielded 8-deoxypumiliotoxin 193H 86, a 6,8-diethyl-5-propenylindolizidine, a tentatively identified 1-ethyl-4-pentenynylquinolizidine and, as a minor component, pumiliotoxin 237A95, confirmed by comparison with a synthetic sample. At least six other alkaloids could not be identified. Although this work did not provide evidence for the dietary transfer of alkaloids from mites to frogs (perhaps with ants forming an intermediate trophic level), it is known that mites form a large component of the diet of dendrobatid frogs.
Pumiliotoxin 237A95 has also been isolated from a collection of the red-bellied toad, Melanophryniscus montevidensis, in south-eastern Uruguay together with the related pumiliotoxins 265D, 267C, 307A, 309A and 323A (96–100, respectively) and several unspecified indolizidines.32 This is the first time that these pumiliotoxins have been found in the species. However, the major component in most cases was pumiliotoxin 251D93; and the surprising constituent hydroquinone, reported for the first time in the species, was also found in high concentrations. The pumiliotoxin content of the toads proved to be highly variable among populations from six different locations as well as among individual specimens from each location, perhaps reflecting variability in dietary sources. However, a parallel investigation of 125 arthropod samples, which may represent a food source for the toads, failed to show the presence of any pumiliotoxins. Geographical and seasonal variation in skin alkaloids was also found to be pronounced in Dendrobates pumilio specimens collected from Isla Bastimentos in the Bocas del Toros archipelago of Panama.33 The most abundant alkaloids in the frogs were disubstituted indolizidines 205A, 207A and 235B″ 101–103, trisubstituted indolizidine 223A 83, pumiliotoxins 307A and 323A and tricyclic alkaloids 191B and 205B, 104 and 105, as well as several others not relevant to this review. Once again, the variations were ascribed to spatial and temporal differences in the arthropod food sources.
Potential arthropod food sources for the alkaloids found in Madagascan frogs of the genus Mantella remained unknown until a recent study by Clark et al. revealed the presence of pyrrolidines and pyrrolizidines, among other classes, in ants endemic to the island.34 Although only one indolizidine alkaloid was identified in an ant species (the myrmicine ant Tetramorium electrum), the same alkaloid, an isomer of the 5,8-disubstituted indolizidine 217B 106, was detected in specimens of M. bernhardi from different locations. Indolizidine 217B itself was identified in most specimens of the same frog species as well as in M. madagascariensis and M. baroni, while individual specimens of M. baroni also contained a third 217B isomer, an unspecified isomer of indolizidine 195B (cf. monomorine, Section 2.3) and pumiliotoxin 307A98. Once again, the pattern of alkaloid distribution was very variable, depending both on geographical location and individual assimilation. That T. electrum was found among the stomach contents of a specimen of M. baroni supports the notion of a dietary origin for the frog alkaloids. The article also suggests that, since the neither the ants nor the frogs found in Madagascar and the Neotropics (Central and South America) are closely related, the evolution of alkaloid sequestration in the two regions must have occurred independently.
It has long been argued that the alkaloids sequestered by frogs and toads serve a defensive function against predators, while peptides produced by the amphibians themselves have antimicrobial activity. However, the alkaloid-containing dendrobatid and mantelline frogs and the bufonid toads appear not to produce peptides, thus prompting the hypothesis that the skin alkaloids of the poison frogs might also function as antimicrobial agents against skin infections. Since this hypothesis has never been investigated, Daly and co-workers recently examined the antimicrobial potential of representative alkaloids from ten of the twenty major classes of amphibian alkaloids against a Gram-positive bacterium (Bacillus subtilis), a Gram-negative bacterium (Escherichia coli) and a fungus (Candida albicans).35 None of the alkaloids relevant to this review affected the growth of E. coli, whereas (−)-indolizidine 235B′ 107 and the synthetic pumiliotoxin 108 inhibited the growth of B. subtilis; and only the latter was active against the fungus. Also effective were certain pyrrolidines, piperidines and decahydroquinolines. These preliminary findings clearly deserve further investigation.
2.2 Synthesis of monosubstituted indolizidine and quinolizidine alkaloids
Two complementary syntheses of (−)-indolizidine 167B109 by Settambolo and co-workers36 (Scheme 7) proceeded viaethyl (S)-5,6-dihydroindolizine-5-carboxylate110, which these workers had previously made by cyclisation of ethyl (S)-5-oxo-2-(pyrrol-1-yl)pentanoate and used in a formal synthesis of the tricyclic ant alkaloid (−)-myrmicarin 21737 (cf. ref. 5e). The first route, an adaptation of their recent synthesis of the racemic compound38 (cf. ref. 5f), entailed conversion of the ester into an aldehyde, chain extension by Wittig olefination and diastereoselective hydrogenation of the alkene intermediate 111 in the presence of 5% rhodium on carbon. This approach suffered from apparent racemisation during formation of the intermediates; while ester110 was essentially a single enantiomer, the optical purity of the aldehyde was 80%, and that of the final product was only 42%. More satisfactorily, 110 could be reduced with lithium aluminium hydride to yield the alcohol 112 in 98% optical purity. The corresponding tosylate113 reacted with diethylcuprate to give the dihydroindolizine114 in undiminished optical purity, after which rhodium-catalysed hydrogenation completed the synthesis of the target 109 in 25% overall yield based on 110.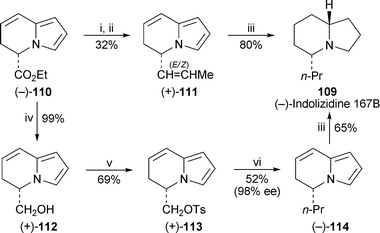 | ||
Scheme 7 Reagents and conditions: i, DIBAL (1 M in hexane), THF–hexane (1 : 4), −78 °C, 1.5 h, then aq. NaK tartrate, −78 °C to rt; ii, EtPPh3+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Br−, NaNH2, THF, −30 °C, 1.5 h, then 0 °C, 17 h, rt, 4 h; iii, H2 (10 atm), 5% Rh/C, Et2O, rt, 1.5 h; iv, LiAlH4, THF, 0 °C, then rt, 1 h; v, p-TsCl, NEt3, CH2Cl2, rt, 5 h; vi, EtMgBr (3 M in Et2O), Li2CuCl4 (0.28 M in THF), THF, −78 °C, warm to rt over 63 h. Br−, NaNH2, THF, −30 °C, 1.5 h, then 0 °C, 17 h, rt, 4 h; iii, H2 (10 atm), 5% Rh/C, Et2O, rt, 1.5 h; iv, LiAlH4, THF, 0 °C, then rt, 1 h; v, p-TsCl, NEt3, CH2Cl2, rt, 5 h; vi, EtMgBr (3 M in Et2O), Li2CuCl4 (0.28 M in THF), THF, −78 °C, warm to rt over 63 h. | ||
A postscript to the previously published synthesis of (±)-indolizidines 167B and 209D, rac-109 and 115, by Baskaran and coworkers39 (cf. ref. 5e) reports a dramatic solvent effect in the last step of their synthesis, in which the tosylate116 was treated with dialkylcuprates at low temperature40 (Scheme 8). In tetrahydrofuran, the reagent prepared from n-butyllithium and copper(I) cyanide underwent nucleophilic addition to the aromatic ring to give the novel sulfonate117, while the reagent made from ethylmagnesium bromide and copper(I) cyanide afforded the bromo product 118. Only when the reaction with alkylmagnesium bromides was conducted in diethyl ether were the desired alkaloids 109 and 115, as well as the 5-pentyl analogue, obtained. Also shown in Scheme 8 is a short synthesis of (±)-indolizidine 209D by Amos et al., in which the anion of pyrrole reacted with γ-decanolactone119 followed by iodomethane to give the ester120.41Cyclisation to the indolizinone121 was effected in 90% yield with boron tribromide, after which catalytichydrogenation produced the racemic target compound 115 in 24% overall yield based on commercially available reactants.
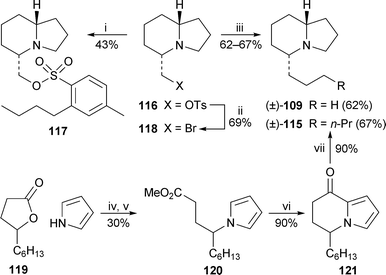 | ||
| Scheme 8 Reagents and conditions: i, n-BuLi, CuCN, THF, −78 °C, 4 h, then −10 °C, 4 h; ii, EtMgBr, CuCN, THF, −78 °C, 30 min, then 0 °C, 2 h; iii, EtMgBr or n-C5H11MgBr, CuCN, Et2O, −78 °C; iv, pyrrole + KH, DMF, 0 °C, 10 min, then lactone118, 160 °C, 4 h; v, add MeI, K2CO3, rt, overnight; vi, BBr3, CH2Cl2, 0 °C, 10 min; vii, H2 (40 psi), 5% Pd/C, AcOH, 10 h. | ||
Concise syntheses of the 5-epi analogues of indolizidines 167B and 209D are shown in Scheme 9. Toyooka and Nemoto applied a Sonogashira coupling to their tried and tested enol triflate intermediate 122 to prepare the alkynyltetrahydropyridine (−)-123, which was carefully reduced to give the 2,6-trans-disubstituted piperidine (−)-124.42 The 5-propyl chain was introduced by Swern oxidation of 124 followed by Wittig chain extension and reduction to yield (−)-125. The bicyclic lactam (−)-126 was obtained from 125 by standard transformations, after which the synthesis of (−)-(5R,9S)-5-epi-indolizidine 167B127 was completed by reduction of the lactam with lithium aluminium hydride. Takahata and Ichinose hydroborated another previously reported intermediate, the 2,6-trans-bis(allyl)piperidine (+)-128, to give the corresponding diol, which was deprotected and cyclised to afford the indolizidine (−)-129.43 The side chain was elaborated by oxidation to the aldehyde, Wittig homologation and reduction, thereby giving (+)-(5S,9R)-5-epi-indolizidine 209D 130, incorrectly named as (+)-indolizidine 209D in the article.
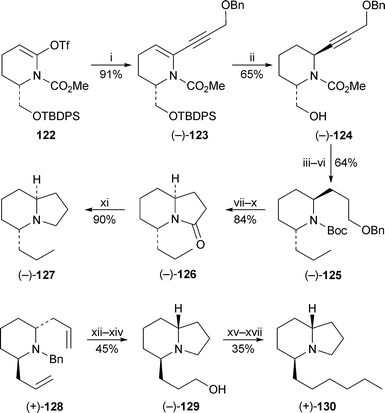 | ||
Scheme 9 Reagents and conditions: i, BnOCH2C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH, (PPh3)4Pd (10 mol%), CuI (1 mol%), Pri2NH–THF (2 : 1), rt, 20 h; ii, NaBH3CN, TFA, CH2Cl2, −78 °C to −10 °C, 1 h; iii, Swern oxidation, then Ph3PCHMe, THF, rt, 4 h; iv, H2 (1 atm), 10% Pd/C, EtOAc, 3 d; v, n-PrSLi, HMPA, THF, 0 °C, then rt, 52 h; vi, Boc2O, NaOH, dioxane–H2O (2 : 1), 0 °C, then rt, 17 h; vii, H2 (4 atm), 20% Pd(OH)2/C, MeOH, 20 h; viii, Swern oxidation, then NaClO2, MeCHCMe2, NaH2PO4·2H2O, ButOH, rt, 1 h; ix, TFA, CH2Cl2, 0 °C, then rt, 5 h; x, (EtO2C)2O, DMF, NEt3, 0 °C, then rt, 1 h; xi, LiAlH4, THF, reflux, 5 h; xii, 9-BBN (0.5 M in THF), THF, 0 °C, then rt, 1 h, then aq. NaOH (3 M), aq. H2O2 (30%), 0 °C, 1 h; xiii, H2, Pd(OH)2, MeOH, rt, 1 h; xiv, CBr4, PPh3, CH2Cl2, rt, overnight, then NEt3, 0 °C, 30 min; xv, Dess–Martin periodinane, CH2Cl2, rt, 3 h; xvi, Ph3PCHEt, THF, 0 °C, overnight; xvii, H2, Pd(OH)2, MeOH, rt, 9 h. CH, (PPh3)4Pd (10 mol%), CuI (1 mol%), Pri2NH–THF (2 : 1), rt, 20 h; ii, NaBH3CN, TFA, CH2Cl2, −78 °C to −10 °C, 1 h; iii, Swern oxidation, then Ph3PCHMe, THF, rt, 4 h; iv, H2 (1 atm), 10% Pd/C, EtOAc, 3 d; v, n-PrSLi, HMPA, THF, 0 °C, then rt, 52 h; vi, Boc2O, NaOH, dioxane–H2O (2 : 1), 0 °C, then rt, 17 h; vii, H2 (4 atm), 20% Pd(OH)2/C, MeOH, 20 h; viii, Swern oxidation, then NaClO2, MeCHCMe2, NaH2PO4·2H2O, ButOH, rt, 1 h; ix, TFA, CH2Cl2, 0 °C, then rt, 5 h; x, (EtO2C)2O, DMF, NEt3, 0 °C, then rt, 1 h; xi, LiAlH4, THF, reflux, 5 h; xii, 9-BBN (0.5 M in THF), THF, 0 °C, then rt, 1 h, then aq. NaOH (3 M), aq. H2O2 (30%), 0 °C, 1 h; xiii, H2, Pd(OH)2, MeOH, rt, 1 h; xiv, CBr4, PPh3, CH2Cl2, rt, overnight, then NEt3, 0 °C, 30 min; xv, Dess–Martin periodinane, CH2Cl2, rt, 3 h; xvi, Ph3PCHEt, THF, 0 °C, overnight; xvii, H2, Pd(OH)2, MeOH, rt, 9 h. | ||
The recently isolated quinolizidine alkaloidepiquinamide131 has elicited much interest because of its selectivity as a nicotinic agonist for the β2 receptor subtype. In view of the minute quantity (240 µg) originally isolated, its absolute stereochemistry was not determined. Syntheses of both enantiomers have now been reported. Blaauw and co-workers commenced with L-allysine ethylene acetal132, which was converted by a reported procedure into the acyliminium ion precursor (2S,5R)-133 in 94% yield44 (Scheme 10). Reaction with allyltrimethylsilane and boron trifluoride etherate followed by removal of the Cbzprotecting group afforded the trisubstituted piperidine134, which was acylated with acryloyl chloride to give the unstable amide135. Immediate treatment with the Grubbs second-generation catalyst followed by hydrogenation produced the bicyclic lactam (−)-136 in a yield of 63% over three steps. The alcohol was replaced by azidevia the corresponding methanesulfonate with inversion of configuration to give (+)-137, from which the estergroup was removed by a Barton radical-mediated decarboxylation. Finally, reduction of the azidolactam (−)-138 with lithium aluminium hydride and acetylation of the primary amine produced (+)-epiquinamide131 in an overall yield of 15.5% based on 133. An alternative approach in which 132 was acylated with crotonoyl chloride prior to piperidine formation, allylation and metathesis was shorter, but suffered from decomposition of intermediates during purification.
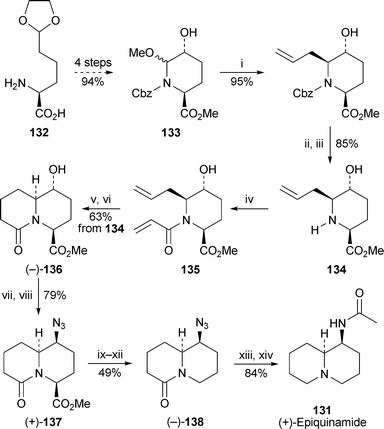 | ||
| Scheme 10 Reagents and conditions: i, H2CCHCH2SiMe3, BF3·Et2O, CH2Cl2, −30 °C, then rt, 2 h; ii, HBr (33% in AcOH), 0 °C, 10 min; iii, K2CO3, MeOH, rt, 2 h; iv, H2CCHCOCl, NEt3, CH2Cl2, −60 °C, 2 h; v, Grubbs II catalyst (10 mol%), CH2Cl2, rt, overnight; vi, H2 (1 atm), Pd/C, MeOH, 2 h; vii, MeSO2Cl, NEt3, CH2Cl2, 0 °C, 1 h; viii, NaN3, DMF, 100 °C, 18 h; ix, aq. NaOH (1 M), THF, 4 h, then aq. HCl (1 M); x, ClCO2Bui, N-methylmorpholine, THF, −15 °C, 5 min; xi, add 2-mercaptopyridine N-oxide, NEt3, THF, 1 h (dark); xii, ButSH, sun lamp (250 W), 2 h; xiii, LiAlH4, THF, 60 °C, overnight; xiv, Ac2O, aq. NaOH (1 M), dioxane, 2 h. | ||
The approach to (−)-epiquinamide by Huang et al. commenced with the addition of Grignard reagent139 to the (S)-3-benzyloxyglutarimide140 to give a mixture of the hydroxylactam141 and its ring-opened keto-amide tautomer 142 in 93% combined yield45 (Scheme 11). The acyliminium ion produced in situ by dehydration of this mixture in the presence of boron trifluoride etherate was reduced with triethylsilane to give the piperidone (+)-143 almost exclusively (96 : 4) as the 5,6-trans-disubstituted isomer. Activation of the alcohol as the tosylate and N-deprotection produced (+)-144, which was quantitatively cyclised to the quinolizidinone (+)-145 under basic conditions. After hydrogenolytic removal of the benzylprotecting group, the scene was set for replacing the alcohol in the intermediate (−)-146 with the target's acetamido substituent with inversion of configuration. The method adopted, similar to that in Blaauw's route (vide supra; Scheme 10), entailed activation as the mesylate prior to displacement with azide ion. However, when the reaction was performed in N,N-dimethylformamide at 65–70 °C, the azide (−)-147 was obtained in only 53% yield, the remainder of the precursor being converted into the elimination product (−)-148 (30%). Elimination was the sole outcome when the reaction was conducted in dimethyl sulfoxide at 70–75 °C. Finally, 147 was transformed into (−)-epiquinamide ent-131 by reducing both the azide and lactamgroups with lithium aluminium hydride and acetylating the primary amine. As a bonus, intermediate 146 could converted into the alcohol (−)-149 in 90% ee by a one-pot Swern oxidation followed by addition of methylmagnesium iodide. Since racemic 149 has previously been converted into (±)-homopumiliotoxin 223G46 (cf. ref. 5g), Huang's approach can be considered to complete a formal synthesis of the (−)-alkaloid150.
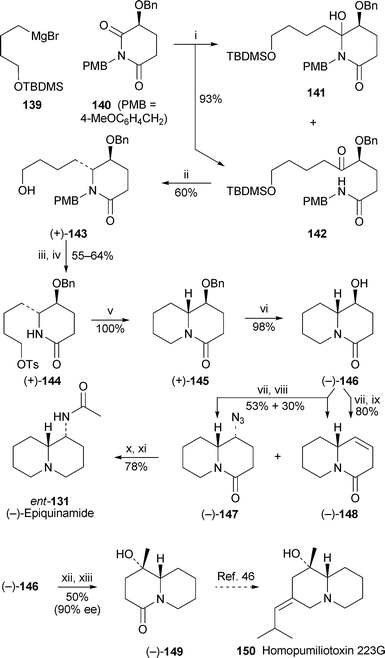 | ||
| Scheme 11 Reagents and conditions: i, 139 (1 M in THF), CH2Cl2, −78 °C, 3 h; ii, Et3SiH, BF3·Et2O, CH2Cl2, −78 °C, 6 h, then rt, 2 d; iii, p-TsCl, py, CH2Cl2, −30 °C, 1 h, then −10 °C, overnight; iv, CAN, MeCN–H2O (9 : 1), 0 °C, 20 min, then rt, 45 min; v, NaH, THF, −40 °C, 1 h, then 40 °C, 1 h; vi, H2 (1 atm), 10% Pd/C, MeOH, rt, 1 d; vii, MeSO2Cl, NEt3, CH2Cl2, −30 °C, 2 h, then −10 °C, overnight; viii, NaN3, DMF, 65 °C, 40 h; ix, NaN3, DMSO, 75 °C, 40 h; x, LiAlH4, THF, 60 °C, 3 h; xi, Ac2O, aq. NaOH (1 M), dioxane, 6 h; xii, (COCl)2, DMSO, CH2Cl2 −78 °C, 1 h, then EtNPri2, warm to −40 °C; xiii, add MeMgI in Et2O, −78 °C, 3 h. | ||
2.3 Synthesis of disubstituted indolizidine and quinolizidine alkaloids
Two of the synthetic routes described in the previous section have also been adapted to complete formal syntheses of 3,5-disubstituted indolizidine alkaloids. In the approach of Amos et al., the indolizinone151, obtained by cyclisation of the corresponding methyl 4-(1H-pyrrol-1-yl)pentanoate152, was defunctionalised at C-8 prior to acylation at C-3 with butanoyl chloride, giving a modest yield of 15341 (Scheme 12). This represents a formal synthesis of racemic monomorine (±)-154, which has previously been obtained from 153 by stereoselective high-pressure catalytichydrogenation over palladium on carbon.47 The new route to the bicyclic lactam (−)-126 (vide supra; Scheme 9) by Toyooka and Nemoto42 effectively also completes a formal synthesis of the (5E,9Z)-diastereomer155 of the bufonid toad alkaloid indolizidine 223AB, since Hart and Tsai previously accomplished the transformation of 126 into 155 with racemic material.48 The important enantioselective synthesis of (−)-indolizidine 223AB156 itself by Smith and Kim via a ‘linchpin’ dithiane, previously reported as a communication49 (cf. ref. 5h), has now been reprised with amplifying discussion and full experimental details50 (also see Section 2.4).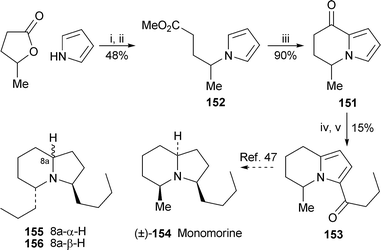 | ||
| Scheme 12 Reagents and conditions: i, pyrrole + KH, DMF, 0 °C, 1 h, then γ-valerolactone, 160 °C, 4 h; ii, add MeI, K2CO3, DMF, rt, overnight; iii, BBr3, CH2Cl2, 0 °C, 10 min; iv, NaBH3CN, ZnI2, CH2Cl2, reflux, 6 h; v, MeCH2CH2COCl, AgOTf, CH2Cl2, rt, overnight. | ||
An enantioselective route to several 3,5-disubstituted indolizidine alkaloids by Remuson and co-workers51 (Scheme 13) proceeded through the known carbamate157, which was prepared in five steps from (S)-pyroglutamic acid. The allylsilanes 158 condensed with the acyliminium ion generated from 157 in the presence of tin tetrachloride to give rather poor yields of the alcohols 159 and 160 as inseparable mixtures of diastereomers at C-5. The 2,5-relative stereochemistry could not be established from the spectroscopic data, and the ratios of the diastereomers were not determined. However, when the corresponding ketone diastereomer 161 was hydrogenated over palladium on carbon, sequential N-deprotection, cyclisation to the bicyclic iminium ion and reduction of the double bonds yielded separable mixtures of (−)-indolizidine 195B162 and (+)-monomorine154 in isolated yields of 36% and 7%, respectively. Similarly, hydrogenation of ketone163 afforded (−)-indolizidine 223A 156 (34%) and its known (5Z,9Z)-diastereomer (−)-164 (13%). The results show that the (5R)-butylgroup in 157 must bias the attachment of the allylsilane to give predominantly the 2,5-trans isomers of 159 and 160.
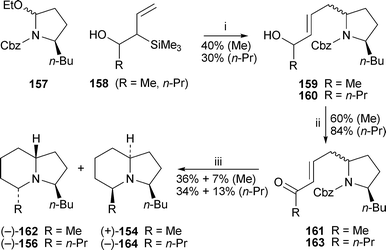 | ||
| Scheme 13 Reagents and conditions: i, 157 + SnCl4 (1 M in CH2Cl2), CH2Cl2, −78 °C, then allylsilane158 (1.1 equiv.), −20 °C, 90 min; ii, pyridinium dichromate, CH2Cl2, 25 °C, 24 h; iii, H2 (3 atm), 10% Pd/C, MeOH, rt, 5 h. | ||
Toyooka, Nemoto and co-workers continue to make valuable contributions to the total synthesis of 5,8-disubstituted indolizidine alkaloids. No fewer than five such compounds have been prepared enantioselectively via a common intermediate, the bicyclic lactam165, as shown in Scheme 14.52 The enol triflate of the known enantiomerically pure piperidinone166 underwent palladium-catalysed carbonylation in methanol to give the unsaturated ester167. Highly stereoselective conjugate addition of methylcuprate to 167—one of the signature reactions of the Toyooka approach—produced the 2,3,6-trisubstituted piperidine168, which contains all three of the target alkaloids’ stereogenic centres with the correct relative and absolute stereochemistry. Reduction of the estergroup produced 169, the unprotected hydroxymethyl group of which was oxidised prior to chain extension by Horner–Emmons reaction to give the enoate170. The pivotal lactam165 was then obtained by hydrogenation, cyclisation and desilylation. For the synthesis of (−)-indolizidine 203A 80, further steps included chain extension by oxidation, Arndt–Eistert homologation and Wittig reaction to give the (Z)-vinyl iodide171, which underwent Sonogashira coupling with trimethylsilylacetylene to afford the desired alkaloid (apparently, the first enantioselective total synthesis) after a final desilylation. This product proved to be spectroscopically identical with natural laevorotatory indolizidine 203A, originally isolated from Dendrobates auratus, although it had a much larger specific rotation. The (5S,8R,8aS)-absolute configuration of the natural product has thus been established. Other chain-extension reactions (not shown) were performed on lactam165 to give the much-synthesised (−)-indolizidine 209B 172, as well as the dendrobatid alkaloids (−)-indolizidines 231C, 233D and 235B″, 173, 174 and 103. Since naturally occurring indolizidine 233D is also laevorotatory, its absolute configuration was deduced to be (5R,8R,8aS); and, while the specific rotations of natural indolizidines 209B and 231C are unknown, the (5R,8R,8aS)-configurations of the synthetic (−)-enantiomers are unambiguous. Remarkably, however, naturally occurring indolizidine 235B″ has a small positive specific rotation, which suggests that the natural product has the opposite absolute configuration, i.e., ent-103.
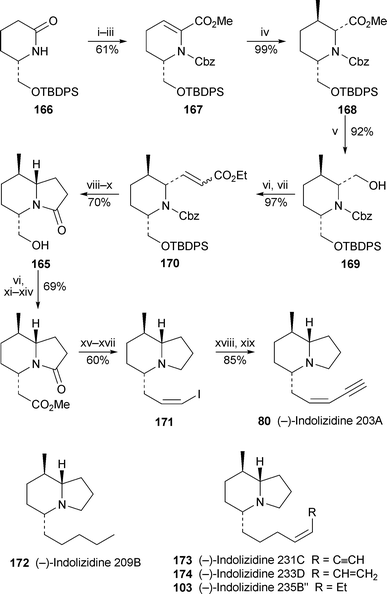 | ||
| Scheme 14 Reagents and conditions: i, n-BuLi, CbzCl, THF, −78 °C to 0 °C; ii, LiHMDS, 5-Cl-2-NTf2-pyridine, THF, −78 °C to −40 °C; iii, (PPh3)4Pd, CO, MeOH, NEt3, DMF, 75 °C; iv, Me2CuLi, Et2O, −78 °C to −10 °C; v, LiBHEt3, THF, 0 °C; vi, Swern oxidation; vii, (EtO)2POCH2CO2Et, NaH, THF, 0 °C to rt; viii, H2 (4 atm), 20% Pd(OH)2/C, MeOH; ix, Me3Al, CH2Cl2, reflux; x, Bu4NF, THF, rt; xi, NaClO2, NaH2PO4, ButOH–H2O, 0 °C to rt; xii, ClCO2Et, NEt3, THF, 0 °C; xiii, CH2N2, Et2O, rt; xiv, PhCO2Ag, NEt3, MeOH, rt; xv, LiAlH4, THF, reflux; xvi, Dess–Martin oxidation, CH2Cl2, rt; xvii, Ph3PCH2I+I−, NaHMDS, HMPA, THF, −78 °C to rt; xviii, HCCSiMe3, CuI, (PPh3)4Pd, Pri2NH, THF, rt; xix, K2CO3, MeOH, rt. | ||
The 5,8-disubstituted indolizidine alkaloids also include 8-ethyl analogues, none of which has previously been synthesised. Toyooka's team has now made two of these compounds by a route similar to that shown in Scheme 14.53Conjugate addition of vinylcuprate to the enoate167 stereoselectively produced the substituted piperidine175 in 94% yield, after which transformations analogous to those shown previously were used to make the bicyclic lactam176 (Scheme 15). Once again, the hydroxymethyl substituent was subjected to a chain extension by oxidation and Wittig reaction to give the primary alcohol177, the corresponding aldehyde from which was transformed into the alkyne (−)-178 with the Seyferth–Gilbert reagent. The GC–MS and GC–FTIR spectra of this product were identical to those of natural indolizidine 219F, obtained from the Madagascan frog Mantella betsileo. Since the optical rotation of the natural product is not known, the synthesis establishes only the relative configuration of the alkaloid. Alternative transformations of lactam176 afforded (−)-179, which was spectroscopically indistinguishable from indolizidine 221I, isolated from M. viridis. However, the chromatographic and spectroscopic properties of a third synthetic product, (−)-180, differed somewhat from those of the expected target, natural indolizidine 193E. Since the Bohlmann bands of the natural product indicate a cis-relationship of the hydrogen atoms at C-5 and C-8a, the authors suggest that the natural product is probably the 8-epimer of 180, i.e., with the C-5 and C-8 substituents cis to each other.
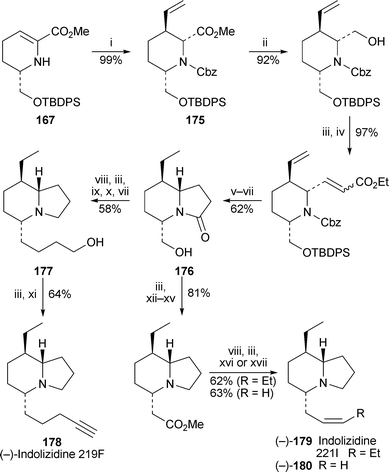 | ||
| Scheme 15 Reagents and conditions: i, (H2CCH)2CuLi, −78 °C to −10 °C; ii, LiBHEt3, THF, 0 °C; iii, Swern oxidation; iv, (EtO)2POCH2CO2Et, NaH, THF, 0 °C to rt; v, H2 (4 atm), 20% Pd(OH)2/C, MeOH; vi, Me3Al, CH2Cl2, reflux; vii, Bu4NF, THF, rt; viii, LiAlH4, THF, reflux; ix, Ph3P(CH2)3OTBDPS+Br−, n-BuLi, THF, 0 °C to rt; x, H2 (1 atm), 10% Pd/C, EtOAc; xi, (MeO)2POCHN2, ButOK, THF, −78 °C to rt; xii, NaClO2, NaH2PO4, ButOH–H2O, 0 °C to rt; xiii, ClCO2Et, NEt3, THF, 0 °C; xiv, CH2N2, Et2O, rt; xv, PhCO2Ag, NEt3, MeOH, rt; xvi, Ph3PCH2CH2Me+I−, NaHMDS, HMPA, THF, −78 °C to rt; xvii, Ph3PMe+I−, n-BuLi, THF, 0 °C to rt. | ||
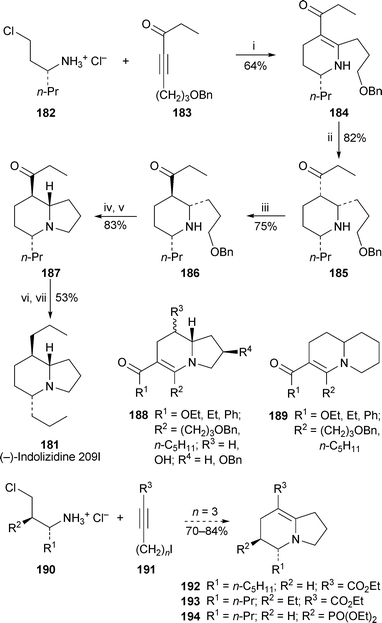
(−)-Indolizidine 209I 181, an 8-propylhomologue of the above alkaloids, has been prepared by Ma and co-workers by the route shown in Scheme 16.54 The key feature is a formal [4 + 2] cycloaddition between the homochiral chloroamine salt 182 and the alkynyl ketone183 to give the piperidine184 in 64% yield. The transformation entails sequential conjugate addition and intramolecular alkylation of the intermediate enaminone. Hydrogenation of 184 produced the all-cis trisubstituted piperidine185, base-catalysed epimerisation of which afforded the diastereomer 186. With all three of the target alkaloid's stereocentres in place, the remaining steps were straightforward, entailing construction of the indolizidinenucleus by debenzylation of 186, cyclisation of the corresponding iodide to give indolizidine187 and defunctionalisation of the 8-propanoyl substituent via the ethylene dithioacetal to complete the synthesis of (−)-181. These authors also provided several additional examples of the formal [4 + 2] cycloaddition process with 2-(2-chloroethyl)pyrrolidines and piperidines to produce a variety of indolizidine and quinolizidine systems of general structure 188 and 189; and they have also published a methodological paper in which numerous examples of the reaction of various chloroamine salts 190 with iodoalkynes 191 (n = 3–5; R3 = CO2Et, Ts, PO(OEt)2, Ac, Bz) produced bicyclic enaminone products, among them the indolizidines 192 (80%), 193 (70%) and 194 (84%), all of which are obvious candidates for conversion into amphibian alkaloids.55
 | ||
| Scheme 16 Reagents and conditions: i, Na2CO3, NaI, PriOH, reflux, 30 h; ii, H2 (1 atm), PtO2, AcOH, rt, 1 h; iii, NaOMe, MeOH, reflux, 12 h; iv, H2 (1 atm), 20% Pd(OH)2/C, MeOH, 25 °C, 20 h; v, PPh3, I2, imidazole, CH2Cl2, 0 °C to rt over 2 h; vi, (CH2SH)2, BF3·Et2O, CH2Cl2, 0 °C, 30 min, then rt, 12 h; vii, Raney Ni, PriOH, 70 °C, 10 h. | ||
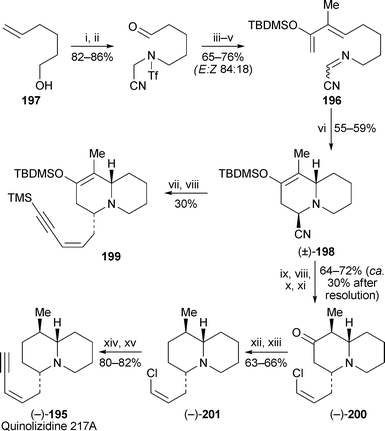
A new synthesis of (−)-quinolizidine 217A195 by Maloney and Danheiser features an intramolecular [4 + 2] cycloaddition of the iminoacetonitrile196 for the construction of the quinolizidine ring system56 (Scheme 17). This precursor was prepared in five steps from 5-hexenol197 as an 84 : 16 mixture of E and Z isomers—not significant, as the isomers apparently interconvert at the elevated temperature (130 °C) required for the cycloaddition. The quinolizidine198, containing an axially orientated cyano group, was the sole product of the reaction. An attempt to introduce the required C-4 enyne substituent directly by alkylation of the anion of 198 gave the product 199 in a disappointing yield of 30% after reductive decyanation. An alternative approach entailing alkylation with (Z)-3-bromo-1-chloropropene gave (±)-200 in approximately 70% yield after reductive decyanation and hydrolysis of the silyl enol ether. As expected, hydride was delivered axially to the intermediate iminium ion to give the equatorially substituted product as the sole isomer. Racemic 200 was resolved with (R)-(−)-1,1′-binaphthyl-2,2′-diylphosphoric acid to give (−)-200 in about 30% overall yield from racemic 198. Removal of the ketonegroup by reduction of the corresponding tosylhydrazone yielded (−)-201, which was converted into (−)-quinolizidine 217A195 by Sonogashira coupling with trimethylsilylacetylene followed by desilylation.
 | ||
| Scheme 17 Reagents and conditions: i, TfNHCH2CN, PPh3, DIAD, THF, rt, 2 h; ii, O3, CH2Cl2, −78 °C, 25 min, then PPh3, rt, 18 h; iii, MeCOC(Me)PPh3, PhMe, 70 °C, 7 h; iv, TBDMSCl, NaI, NEt3, MeCN, rt, 18 h; v, Cs2CO3, THF, 55 °C, 1.5 h; vi, BHT (3 equiv.), PhMe, 130 °C, 36 h; vii, LDA, (Z)-BrCH2CHCHCCTMS, THF, −78 °C, 1.5 h; viii, NaBH3CN, AcOH, MeCN, rt, 2 h; ix, LiHMDS, THF, −78 °C, 3.5 h, then (Z)-BrCH2CHCHCl, LiHMDS, THF, −78 °C, 1 h, warm to 0 °C, 1 h; x, Bu4NF, THF, −78 °C, 1.5 h; xi, resolution with (R)-(−)-1,1′-binaphthyl-2,2′-diylphosphoric acid; xii, TsNHNH2, p-TsOH (cat.), DMF–sulfolane (1 : 1), 110 °C, 2 h; xiii, add NaBH3CN (4 equiv.), ButSH (15 equiv.), C6H12, 110 °C, 5 h; xiv, HCCSiMe3 (2 equiv.), PdCl2(PhCN)2 (cat.), CuI (cat.), piperidine, rt, 2 h; xv, K2CO3 (1 equiv.), MeOH, rt, 2 h. | ||
2.4 Synthesis and reactions of tricyclic alkaloids
In a novel route to the tricyclic dendrobatid alkaloid (−)-205B 105, Smith and Kim have reported a three-component approach involving the solvent-controlled sequential attachment of the chiral intermediates (−)-202 and (+)-203 to the ‘linchpin’ dithioacetal20450,57 (Scheme 18). The reaction entails initial alkylation of the anion of 204 with 202 followed by a 1,4-Brook rearrangement of the silyl substituent, after which the resulting anion formed in situ undergoes a second alkylation with 203. The product (+)-205 was obtained in 53% overall yield, although accompanied by a large amount of the mono-alkylated product (+)-206 (31%).50 Once the silylprotecting groups had been removed and the liberated alcohols converted into mesylates, cleaving of the sulfonamide with sodium amalgam resulted in a double cyclisation to give the indolizidine (+)-207, containing four of the target's five stereogenic centres with the correct absolute stereochemistry. After hydrolysis of the ethylene ketal, the third ring was created by ring-closing metathesis of the corresponding enol silyl ether derived from (+)-208 with the Grubbs second-generation metathesiscatalyst to give the 8b-azaacenaphthylen-5-one (+)-209 in 81% yield. Both the structure and the relative stereochemistry of this product were confirmed by X-ray crystallography. However, the introduction of the alkaloid's axial methyl group proved to be problematic. After several unsuccessful attempts, the task was accomplished by Wittig reaction of 209 with methoxymethyltriphenylphosphorane followed by acidic hydrolysis to the aldehyde and reduction with sodium borohydride. The two alcohols (+)-210 and (+)-211 were formed in isolated yields of 18% and 74%, respectively. The stereoselectivity is probably due to protonation of the intermediate enol ether from the equatorial position, as axial delivery would be disfavoured by electrostatic repulsion from the protonated tertiary amine. Defunctionalisation of (+)-211 via its mesylate produced (+)-212 containing the desired axial methyl group, after which removal of the dithioketal, Wittig methylenation of the corresponding ketone (−)-213 and isomerisation of the double bond into the endocyclic position completed the synthesis of (−)-alkaloid 205B 105.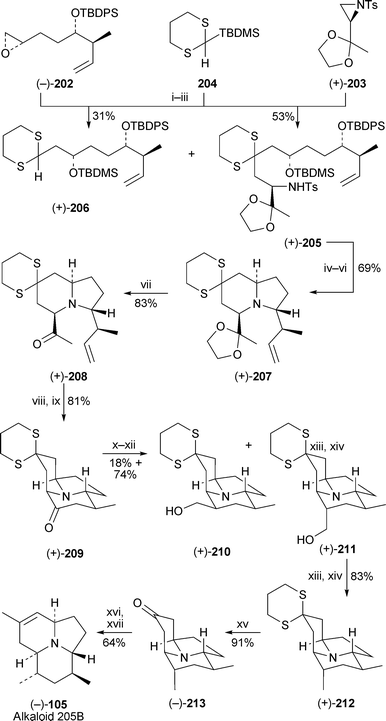 | ||
| Scheme 18 Reagents and conditions: i, 204, ButLi (1.7 M in pentane), Et2O, −78 °C to −45 °C over 1 h; ii, cool to −78 °C, add (−)-202 (1.0 equiv.), Et2O, −78 °C to −20 °C over 30 min, then 1.5 h; iii, cool to −78 °C, add (+)-203 (1.3 equiv.), DME (3.2 equiv.), THF, −78 °C, 10 min, then 0 °C, 2 h; iv, Bu4NF, THF, rt, 20 h; v, MeSO2Cl, NEt3, CH2Cl2, 0 °C, then rt, 30 min; vi, K2CO3, MeOH, rt, 12 h, then 5% Na–Hg, Na2HPO4, 30 h; vii, aq. HCl (2 M), Me2CO, reflux, 24 h; viii, LiHMDS, Me3SiCl, THF, −78 °C, 2 h; ix, Grubbs II catalyst (0.1 equiv.), C6H6, 65 °C, overnight; x, Ph3PCH2OMe+Cl−, ButOK, THF, 20 min; xi, aq. HCl (6 M), THF, 0 °C, 24 h; xii, NaBH4, MeOH, 0 °C, 40 min; xiii, MeSO2Cl, NEt3, THF, 0 °C, then rt, 20 min; xiv, LiBHEt3 (1 M in THF), THF, reflux, 36 h; xv, PhI(O2CCF3)2 (3 equiv.), TFA (10 equiv.), MeCN–H2O (1 : 1), rt, 4 h; xvi, Ph3PMe+Br−, n-BuLi, THF, 0 °C, then rt, 40 min; xvii, p-TsOH·H2O, C6H6, reflux, 20 h. | ||
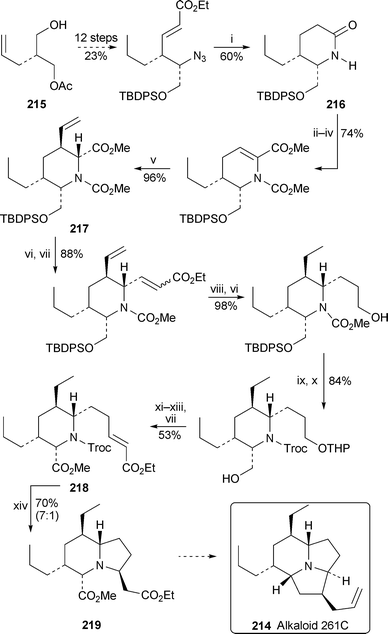
The strategy devised by Toyooka et al. for synthesising the highly substituted indolizidine core of the tricyclic frog alkaloid 261C 214 (Scheme 19) provides yet another example of this productive team's contributions to the chemistry of amphibian alkaloids.58 Starting from the enantiopure desymmetrised diol derivative 215, they prepared the lactam216 in several steps before converting it into the stereodefined 2,3,5,6-tetrasubstituted piperidine217 by methods analogous to those described above (see Schemes 14 and 15, Section 2.3). Chain extensions by Horner–Wadsworth–Emmons methodology thereafter featured in the production of 218, the pivotal precursor for the indolizidinenucleus. Removing the N-protecting group from 218 by treatment with a cadmium–lead alloy preceded cyclisation by intramolecular conjugate addition to give the bicyclic product as a 7 : 1 mixture of isomers; 219 was shown to be the major diastereomer by NOE experiments. The conversion of 219 into alkaloid 261C has yet to be completed; but, with five of the tricyclic target's six stereogenic centres in place, the final transformations should be relatively easy to accomplish.
 | ||
| Scheme 19 Reagents and conditions: i, H2 (1 atm), 10% Pd/C, EtOAc; ii, n-BuLi, ClCO2Me, THF, −78 °C to 0 °C; iii, LiHMDS, 5-Cl-2-NTf2-pyridine, THF, −78 °C to −40 °C; iv, Pd(PPh3)4, CO, MeOH, NEt3, DMF, 75 °C; v, (H2CCH)2CuLi, Et2O, −78 °C to −10 °C; vi, Super-Hydride, THF, 0 °C; vii, Swern oxidation, then (EtO)2POCH2CO2Et, NaH, THF, 0 °C to rt; viii, H2 (4 atm), 10% Pd/C, EtOAc; ix, dihydropyran, PPTS, CH2Cl2, rt; x, KOH (2.2 M in PriOH), 120 °C (sealed tube), then TrocCl, K2CO3, CH2Cl2–H2O; xi, Swern oxidation, then NaClO2, NaH2PO4, ButOH–H2O; xii, CH2N2, EtOAc; xii, PPTS, EtOH, 60 °C; xiv, 10% Cd–Pb; NH4OAc (1 M in THF), rt. | ||
The tricyclic myrmicarins, air- and temperature-sensitive alkaloids isolated from the poison gland secretions of the African ant species Myrmicaria opaciventris, have been synthesised by Movassaghi and Ondrus in a convergent enantioselective route that proceeds through a key dihydroindolizidine intermediate 22059 (Scheme 20). Claisen condensation between the acetal-containing ester221 and the lithium enolate of tert-butyl acetate followed by trapping of the resulting β-keto ester as the (Z)-enol triflate afforded 222 with high selectivity (Z/E >20 : 1). This compound underwent a palladium-catalysed coupling with the substituted pyrrole223 (made in 66% yield from hept-4-en-3-one and tosylmethyl isocyanide) in the presence of the ligand 2-dicyclohexylphosphino-2′,4′,6′-triisopropyl-1,1′-biphenyl (XPhos) to give the (Z)-β-pyrrolyl enoate224 in 95% yield. This reaction, apparently the first example of a palladium-induced N-vinylation of an azaheterocycle, could be performed on a multigram scale (7 g). The enantioselective conjugate reduction of 224, carried out with polymethylhydrosiloxane in the presence of (S)-BINAP and copper(II) acetate, could also be done on a large scale, and gave (R)-225 in 89% yield (85% ee). In addition, changing the chiral ligand to (R)-BINAP gave (S)-225 (89% yield, 81% ee). The subsequent Friedel–Crafts cyclisation of 225 under mildly acidic conditions afforded a quantitative yield of the pivotal dihydroindolizine220 and its regioisomer (>10 : 1). The tricyclic core of the target alkaloids was then made by first protecting the ketonegroup of 220 as a silyl ether, reducing the ester, hydrolysing the silyl ether and finally converting the alcohol into the primary iodide226, cyclisation of which was effected by treatment with silver tetrafluoroborate. Reduction of the product (+)-227 with lithium aluminium hydride in refluxing dioxane effected defunctionalisation of the ketone, giving (R)-(+)-myrmicarin 217 228 in 85% yield. The properties of the product matched those of the same enantiomer previously made from D-glutamic acid by Vallée and co-workers60 (cf. ref. 5i). For the synthesis of (−)-myrmicarin 215A 229, dehydration of 227 with 2-chloro-3-ethylbenzoxazolium tetrafluoroborate gave an especially sensitive alkyne intermediate, which could be isolated and purified only if the conversion was stopped at less than 30%; partial hydrogenation over a Lindlar catalyst to the cis-alkene then completed the synthesis of (−)-228. The overall yield of 9% from 227 reflects the partial conversion; if recovered 227 is taken into account, then the overall yield was about 60%. Finally, mild reduction of 227 with lithium aluminium hydride followed by acid-catalysed dehydration produced (+)-myrmicarin 215B 230. This appears to be the first time that the two isomers 229 and 230 have been obtained in pure form; the natural products were isolated as mixtures. It was also shown that the (S)-enantiomer of 225 could be taken through the same sequence of transformations with similar efficiencies to afford the (R)-enantiomers of the three alkaloids.
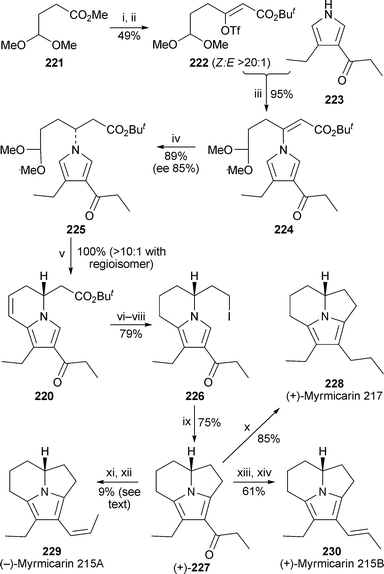 | ||
| Scheme 20 Reagents and conditions: i, 221 + LiOBut (2.5 equiv.), THF, −30°, add H2CC(OLi)OBut (2.2 equiv.), THF, −78 °C over 1.5 h, then −40 °C, 20 min; ii, NaH, 5-Cl-2-NTf2-pyridine, THF, −78 °C to 0 °C over 30 min, then 0 °C to 23 °C over 1 h; iii, 223 + Pd2dba3 (0.075 equiv.), Xphos (0.15 equiv.), K3PO4, PhMe, 23 °C, 30 min, then add 222, PhMe, 65 °C, 12 h; iv, Cu(OAc)2·H2O (0.2 equiv.), (S)-BINAP (0.2 equiv.), polymethylhydrosilane (6.5 equiv.), ButOH (6.5 equiv.), THF, 23 °C, 5.5 h; v, Me2CO–H2O–AcOH (2 : 1 : 1), 40 °C, 12 h; vi, H2 (1 atm), 5% Pd/C, EtOAc, 45 min; vii, TIPSOTf, NEt3CH2Cl2, −78 °C, 15 min, then LiAlH4, Et2O, −78 °C, 15 min, then H2O, warm to 23 °C, then aq. HCl (1 M); viii, I2, PPh3, imidazole, CH2Cl2, 0 °C, 45 min; ix, AgBF4 (2.5 equiv.), CH2Cl2–C6H6 (3 : 1), 23 °C, 2.5 h; x, LiAlH4, dioxane, reflux, 30 min; xi, 2-chloro-3-ethylbenzoxazolium tetrafluoroborate (1.25 equiv.), NEt3, CH2Cl2, 0 °C to 23 °C over 2.5 h; xii, H2 (1 atm), Lindlar catalyst, EtOAc–pyridine (4 : 1), 23 °C, 2.5 h; xiii, LiAlH4, Et2O, −78 °C to 0 °C, then 0 °C, 40 min; xiv, aq. NH4Cl–aq. HCl (pH 2), Et2O. | ||
Ondrus and Movassaghi, working on the hypothesis that suitable tricyclic myrmicarins might conceivably serve as precursors for naturally occurring dimers such as myrmicarin 430A231, undertook a painstaking investigation of the acid-induced dehydration of alcohol 232, the penultimate intermediate in their synthesis of myrmicarin 215B (vide supra; Scheme 20).61 In a series of NMR-scale experiments, 232 was shown to convert slowly into myrmicarin 215B230via a detectable acetate when treated with acetic acid at ambient temperature; however, the putative and no doubt short-lived intermediate azafulvenium ion 233 could not be detected (Scheme 21). By contrast, similar exposure of 232 to trifluoroacetic acid resulted in the almost immediate conversion into myrmicarin 215B, which was then completely converted within 45 minutes into a single new product, the NMR spectra of which indicated a dimeric structure. The same highly unstable new product was also detected when a stoichiometric amount of trifluoroacetic acid was added to a dilute solution of myrmicarin 215B itself (ca. 0.005 M in benzene-d6). The new product could not be isolated, but an azafulvenium or iminium ion structure 234 was suggested when subsequent treatment of a benzene solution with sodium triacetoxyborohydride produced the isolable oxygen-sensitive dimer 235 in 68% yield. Alternatively, exposure of a benzene solution of the intermediate ion to a resin-bound 1,3,2-diazaphosphorine base under oxygen-free conditions resulted in the detection of the exceedingly air-sensitive dimer 236, which could in turn be hydrogenated over palladium on carbon to give the isolable product 237 as a single diastereomer in 87% yield based on alcohol 232. Intermediate 236, named isomyrmicarin 430A, could also be trapped with 4-bromobenzoyl chloride to give 238. All the accumulated evidence, including comprehensive one- and two-dimensional NMR spectroscopic data, supported structure 234 for the intermediate dimeric ion. The authors proposed convincing mechanisms for the dimerisation, detailed discussion of which is beyond the scope of this review.
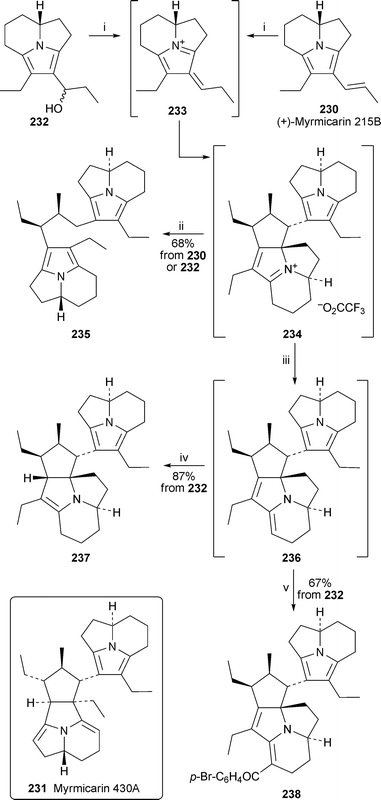 | ||
| Scheme 21 Reagents and conditions: i, 230 or 232 in C6D6 (0.02–0.04 M), TFA (1.1 equiv), 4.5 h, rt; ii, add NaBH(OAc)3, 3.5 h; iii, add resin-bound 2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine (10 equiv.), 30 min; iv, H2 (1 atm), 5% Pd/C, 35 min; v, 4-BrC6H4COCl, EtNPri2, 30 min. | ||
The formal total synthesis of precoccinelline94, originally isolated from coccinellid beetles but now also recognised as a frog skin alkaloid (see Section 2.1), by Takahata and co-workers was described in a prior communication62 (cf. ref. 5j). The full paper containing experimental details and related syntheses of Poranthera alkaloids (cf. Section 5) has now been published.63
3 Polygonatum alkaloids
Polygonatines A and B, 239 and 240, two 5,6,7,8-tetrahydroindolizine alkaloids isolated from the rhizomes of Polygonatum sibiricum, have been synthesised by the simple route shown in Scheme 22.64Cyclisation of 4-(pyrrol-1-yl)butanoic acid241 (R = H) in polyphosphoric acid was found to be more efficient for making the alkaloids’ heterocyclic core 242 than a reported cyclisation of the ethyl ester 241 (R = Et) with boron tribromide. Vilsmeier–Haack formylation of 242 was expected to introduce an aldehyde substituent at C-3, but under standard conditions the C-8 ketone was also converted into a vinyl chloride, giving 243 in a moderate yield of 54%. This unwanted transformation could be reversed in quantitative yield by hydrolysis with a mixture of perchloric and formic acids to give 244. Chemoselective reduction of the aldehyde was achieved with zinc borohydride to complete the synthesis of polygonatine A 239. The transformation of 239 into polygonatine B240, although shown to occur by simply stirring in ethanol with a catalytic quantity of acid, was more satisfactorily accomplished by proceeding through the acetate245, extended heating of which in ethanol gave a good yield of the desired alkaloid240, probably through an azafulvenium ion intermediate. This result gives credence to a prior speculation that 240, with its unusual ethyl ether, might be an artefact of the originally reported isolation procedure, in which ethanol was used as an extractionsolvent (cf. ref. 5k); the naturally occurring metabolite is more likely to be the hydroxymethyl compound 239 or an ester thereof.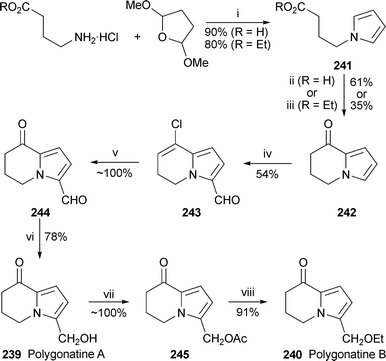 | ||
| Scheme 22 Reagents and conditions: i, NaOAc, H2O–AcOH–(ClCH2)2, 90 °C, 16 h; ii, PPA, rt, 16 h; iii, BBr3, CH2Cl2, 5 °C, 20 min; iv, DMF, POCl3, 0 °C, then add 242, PhMe, reflux, 3 h; v, aq. HClO4 (60%)–HCO2H (1 : 10), 80 °C, 1.5 h; vi, Zn(BH4)2, THF, −10 °C, 0.5 h; vii, Ac2O, py, 80 °C, 2 h; viii, EtOH, reflux, 2 d. | ||
4 Prosopis alkaloids
Juliflorine (juliprosopine) 246, a principal alkaloid of Prosopis juliflora, has been found to inhibit acetylcholinesterase (AChE) and butytylcholinesterase (BChE) in a concentration-dependent manner (IC50 0.42 and 0.12 µM, respectively).65 The inhibition was of the non-competitive type (Ki 0.4 and 0.1 µM, respectively). Since the preferred conformation of AChE when bound to various inhibitors is known from several X-ray crystal structure determinations, the authors performed molecular docking studies to probe the interaction of the alkaloid with the enzyme. This revealed that the dehydroindolizidine section penetrates well into the ‘aromatic gorge’ of the enzyme, at the bottom of which (ca. 20 Å from the protein surface) lies the catalytic site. Rigidity in the molecule appears to be enhanced by π-stacking between the unsaturated ring C and tyrosine residues in the gorge, while an aspartic acid residuehydrogen-bonds to the bridgehead nitrogen. Hydrophobic contacts help to keep the alkyl chains in close enough proximity to be accommodated in the peripheral anionic sites along the walls of the cavity, while the hydroxy substituent in ring A is hydrogen-bonded to an asparagine residue and, like ring B, remains at the entrance to the gorge. Since alkaloid246 also showed dose-dependent spasmolytic and calcium-channel blocking activity, but did not significantly affect the viability of human neutrophils at low concentrations, it might well prove to be a suitable drug candidate for treating Alzheimer's disease.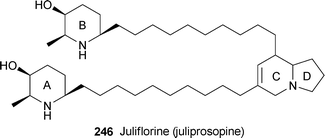
5 Poranthera alkaloids
A prior communication by Tahakata and co-workers describing the synthesis of (−)-porantheridine24762 (cf. ref. 5j) has now been published with complete experimental details as well as a complementary synthesis of (−)-2-epi-porantheridine24863 (Scheme 23). Allylation of pentanedial249 with (−)-B-allyldiisopinocamphenylborane produced an inseparable mixture of the (R,R)-diol250 and its meso-diastereomer, but the subsequently formed piperidine products could be separated by chromatography. Among several methods attempted for the conversion of 2,6-trans-disubstituted piperidine251 into the enantioselectively functionalised diol253, the best entailed initial protection of one alkene bond by formation of the iodo-oxazinone252, Sharpless asymmetric dihydroxylation of the remaining alkene with AD-mix-α, reductive deprotection of the alkene with zinc, and reprotection of nitrogen as a carbamate. The epoxide254, readily prepared from 253, was opened with ethylcuprate to give 255, thereby creating the propyl side chain of the target alkaloids. Elaboration of the remaining allyl substituent by cleavage to the aldehyde, Wittig chain extension and hydrogenation of the resulting double bond produced hydroxy-ketone256. A further two steps yielded the ketal257, thus converging with a previously published route by Comins and Hong66 (cf. ref. 5l) and completing a formal synthesis of (−)-porantheridine247. Alternatively, epimerisation and cyclisation of 256 with iodotrimethylsilane followed by ketal formation afforded 258, which cyclised to (−)-2-epi-porantheridine248 after hydrolysis of the cyclic carbamate and treatment with acid.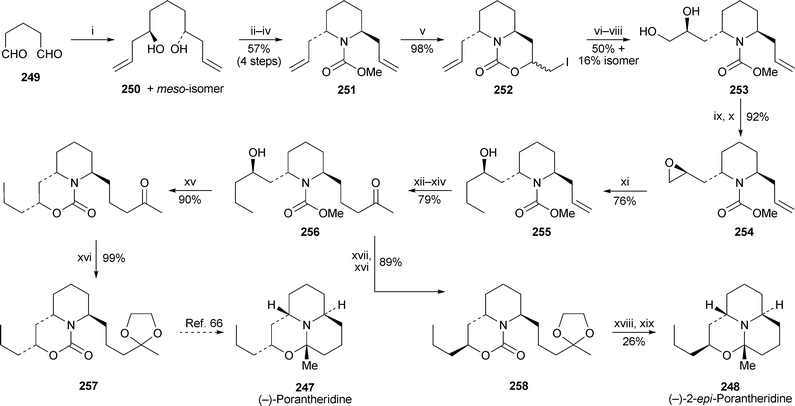 | ||
| Scheme 23 Reagents and conditions: i, (−)-Ipc2BOMe, H2CCHCH2MgBr, Et2O, −78 °C, 15 min, then 249, −78 °C, 1 h, then aq. NaOH (3 M), aq. H2O2 (30%), 0 °C, then reflux, 1 h; ii, p-TsCl, NEt3, DMAP (cat.), rt, 2 d; iii, PhCH2NH2, 75 °C, 2 d; iv, ClCO2Me, CHCl3, 90 °C (sealed tube), 2 d; v, I2, CH2Cl2, rt, overnight; vi, AD-mix-α, ButOH–H2O (1 : 1), 0 °C, 17 h; vii, Zn, THF–AcOH–H2O, rt, 1.5 h; viii, ClCO2Me, K2CO3, THF–H2O, rt, overnight; ix, n-Bu2SnO, p-TsCl, NEt3, CH2Cl2, rt, 1.5 h; x, K2CO3, MeOH, rt, 1 h; xi, EtMgBr, CuBr·SMe2, THF, −45 °C, 1 h, then 254, −45 °C to −30 °C, 1 h; xii, OsO4 (cat.), NaIO4, dioxane–H2O (1 : 1), rt, 1.25 h; xiii, Ph3PCHCOMe, CH2Cl2, reflux, 15 h; xiv, H2 (1 atm), Pd(OH)2, EtOAc, rt, 1 h; xv, n-PrSLi, HMPA, THF, rt, 5 d; xvi, HOCH2CH2OH, p-TsOH·H2O, C6H6, reflux (Dean–Stark), overnight; xvii, Me3SiI, MeCN, 0 °C, 2 h; xviii, KOH (2 M in PriOH), 120 °C (sealed tube), 3 d; xix, p-TsOH·H2O, C6H6, reflux, 3 h. | ||
6 Phenanthroindolizidine and phenanthroquinolizidine alkaloids and seco analogues
The three known alkaloids (−)-antofine 259, (−)-(10S,13aR)-antofine N-oxide260 and (−)-14β-hydroxyantofine N-oxide261 have been isolated from the roots and aerial parts of Vincetoxicum pumilum (Apocynaceae—Asclepiadoideae), a perennial herb endemic to Central Asia.67 Worth noting is the complex taxonomic history of the genus; this particular species has variously been classified as Alexitoxicon pumilum, Antitoxicum pumilum and Cynanchum pumilum.Ficus septica (Moraceae), a subtropical tree found in the low-altitude forests of Taiwan, is used traditionally for the treatment of colds, fever and various fungal and bacterial ailments. Bioassay-guided fractionation of a methanolic extract obtained from the stems of this well-investigated source of alkaloids against two human cancer cell lines has led to the isolation of eight new phenanthroindolizidine alkaloids and another six known metabolites, all in quantities too small for reliable 13C NMR spectroscopic characterisation.68 The structures of the compounds were elucidated on the basis of 1H NMR spectroscopy, high-resolution mass spectrometry, as well as UV and IR spectroscopy. The new alkaloids include (−)-ficuseptines B–D 262–264, (−)-(10R,13aR)-tylophorine N-oxide265, (−)-(10R,13aR)-tylocrebrine N-oxide266, (−)-(10S,13aR)-tylocrebrine N-oxide267, (−)-(10S,13aR)-isotylocrebrine N-oxide268 and (+)-(10S,13aS)-isotylocrebrine N-oxide269. The absolute configurations were assigned by analogy with known compounds on the basis of observed Cotton effects in their CD spectra. The known alkaloids (10S,13aR)-antofine N-oxide 260 and dehydrotylophorine270 have not been reported previously from this species, while the stereochemistry of a third known alkaloid, (10S,13aR)-tylophorine N-oxide271, has previously been unspecified. The remaining three alkaloids detected in this study (tylophorine272, tylocrebrine273 and isotylocrebrine274) have been known for decades. In vitro tests for cytotoxicity towards the and NUGC cancer cell lines showed that 267, 268 and tylophorine inhibited the HONE-1 cancer cell line by 92%, 87% and 80%, respectively, at a concentration of 10 µM, while the inhibition of the NUGCcell line was 94%, 93% and 85%, respectively. In an unrelated study, the anti-inflammatory mechanisms by which dehydrotylophorine, tylophorine and ficuseptine-A 275 (all isolated from F. septica) and the artefact 276, a decomposition product of ficuseptine-A, were investigated.69 Although tylophorine, ficuseptine-A and 276 all inhibited the growth of NUGC and HONE-1 cancer cells by similar amounts, tylophorine and ficuseptine A, which contain non-planar indolizidine components, were notably more effective in suppressing the production of nitric oxide in murine macrophagecells than their ‘planar’ analogues 270 and 276. For tylophorine in particular, anti-inflammatory activity could be linked to the inhibition of several specific molecular targets and the resulting effects on signalling pathways.
A new total synthesis of (±)-tylophorine 272 by Wu and co-workers70 commenced with the phenanthrene-9-carbaldehyde277, which was converted in four steps into the acyl azide278 (Scheme 24). Thermolysis in the presence of mercury(II) acetate effected sequential Curtius rearrangement, electrocyclic ring closure and hydrogenrearrangement to give the dibenzo[f,h]isoquinolin-1(2H)-one279 in 85% yield. N-Alkylation with 1-bromo-3-chloropropane then yielded 280, which was cyclised with tributyltin hydridevia the corresponding iodide to give the phenanthroindolizidinone281 in an excellent yield of 87% together with a small quantity of the 13a,14-dehydro analogue (5%). The synthesis was completed by careful hydridereduction of 281 to give tylophorine in 49% overall yield based on aldehyde277. Similar routes were employed to make tetrademethoxytylophorine282 and the phenanthroquinolizidine congener 283 (surprisingly, not a known natural product) in comparable overall yields. Interestingly, NOE studies showed the indolizidine ring junction in 272 and 282 to be cis-fused, while the quinolizidine junction in 283 was trans. The three products were found to be unstable to light, decomposing to the dehydroiminium salts 270, 284 and 285, respectively, when left standing in chloroform for a day. The dehydrogenation could be promoted by treatment with N-bromosuccinimide. When the compounds prepared in this work were tested for cytotoxicity towards three human cancer cells lines, tylophorine itself showed pronounced activity (IC50 489–1764 nM), which was exceeded by that of the phenanthroquinolizidine283 (104–130 nM). Compound 282 and the dehydro products 284 and 285 were substantially less active.
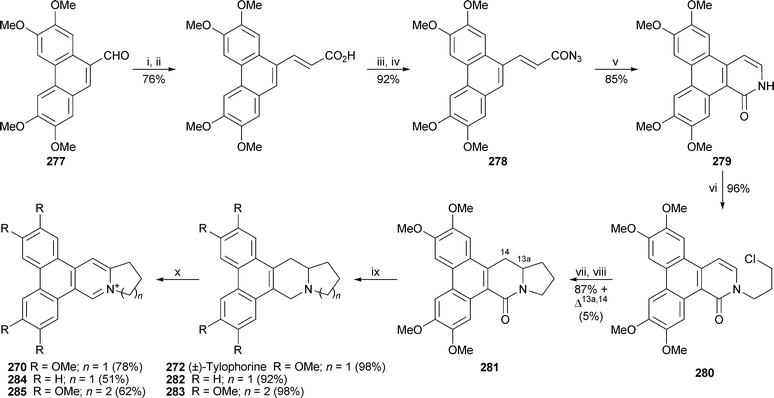 | ||
| Scheme 24 Reagents and conditions: i, Ph3PCHCO2Et, PhMe, reflux, 4 h; ii, KOH (1 M), EtOH, reflux, 3 h; iii, (COCl)2, PhMe, 80 °C, 5 h; iv, NaN3, Me2CO, rt, 2 h; v, Hg(OAc)2, 1,2-Cl2C6H4, reflux, 1 h; vi, NaH, DMF, 0 °C, then rt, 30 min, then Br(CH2)3Cl, DMF, rt, overnight; vii, NaI, MeCN, 125 °C (sealed tube), 12 h; viii, Bu3SnH, AIBN, PhMe, reflux, 6 h; ix, NaAl(OCH2CH2OMe)2H2 (3.5 M in PhMe), dioxane, reflux, 2 h (dark conditions); x, NBS, CHCl3, rt, 1 h. | ||
The phenanthroquinolizidinealkaloid cryptopleurine 286 and its cis-stilbene analogue julandine 287 exhibit a range of biological activities, including antiviral, antifungal and anticancer effects. Intrigued by structural similarities between these alkaloids and the potent anti-mitotic and anti-angiogenic agent combretastatin 288, Banwell and co-workers prepared the simple phenanthrenes 289 and 290 as well as both enantiomers of the four monoseco variants of the parent alkaloids, 291–294,71 by adapting a published approach in which related seco compounds had featured72 (cf. ref. 5m). Compounds 291 and 292 proved to be about three orders of magnitude less cytotoxic towards a panel of human cancer cell lines than (−)-cryptopleurine itself (IC50 3 nM), while the cis-stilbenes 293 and 294 were essentially inactive. The absolute configuration at the single stereogenic centre had virtually no effect on activity. In addition, all the new compounds showed powerful anti-angiogenic properties; in in vitro assays with rat aorta, most of them completely inhibited the growth of blood vessels at concentrations of 100 µg ml−1, and even maintained significant activity down to concentrations of 0.1 µg ml−1. Once again, the absolute configuration had little or no impact on activity. Both the phenanthrene and aminomethyl units appear to contribute to the activity, since compounds 289 and 290 also inhibited the growth of blood vessels. The capacity for bringing about vascular shut-down in combination with low cytotoxicity makes the monoseco alkaloid analogues very interesting leads to explore for potential therapeutic applications.
7 Nuphar alkaloids
The use of chiral N-sulfinylimines as pivotal intermediates is a hallmark of the generalised approach to alkaloid synthesis devised by Davis and his team. Davis and Santhanaraman have now reported a synthesis of the Nupharindolizidine (−)-295 by a route in which the chiral amine salt 296, derived from the sulfinimine (R)-(−)-297, underwent intramolecular Mannich reaction with crotonaldehyde to give, via the imine298, the piperidinone (−)-299 as a single diastereomer73 (Scheme 25). After formation of the N-allyl product (−)-300, ring-closing metathesis followed by hydrogenation afforded the indolizidinone (−)-301. Defunctionalisation of the ketonevia the dithioacetal302 completed this short synthesis of the target alkaloid (−)-295. An alternative Mannich cyclisation with ethyl (E)-4-oxobut-2-enoate produced the piperidinone (−)-303 (53% yield from 297), the ethylene dithioacetal of which was reduced with Raney nickel to give the trisubstituted piperidine (−)-304 in 73% overall yield. This compound previously appeared in the previous (and hitherto only) asymmetric synthesis of (−)-295 by Barluenga et al.74 (cf. ref. 5n), and thus this alternative route also completes a formal synthesis of the alkaloid.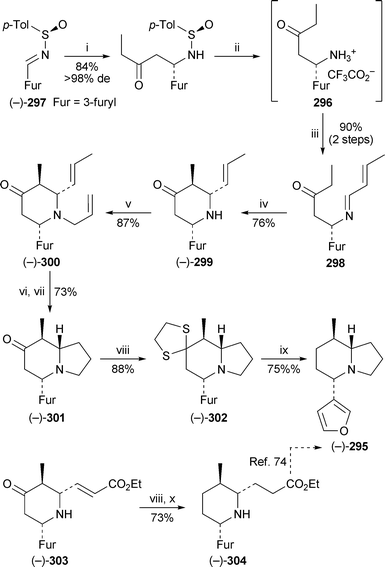 | ||
| Scheme 25 Reagents and conditions: i, EtCOCH2K, Et2O, −78 °C, 30 min; ii, TFA, MeOH, 0 °C, 2 h; iii, MeCHCHCHO, CH2Cl2, MgSO4 (anhyd.), rt, 1.5 h; iv, p-TsOH·H2O, C6H6, 60 °C, 6 h; v, H2CCHCH2Br, Na2CO3, EtOH, reflux, 2 h; vi, (PCy3)2Cl2RuCHPh (5 mol%), CH2Cl2, reflux, 2 h; vii, H2 (1 atm), 5% Pd/C, MeOH, rt, 2 h; viii, HSCH2CH2SH, BF3·OEt2, CH2Cl2, rt, 2 h; ix, Bu3SnH, AIBN, C6H6, reflux, 2 h; x, Raney Ni, EtOH, reflux, 2 h. | ||
Honda and co-workers employed the known (R)-4-methylpyroglutamic ester305 in a new synthesis of (−)-deoxynupharidine306 and its naturally occurring 7-epimer30775 (Scheme 26). The carbon substituent at C-5 was introduced by Horner–Emmons reaction of the corresponding aminal of 305 with diethyl (N-methoxy-N-methylcarbamoylmethyl)phosphonate, after which the Weinreb amide308 was reduced to the aldehyde and converted by Wittig reaction into the 5-allyl product 309. Removal of the N-Bocprotecting group was followed by a crucial reductive deamination with samarium(II) iodide, spontaneous recyclisation then giving the piperidin-2-one310 stereoselectively in 90% overall yield. Alkylation with 3-bromo-2-methylpropene paved the way for a subsequent ring-closing metathesis with the Grubbs second-generation catalyst, which converted the precursor 311 into the bicyclic lactam312 in 96% yield. The furyl substituent was introduced by treating the lactam with 3-lithiofuran followed by reduction with sodium borohydride to give a 5 : 1 mixture of 313 and its C-4 diastereomer. Finally, hydrogenation over palladium hydroxide produced a mixture of (−)-deoxynupharidine306 and (−)-7-epideoxynupahridine307 in yields of 68% and 11%, respectively.
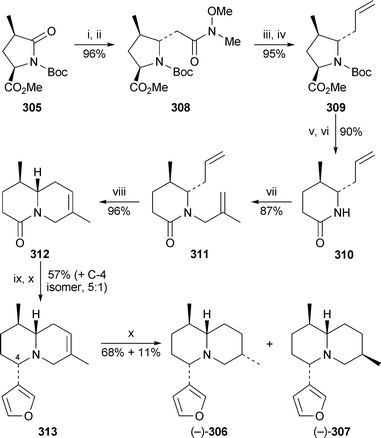 | ||
| Scheme 26 Reagents and conditions: i, LiBHEt3, THF, −78 °C; ii, (EtO)2P(O)CH2CON(Me)OMe, NaH, THF, rt; iii, DIBAL, THF, −78 °C; iv, Ph3PCH2, THF, −78 °C to rt; v, TFA, CH2Cl2, 0 °C to rt; vi, SmI2, HMPA, THF, MeOH, 0 °C to rt; vii, BrCH2C(Me)CH2, NaH, DMF, 0 °C to rt; viii, Grubbs II catalyst (2 mol%), C6H6, 60 °C; ix, 3-lithiofuran, THF −78 °C; x, NaBH4, MeOH, rt; xi, H2, Pd(OH)2, MeOH, rt. | ||
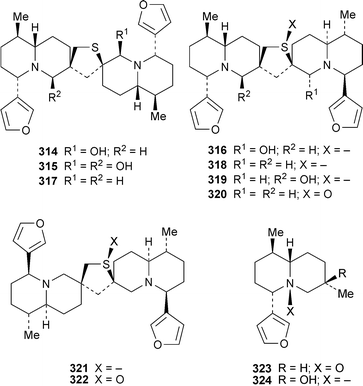
The cytotoxicity of the alkaloid fraction obtained from the rhizomes of Nuphar pumilum towards three cancer cell lines appears to be due almost entirely to three dimeric alkaloids, 6-hydroxythiobinupharidine314, 6,6′-dihydroxythiobinupharidine315 and 6-hydroxythionuphlutine B316, all of which showed high levels of inhibition at a concentration of 10 µM.76 By contrast, dimeric alkaloids lacking the 6-hydroxygroup (thiobinupharidine317, thionuphlutine B318, 6′-hydroxythionuphlutine B 319, thionuphlutine B β-sulfoxide320, neothiobinupharidine321 and neothiobinupharidine β-sulfoxide322) and monomeric alkaloids (7-epi-deoxynupharidine307, nupharidine323 and nupharolutine 324) showed little activity. Morphological examination and DNA fragmentation assays showed that 6-hydroxythiobinupharidine, a principal alkaloid, induced apoptosis of human leukaemia U927 cells remarkably rapidly; cell death was noted within one hour at alkaloid concentrations of 2.5–10 µM. The mechanism of apoptosis appears to involve activation of caspase-8 and caspase-3, two of the enzymes that play a central role in cleaving various cellular components.
8 Alkaloids of the lupinine–cytisine–sparteine–matrinegroup
8.1 Occurrence, analysis and chemical ecology
Very few phytochemical studies on the lupine alkaloids were reported during the period under review. These studies include investigations of the alkaloid content and profile of populations of Calia secundiflora from two different Mexican sites;77GC–MS analysis of alkaloids from the aerial parts of Genista tenera;78 and the isolation of lupinine325 and sparteine326 from Capsella bursa-pastoris (shepherd's purse)79—unusual because the plant belongs to the Brassicaceae rather than the Fabaceae (Leguminosae), the principal source of the lupine alkaloids. The only new alkaloid to be described was (+)-12α-hydroxysophocarpine327, obtained together with ten known alkaloids from the roots of the otherwise well-explored species Sophora flavescens.80 The customary spectroscopic evidence for the structure was supplemented by single-crystal X-ray crystallography of its monohydrate. All the alkaloids isolated in this study exhibited activity against hepatitis B virus at sub-micromolar concentrations; (−)-sophocarpine328, (+)-lehmannine329, (−)-13,14-dehydrosophoridine330 and (+)-oxysophocarpine331 in particular inhibited the HBsAgsecretion by 48–79%, and showed greater activity than matrine332 and its N-oxide333, which have apparently been used clinically in China for treating hepatitis B. Another report describing the isolation of sophocarpine from a common source, S. alopecuroides, included full assignment of its 1H and 13C NMR spectra.81
Numerous studies on the analysis of quinolizidine alkaloids from herbal remedies continue to appear in the Chinese literature, but few make it into more accessible journals. Among the latter are publications on the separation and quantitative determination of alkaloids in Sophora tonkinensis by high performance capillary electrophoresis;82 and the use of LC–MS/MS in analysing the matrine content of remedies made from Sophora roots.83GC and GC–MS have been used in assessing the quinolizidine alkaloid content in seeds of various Lupinus species used in food products.84
Many prior studies have shown that the toxic alkaloid sparteine 326 acts as a defensive chemical to deter herbivory. Its unpalatability was confirmed in a recent study in which the response of gypsy moth larvae to various allelochemicals was investigated.85 A more unusual investigation showed that the production of sparteine in Lupinus arcticus, native to the Yukon, followed a significant daily cycle, with maximum concentrations occurring during the night; this appears to be a response to the feeding patterns of snowshoe hares (Lepus americanus), which are most active at night.86
8.2 Structural, spectroscopic and computational studies
(−)-Sparteine326, the prototypical bis(quinolizidine)alkaloid, assumes the chair–chair–boat–chair conformation 334 because of lone pair repulsions, unless bidentate chelation to metal ions facilitates adoption of the all-chair conformation, as in 335. In the former, both quinolizidine ring junctions are trans, while the C/D ring junction in the all-chair form is cis. Recent single-crystal X-ray diffraction studies revealing the all-chair arrangement have been undertaken on sparteine complexes with mercury(II) chloride,87diethylzinc88 and Cu(II)Br(PhCO2).89 The last-named complex, which showed weak through-space Cu–Cu ferromagnetic interactions, has tetragonally distorted square pyramidal geometry around the metal ion; it was further characterised by far-IR, UV-Vis, NMR and ESR spectroscopies. Crystal structures of zinc(II) methacrylate with sparteine and α-isosparteine (which has a relatively rigid all-chair conformation with trans-A/B and C/D ring junctions by virtue of its inherent C2 symmetry; cf. structure 336) were further stabilised by intramolecular C–H⋯O hydrogen bonds; these complexes, too, were characterised by IR and NMR spectroscopy.90 Another publication in which crystal structures of the sparteine complexes with zinc(II) acrylate or methacrylate were described also included interesting observations on the transfer of chirality from the complexes to the (meth)acrylatecopolymers formed by solution polymerisation with styrene or 2-vinylnaphthalene in the presence of azoisobutyronitrile as initiator.91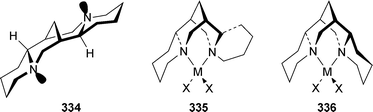
Monoprotonation of sparteine, like complexation with metals, is also accompanied by conformational inversion of ring C to give an all-chair arrangement, with the proton forming a hydrogen-bonded bridge between the two nitrogen atoms. An NMR spectroscopic comparison of the monoperchlorate salts of sparteine, 2-methylsparteine and α-isosparteine with the corresponding zinc(II) complexes containing a variety of counter-ions (Cl, Br, CN, OAc, methacrylate) revealed conformational similarities in solution that agreed well with reported solid-state conformations.92 Interestingly, however, the boat-like conformation of ring C was preserved in N(16)-benzylsparteinium bromide337 even though the benzyl group was attached to the endo-face of the molecule to give a cis-fused C/D ring junction.93
The 1 : 1 complexes of (−)-sparteine with phenyllithium and phenoxylithium proved to be bridged dimers of general structure 338, a structural type that extended to the energetically more favourable mixed aggregate [PhLi·PhOLi·2(−)-spa], the preference for which was supported by quantum chemical calculations.94 By contrast, the complex formed from (−)-sparteine and lithiated 2-bromoindene was monomeric, probably for steric reasons; the interaction between the metal and the indenyl ligand was essentially ionic.95
In view of the importance of (−)-sparteine–alkyllithium complexes in enantioselective synthesis, crystal structures of such complexes are of especial interest. The monomeric complex 339 obtained from the sparteine-assisted lithiation of the corresponding thiocarbamate with n-butyllithium was found to have (S)-absolute configuration both at the tetragonally coordinated lithium cation and at the deprotonated carbon site, from which the pro-Shydrogen of the precursor had been removed.96 The crystal structures of two monomeric sparteine-complexed lithiosilanes have also been determined; that of [Ph2(NEt2)SiLi·(−)-spa] contained a three-coordinate lithium centre, whereas that formed from phenyldimethylsilane also incorporated a molecule of tetrahydrofuran, thus giving a stereogenic four-coordinate lithium centre with (S)-absolute configuration.97 DFT calculations showed that the Li(S) diastereomer 340 was favoured above the Li(R) isomer by 2.9 kJ mol−1; its formation may be the result of thermodynamically controlled epimerisation in solution.
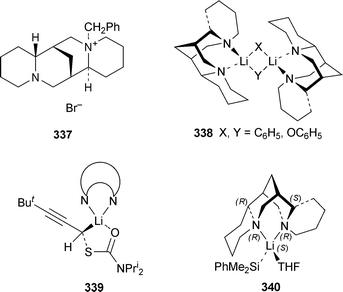
Not unexpectedly, oxo or thiono substituents on the sparteine skeleton introduce conformational changes resulting from flattening around the sp2carbon site. This is the case for ring A in both 13α-hydroxylupanine341 and 13β-hydroxylupanine342; but, surprisingly, the crystal structures of both alkaloids indicated that ring C was a distorted chair—a consequence of intermolecular OH⋯OC hydrogen bonding, the geometry of which was the same despite the different orientations of the hydroxy groups.98 In solution, however, 1H and 13C NMR spectroscopic studies on the free bases revealed a conformational equilibrium for ring C in favour of the boat conformer (85–90%); and the IR spectra in CDCl3 showed that OH⋯OC hydrogen bonding largely gave way to intermolecular deuterium bonding between the solvent and both the carbonyl group and N-16. In a related study, the rigidity of the all-chair skeleton of α-isosparteine permitted the conformational consequences of introducing carbonyl or thione substituents to be evaluated rather precisely for the 2-oxo, 2-thiono, 2,13-dioxo and 2-thiono-13-oxo analogues on the basis of 1H and 13C NMR spectroscopic data.99
As a follow-up to a previous investigation of all eleven possible mono-oxo derivatives of sparteines by DFT methods100 (cf. ref. 5o), Galasso and co-workers have compared the conformations derived from DFT calculations on twelve sparteine derivatives (including 2-, 10-, 15- and 17-monothionosparteines, and several dioxo-, dithiono- and oxothiono-sparteines) with experimental evidence obtained from NMR and He(I)photoelectron spectroscopies.101 In most cases the compounds adopted the standard low-energy chair–chair–boat–chair conformation of the parent alkaloid, with the functionalised ring distorted to a sofa or half-chair conformation; only 2-thionosparteine took on the all-chair conformation, while the data for 2,13-dioxosparteine (= 13-oxolupanine343, a known natural product) showed a nearly 1 : 1 equilibrium mixture of ring C boat and chair conformers in solution. The calculated conformations compared well with those found by X-ray crystallography, where available. A similar comparative study on cytisine344, N-methylcytisine and several unsaturated keto-sparteines (including the alkaloids multiflorine 345, 5,6-dehydromultiflorine, 5,6-didehydrolupanine, aphyllidine346, anagyrine 347 and thermopsine 348) showed that most of the compounds exhibited a marked preference for the rigid cis-C/D twin-chair conformation; an exception was multiflorine, which existed as a mixture of ring C chair and boat forms both in the gas phase and in solution.102 This study also showed that a previously reported X-ray crystal structure for anagyrine103 was, in fact, for its 11β-H isomer thermopsine, which possesses the rigid all-chair trans,trans-fused skeleton of α-isosparteine; a new crystal structure for anagyrine hydrochloride monohydrate unambiguously showed the cis-C/D ring fusion expected of the twin-chair arrangement. Other results to emerge indicated that the most stable ring C chair form of aphyllidine showed significant conformational flexibility in ring A, while the preferred ring C chair conformer of cytisine344 exhibited endo/exo conformational equilibrium of its NH group. The latter feature is associated with an energy barrier calculated by DFT methods to be about 3.6 kcal mol−1 (= 1259 cm−1)—high enough to permit deconvolution of the CO stretching vibrations in the FT-IR spectrum, and hence estimation of the abundances of the two conformers 344′ and 344″ in solution (ca. 40 : 60, respectively).104
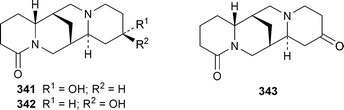
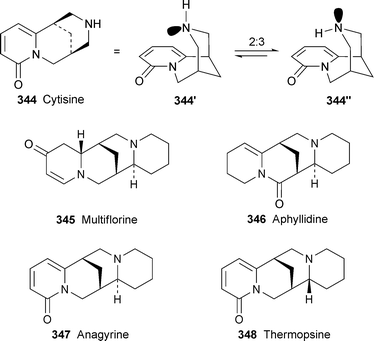
8.3 Synthesis and other chemical transformations
Two short syntheses of racemic tashiromine (±)-349 are shown in Scheme 27. The approach of McElhinney and Marsden105 made use of a cross-metathesis between N-(pent-4-en-1-yl)succinimide350 and allyltrimethylsilane to produce the new allylsilane351 as a 3 : 1 mixture of (E)- and (Z)-isomers. Partial reduction of the imide gave 352, a masked acyliminium ion, the allylsilane then functioning as an intramolecular nucleophile in a highly stereoselective cyclisation to produce the indolizidinone353 as a 96 : 4 mixture of diastereomers. Ozonoloysis and reductive workup yielded 354, after which reduction of the lactam completed the synthesis of the target alkaloid. The overall yield of this six-step synthesis was 19% based on succinimide. The route adopted by Bélanger et al. was part of a more extensive investigation into the intramolecular addition of π-bonded nucleophiles to activated amides—effectively, cyclisations by Vilsmeier–Haack reaction.106 In the example of interest, the lactam355 was treated with trifluoromethanesulfonic anhydride, the intermediate iminium ion then undergoing attack by the strategically placed silyl enol ether to form the unstable enaminal 356 in 93% yield. Hydrogenation under pressure reduced both the alkene bond and the aldehyde to give a 20 : 1 mixture of tashiromine and its 8-epimer in 73% yield. In this case, the overall yield from pyrrolidin-2-one, the precursor of 355, was 26%. The range of reported examples suggests that this method should be suitable for preparing other polycyclic alkaloids containing bridgehead nitrogen.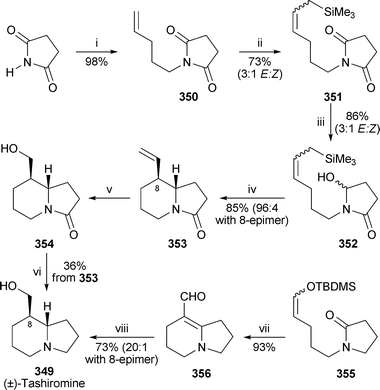 | ||
| Scheme 27 Reagents and conditions: i, H2CCH(CH2)3Br, NaH, DMF, rt; ii, Grubbs II catalyst (5 mol%), H2CCHCH2SiMe3 (4 equiv.), CH2Cl2, reflux, 20 h; iii, NaBH4, aq. HCl (2 M), EtOH; iv, TFA, CH2Cl2, rt; v, O3, MeOH–CH2Cl2, then NaBH4; vi, LiAlH4, THF, reflux; vii, Tf2O, 2,6-di-tert-butyl-4-methylpyridine, CH2Cl2, 0 °C, 20 min, then aq. NaOH (1 M), THF, rt, overnight; viii, H2 (1000 psi), 10% Pd/C, EtOH, Na2CO3, rt, 24 h. | ||
The recent synthesis by Gallagher and associates of the topical alkaloid cytisine 344 in racemic form107 (cf. ref. 5p) has been improved and extended to include the synthesis of two tetracyclic lupine alkaloids.108 This ‘second-generation’ route to cytisine commenced, as in the prior report, with the synthesis of the ester-substituted piperidin-2-one (±)-357; but at this stage an enzymatic resolution was effected by treating the racemic intermediate with α-chymotrypsin, which hydrolysed only the (5S)-enantiomer and returned (5R)-357 in 42% yield and an enantiomeric excess of greater than 98% (Scheme 28). Reduction of (5R)-357 and bromination produced the primary bromide (5R)-358, which was used to alkylate 2-pyridone on nitrogen, thereby giving product 359 (66%) and creating the target alkaloid's N-1–C-10 bond. The 2-alkoxypyridine was also isolated as a minor product (15%). The third ring was formed by an intramolecular 1,6-addition of the enolate formed from the saturated lactam to the newly installed pyridone—a far simpler strategy than in the earlier route, where a palladium(0)-mediated arylation of the lactam was employed. Mild oxidation of the product 360, obtained as a single diastereomer, afforded the tricyclic pyridone361, which was converted into (+)-cytisine344, the unnatural enantiomer, in a further two standard steps. The overall yield of this 10-step synthesis was 3.7%. A similar strategy underlies the synthesis of the tetracyclic alkaloids (±)-anagyrine 347 and (±)-thermopsine 348. For anagyrine, the homopipecolic ester362 was converted into the acrylate analogue 363, conjugate addition of the anion of tert-butyl acetate followed by reduction and cyclisation then giving the quinolizidinone364 containing the correct relative stereochemistry for rings C and D of the target (Scheme 29). The remaining steps were essentially identical to those in the synthesis of cytisine. Thermopsine differs from anagyrine in the stereochemistry at the bridgehead position C-11; in this case, addition of the anion of the homopipecolic ester365 to diethyl methylenemalonate gave adduct 366 as a single diastereomer. The required relative stereochemistry was established by single-crystal X-ray analysis of the quinolizidinone367, readily prepared from 366 by standard methods. The steps required to complete the synthesis of (±)-thermopsine 348 were once again virtually identical to those developed for the synthesis of cytisine.
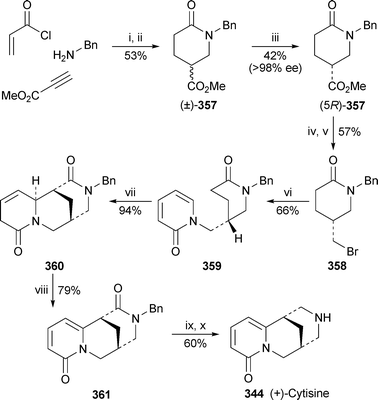 | ||
| Scheme 28 Reagents and conditions: i, HCCCO2Me + BnNH2, Et2O, rt, then H2CCHCOCl, THF, 70 °C; ii, H2 (1 atm), 10% Pd/C, Na2CO3, EtOH; iii, α-chymotrypsin, phosphate buffer (0.1 M), Me2CO, pH 7.4; iv, LiAlH4, THF; v, PBr3, PhMe; vi, 2-pyridone, K2CO3, Bu4NBr, H2O, PhMe, reflux; vii, LiHMDS (2 equiv.), THF, 70 °C (sealed tube), 15 h; viii, MnO2, CH2Cl2; ix, BH3·THF, 0 °C to rt; x, H2 (1 atm), Pd(OAc)2, MeOH, HCl. | ||
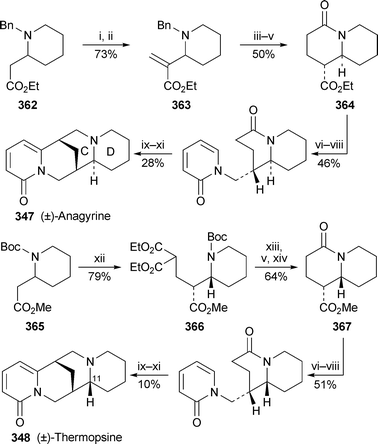 | ||
| Scheme 29 Reagents and conditions: i, LiHDMS, EtOCH2Cl, THF, −78 °C to rt; ii, KOBut, THF, −78 °C; iii, LDA + MeCO2But, THF, −78 °C; iv, H2 (1 atm), 10% Pd/C; v, AcOH, PhMe, reflux; vi, LiAlH4, THF; vii, PBr3, PhMe; viii, 2-pyridone, K2CO3, Bu4NBr, H2O, PhMe, reflux; ix, LDA (2 equiv.), THF, rt, 3 h; x, MnO2, CH2Cl2; xi, BH3·THF, 0 °C to rt; xii, LiHMDS, H2CC(CO2Et)2, THF, −78 °C to rt; xiii, TFA, CH2Cl2, THF, 0 °C; xiv, NaCl, H2O, DMSO, 130 °C, 72 h. | ||
What distinguishes the new synthetic approach to cytisine344 by O'Brien and co-workers from previously reported routes is its early construction of the bispidine core.109 Double Mannich reaction between the piperidin-4-one368, benzylamine and paraformaldehyde gave the bicyclic ketone369, defunctionalisation of which was accomplished by reduction of the corresponding tosylhydrazone to give the key bispidine370 in 47% overall yield (Scheme 30). Allylation of 370 was accomplished after much experimentation by regioselective lithiation with sec-butyllithium and tetramethylethylenediamine, transmetallation to an organocopper(I) intermediate, and alkylation with an allylphosphate; the equatorially allylated bispidine 371 was obtained in 60% yield (effectively 88%, based on recovered starting material). Efficient exchange of the N-Boc substituent by an acryloyl group produced 372, thus setting the scene for the introduction of the third ring, which was rapidly effected by metathesis with the Grubbs first-generation catalyst. The structure of product 373, isolated in 89% yield, was confirmed by X-ray crystallography. The final stages of the synthesis were rather troublesome, because oxidation to the pyridone374, which would have completed a formal synthesis of the alkaloid, proved to be inefficient. However, treatment with palladium on carbon in the presence of cyclohexene as hydrogen transfer reagent not only introduced the additional double bond but also cleaved the N-benzylprotecting group, thereby giving the target alkaloid in 76% yield. The overall yield of (±)-cytisine344 by this interesting new approach was 19% based on 368.
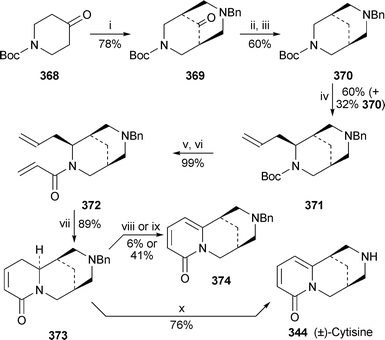 | ||
| Scheme 30 Reagents and conditions: i, BnNH2, (CH2O)n (4 equiv.), AcOH, MeOH, reflux, 5 h; ii, TsNHNH2, EtOH, reflux, 2 h; iii, NaBH4, THF–H2O, rt, 16 h, then reflux, 3 h; iv, sec-BuLi (1.6 equiv.), TMEDA, Et2O, −78 °C, 5 h, then CuCN·2LiCl (1 equiv.), THF, then H2CCHCH2OP(O)(OPh)2; v, TFA–CH2Cl2 (1 : 1), 0 °C, 2 h; vi, H2CCHCOCl, aq. NaOH (10%), CH2Cl2, rt, 1 h; vii, (PCy3)2Cl2RuCHPh (10 mol%), PhMe, reflux, 15 min; viii, MnO2 (2 equiv.), C6H6, reflux, 3 h; ix, 10% Pd/C, dioxane–cyclohexene (3 : 1), 100 °C, 8 h; x, 10% Pd/C, PhMe–cyclohexene (2 : 1), 100 °C, 12 h. | ||
Bispidines also played a starring role in the synthesis of (±)-α-isosparteine375 by Blakemore et al.110 Recognising the C2 symmetry of the alkaloid, the authors devised a bidirectional route in which the bispidine376, easily prepared from dimethyl malonate, paraformaldehyde and ammonia, was elaborated simultaneously in both directions from the central ring B/ring C core (Scheme 31). Bis(N-allylation) of 376 gave 377, the structure of which was confirmed by X-ray crystallography. Reaction of 377 with allylmagnesium bromide occurred regioselectively and apparently at the exo-faces of the carbonyl groups to give the tetraene378, which was shown by NMR spectroscopy to have retained C2 symmetry. However, after a sluggish ring-closing metathesis, the tetracyclic product 379 had lost symmetry, as revealed by single-crystal X-ray analysis. The loss of symmetry persisted after hydrogenation of the double bonds; but the final step, entailing exhaustive reduction with borane, re-established symmetry, probably because the acyliminium ions generated in situ by elimination of the bridgehead hydroxy groups were attacked at the less hindered exo-face by the hydride species. The target alkaloid (±)-375 was isolated as a crystalline hydrate. Remarkably, the entire synthesis was performed without any chromatographic purification; all intermediates were obtained by precipitation, trituration or recrystallisation, and all operations could be performed on multi-gram scales.
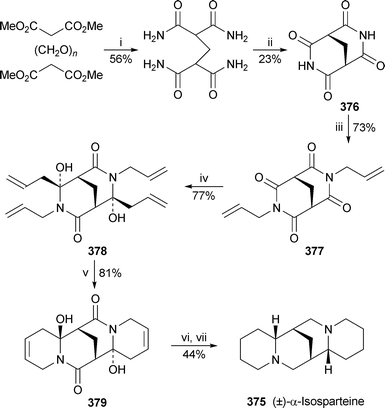 | ||
| Scheme 31 Reagents and conditions: i, KOH (cat.), 90 °C, then aq. NH3, rt; ii, MeSO3H, 150 °C; iii, H2CCHCH2Br, NaH, DMF, rt; iv, H2CCHCH2MgBr, THF, −78 °C; v, (PCy3)2Cl2RuCHPh (4 mol%), CH2Cl2, rt, 24 h; vi, H2, Pd/C, MeOH–H2O; vii, BH3·THF, THF, 0 °C. | ||
Of some interest is a new synthesis of O'Brien's ‘(+)-sparteine surrogate’ 380, reported by Lesma and co-workers.111 The route, a modification of one that they had formerly devised for the synthesis of (−)-cytisine112 (cf. ref. 5q), entailed conversion of the previously reported azide381 into the N-allyl intermediate 382, ring-closing metathesis of which gave the bis(piperidine)383 in 89% yield (Scheme 32). Functional group manipulation afforded 384, cyclisation of which produced the tricyclic compound 385. Reduction of the alkene and reductive methylation then completed the synthesis of (+)-380.
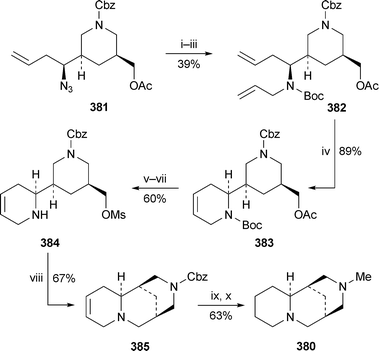 | ||
| Scheme 32 Reagents and conditions: i, PPh3, THF, then H2O; ii, (Boc)2O, NEt3, CH2Cl2; iii, H2CCHCH2Br, NaH, DMF; iv, (PCy3)2Cl2RuCHPh, CH2Cl2, rt; v, TFA in CH2Cl2 (10%); vi, aq. NaOH (0.5 M), THF; vii, MeSO2Cl, NEt3, CH2Cl2; viii, NEt3, CH2Cl2, reflux; ix, H2, 10% Pd/C, EtOAc; x, NaBH3CN, aq. CH2O, THF. | ||
Many synthetic lupinealkaloid derivatives have been reported, in most cases having been prepared for biological studies. New O-aryl analogues of lupinine include 2-hydroxy-4-lupinylchalcones, which were cyclised produce to 7-lupinylflavanones and 7-lupinylflavones.113 The cytisinium salts of xylosyl- and glucosyl-benzyldithiocarbamic acids showed good in vitro fungicidal activity against Fusarium oxysporum.114 Novel N(12)-substituted derivatives of cytisine include (β-chloro-β-phenylvinyl)phosphinic esters,115 monothiooxamides,116 N-propargyl and related compounds117 and, more exotically, numerous N-coumarinyloxyacetyl and furocoumarinalkanoyl derivatives.118,119 The C-substituted cytisine derivatives 386 and 387, prepared for testing as agonists for neuronal nicotinic acetylcholine receptors (nAChRs), showed substantial increases in selectivity for the α4β2 over the α3β4 subtype (3000-fold and 900-fold, respectively) compared with cytisine itself.120 The vinyl derivative 388 showed similar activity to cytisine but may have an advantage in being able to penetrate the blood–brain barrier more easily; however, various aryl and acyl derivatives 389 and 390 showed little or no nAChRagonist activity.120 3-Halogenated cytisines 391 were better ligands for nAChRs than cytisine, especially at α7 receptor sites, and were also better at stimulating the release of various neurochemicals; but halogenation at C-5 proved to be detrimental.121
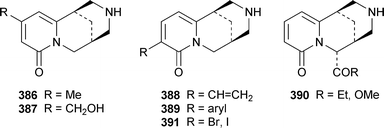
Methodological studies and total syntheses employing (−)-sparteine326 and, more recently, O'Brien's (+)-sparteine surrogate 380 as chiral ligands to induce asymmetric transformations, have become too numerous to describe individually. However, several reviews in which sparteine-mediated transformations are featured merit mention. Hodgson has provided a personal account of his researches into novel reactions of lithiated epoxides and aziridines, many of which proceed enantioselectively in the presence of the alkaloid or its diastereomer α-isosparteine.122 Another worthy review includes examples of (−)-sparteine-mediated addition of organolithiums to aldehydes, ketones, epoxides and acetals.123 Sigman has reviewed his contributions to ligand-modulated palladium-catalysed aerobic oxidative kinetic resolution of racemic alcohols, first discovered with (−)-sparteine as the ligand;124 in addition, his group has probed the origin of the enantioselection with the (−)-sparteine complex of palladium(II) chloride as the catalyst.125 The mechanism for direct insertion of molecular oxygen into the palladium–hydride bond of the complex Pd[(−)-(spa)(Cl)(H)] has also been investigated by means of quantum chemical methods.126
9 Alkaloids from marine sources
The sponge metabolitehalichlorine392 has attracted considerable attention from synthetic organic chemists since its discovery a decade ago because of its potent and selective inhibition of vascular cell adhesion molecule-1 (VCAM-1), implicated in various allergic reactions as well as inflammatory diseases such as rheumatoid arthritis. A timely review by Clive and co-workers focuses on the methodological advances in the synthesis of key components of the halichlorine structure as well as on the successful total syntheses by the groups of Danishefsky and Heathcock.127 Andrade and Martin have subsequently achieved a formal synthesis of the alkaloid128 starting from 393, reported by Wallace and Heathcock as an intermediate in prior synthetic studies on Daphniphyllum alkaloids129 (Scheme 33). After selective protection of the more accessible primary alcohol and oxidation of the other to a carboxylic acid, the product 394 underwent Curtius rearrangement with diphenylphosphoryl azide and tert-butyl alcohol to give the carbamate395. A key cross-metathesis with crotonaldehyde and the Grubbs second-generation catalyst produced the trans-enal396 in an excellent yield of 89% and an E/Z ratio of better than 20 : 1. Once the Bocprotecting group had been removed, cyclisation by aza-Michael reaction took place to give the spiro-piperidine397 in 80% yield and excellent diastereoselectivity (dr >20 : 1). However, the conversion of 397 into the enoate398 proceeded poorly under Baylis–Hillman conditions. A vinylalumination with ethyl propargylate and diisobutylaluminium hydride, although rather better, produced 398 as a 2 : 1 mixture of diastereomers; but under the subsequent acetylation–cyclisation conditions the desired isomer of tricyclic compound 399 could be isolated in 40% yield based on 397. Both 399 and its desilylated analogue 400 gave spectra consistent with those reported by Kibayashi's team in their formal synthesis of halichlorine;130 this work thus also completes a formal synthesis of the alkaloid.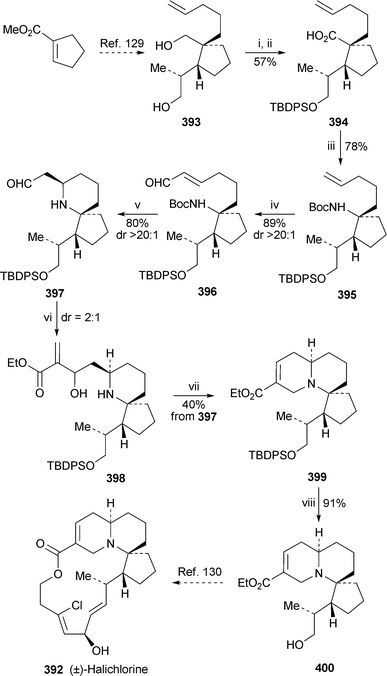 | ||
| Scheme 33 Reagents and conditions: i, TBDPSCl, NEt3, DMAP (cat.), CH2Cl2, rt, 14 h; ii, Jones reagent, Me2CO, 0 °C, 30 min, then rt, 5 h; iii, Ph2P(O)N3, NEt3, C6H6, reflux, 3 h, then ButOH, CH2Cl2, Me3SiCl, rt, 5 h; iv, MeCHCHCHO, Grubbs II catalyst, CH2Cl2, reflux, 3 h; v, TFA, CH2Cl2, rt, 3 h, then K2CO3; vi, DIBAL, NMO, THF, 0 °C, 30 min, then HCCCO2Et, 1 h, then aldehyde397, rt, 4 h; vii, Ac2O, NEt3, DMAP (cat.), CH2Cl2, 0 °C, then rt, 14 h; viii, NEt3·3HF, NEt3, MeCN, rt, 16 h. | ||
10 Acknowledgements
This material is based upon work supported by the National Research Foundation under grant number 2053652. Financial support from the University of the Witwatersrand is also gratefully acknowledged.11 References
- F. Cardona, G. Moreno, F. Guarna, P. Vogel, C. Schuetz, P. Merino and A. Goti, J. Org. Chem., 2005, 70, 6552 CrossRef CAS.
- V. D. Chaudhari, K. S. A. Kumar and D. D. Dhavale, Tetrahedron, 2006, 62, 4349 CrossRef CAS.
- V. D. Chaudhari, K. S. A. Kumar and D. D. Dhavale, Tetrahedron Lett., 2004, 45, 8363 CrossRef CAS.
- T. Ayad, Y. Génisson, M. Baltas and L. Gorrichon, Chem. Commun., 2003, 582 RSC.
- (a) J. P. Michael, Nat. Prod. Rep., 2004, 21, 626 Search PubMed; (b) J. P. Michael, Nat. Prod. Rep., 2007, 24, 192 Search PubMed; (c) J. P. Michael, Nat. Prod. Rep., 2005, 22, 606 Search PubMed; (d) J. P. Michael, Nat. Prod. Rep., 2005, 22, 612 Search PubMed; (e) J. P. Michael, Nat. Prod. Rep., 2004, 21, 631 Search PubMed; (f) J. P. Michael, Nat. Prod. Rep., 2007, 24, 197 Search PubMed; (g) J. P. Michael, Nat. Prod. Rep., 2003, 20, 465 Search PubMed; (h) J. P. Michael, Nat. Prod. Rep., 2005, 22, 608 Search PubMed; (i) J. P. Michael, Nat. Prod. Rep., 2001, 18, 525 Search PubMed; (j) J. P. Michael, Nat. Prod. Rep., 2004, 21, 647 Search PubMed; (k) J. P. Michael, Nat. Prod. Rep., 2007, 24, 202 Search PubMed; (l) J. P. Michael, Nat. Prod. Rep., 1995, 12, 545 Search PubMed; (m) J. P. Michael, Nat. Prod. Rep., 2005, 22, 613 Search PubMed; (n) J. P. Michael, Nat. Prod. Rep., 2000, 17, 593 Search PubMed; (o) J. P. Michael, Nat. Prod. Rep., 2005, 22, 619 Search PubMed; (p) J. P. Michael, Nat. Prod. Rep., 2007, 24, 214 Search PubMed; (q) J. P. Michael, Nat. Prod. Rep., 2005, 22, 621 Search PubMed.
- T. Ayad, Y. Génisson and M. Baltas, Org. Biomol. Chem., 2005, 3, 2626 RSC.
- R. C. Barbosa, F. Riet-Correa, R. M. T. Medeiros, E. F. Lima, S. S. Barros, E. J. Gimeno, R. J. Molyneux and D. R. Gardner, Toxicon, 2006, 47, 371 CrossRef CAS.
- I. M. Hueza, J. L. Guerra, M. Haraguchi, N. Asano and S. L. Górniak, Exper. Toxicol. Pathol., 2005, 57, 53 Search PubMed.
- H. Guo and G. A. O'Doherty, Org. Lett., 2006, 8, 1609 CrossRef CAS.
- A. J. Murray and P. J. Parsons, Synlett, 2006, 1443 CAS.
- M. Nath, R. Mukhopadhyay and A. Bhattacharjya, Org. Lett., 2006, 8, 317 CrossRef CAS.
- N. S. Kumar and B. M. Pinto, J. Org. Chem., 2006, 71, 1262 CrossRef CAS.
- N. S. Kumar and B. M. Pinto, Carbohydr. Res., 2006, 341, 1685 CrossRef CAS.
- N. S. Karanjule, S. D. Markad, V. S. Shinde and D. D. Dhavale, J. Org. Chem., 2006, 71, 4667 CrossRef CAS.
- D. D. Dhavale and S. M. Jachak, Molecules, 2005, 10, 893 Search PubMed.
- L. Song, E. N. Duesler and P. S. Mariano, J. Org. Chem., 2004, 69, 7284 CrossRef CAS.
- Z. Zhao, L. Song and P. Mariano, Tetrahedron, 2005, 61, 8888 CrossRef CAS.
- P. Díaz Pérez, M. I. García-Moreno, C. Ortiz Mellet and J. M. García Fernández, Eur. J. Org. Chem., 2005, 2903 CrossRef.
- V. M. Díaz Pérez, M. I. García-Moreno, C. Ortiz Mellet, J. Fuentes, J. C. Díaz Arribas, F. J. Cañada and J. M. García Fernández, J. Org. Chem., 2000, 65, 136 CrossRef CAS.
- K. Paśniczek, D. Socha, M. Jurczak, J. Solecka and M. Chmielewski, Can. J. Chem., 2006, 84, 534 CrossRef CAS.
- N. Langlois, B. K. L. Nguyen, P. Retailleau, C. Tarnus and E. Salomon, Tetrahedron: Asymmetry, 2006, 17, 53 CrossRef CAS.
- F. Pisaneschi, M. Piacenti, F. M. Cordero and A. Brandi, Tetrahedron: Asymmetry, 2006, 17, 292 CrossRef CAS.
- M. I. Torres-Sánchez, P. Borrachero, F. Cabrera-Escribano, M. Gómez-Guillén, M. Angulo-Álvarez, M. J. Diánez, M. D. Estrada, A. López-Castro and S. Pérez-Garrido, Tetrahedron: Asymmetry, 2005, 16, 3897 CrossRef CAS.
- T. Matsumura, M. Kasai, T. Hayashi, M. Arisawa, Y. Momose, I. Arai, S. Amagaya and Y. Komatsu, Pharmaceut. Biol., 2000, 38, 302 Search PubMed; Chem. Abstr., 2001, 135, 127011 Search PubMed.
- A. S. Davis, S. G. Pyne, B. W. Skelton and A. H. White, J. Org. Chem., 2004, 69, 3139 CrossRef CAS.
- A. A. Bell, L. Pickering, A. A. Watson, R. J. Nash, R. C. Griffiths, M. G. Jones and G. W. J. Fleet, Tetrahedron Lett., 1996, 37, 8561 CrossRef CAS.
- J. W. Daly, H. M. Garraffo and T. F. Spande, in Alkaloids: Chemical and Biological Perspectives, ed. S. W. Pelletier, Pergamon Press, Amsterdam, 1999, vol. 13, pp. 1–161 Search PubMed.
- J. W. Daly, T. F. Spande and H. M. Garraffo, J. Nat. Prod., 2005, 68, 1556 CrossRef CAS.
- P. Armstrong, S. Feutren, H. McAlonan, G. O'Mahony and P. J. Stevenson, Tetrahedron Lett., 2004, 45, 1627 CrossRef CAS.
- N. R. Andriamaharavo, M. Andriantsiferana, P. A. Stevenson, G. O'Mahony, H. J. C. Yeh, T. Kaneko, H. M. Garraffo, T. F. Spande and J. W. Daly, J. Nat. Prod., 2005, 68, 1743 CrossRef CAS.
- W. Takada, T. Sakata, S. Shimano, Y. Enami, N. Mori, R. Nishida and Y. Kuwahara, J. Chem. Ecol., 2005, 31, 2403 CrossRef CAS.
- D. Mebs, W. Pogoda, R. Maneyro and A. Kwet, Toxicon, 2005, 46, 641 CrossRef CAS.
- R. A. Saporito, M. A. Donnelly, H. M. Garraffo, T. F. Spande and J. W. Daly, J. Chem. Ecol., 2006, 32, 795 CrossRef CAS.
- V. C. Clark, C. J. Raxworthy, V. Rakotomalala, P. Sierwald and B. L. Fisher, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11617 CrossRef CAS.
- C. Macfoy, D. Danosus, R. Sandit, T. H. Jones, H. M. Garraffo, T. F. Spande and J. W. Daly, Z. Naturforsch., C, 2006, 60, 932.
- G. Guazzelli, R. Lazzaroni and R. Settambolo, Synthesis, 2005, 3119 CAS.
- R. Settambolo, G. Guazzelli and R. Lazzaroni, Tetrahedron: Asymmetry, 2003, 14, 1447 CrossRef CAS.
- R. Settambolo, G. Guazzelli and R. Lazzaroni, Lett. Org. Chem., 2005, 2, 176 Search PubMed.
- P. G. Reddy and S. Baskaran, J. Org. Chem., 2004, 69, 3093 CrossRef CAS.
- P. G. Reddy, M. G. Sankar and S. Baskaran, Tetrahedron Lett., 2005, 46, 4559 CrossRef CAS.
- R. I. J. Amos, B. S. Gourlay, P. P. Molesworth, J. A. Smith and O. R. Sprod, Tetrahedron, 2005, 61, 8226 CrossRef CAS.
- N. Toyooka and H. Nemoto, Heterocycles, 2005, 66, 549 CrossRef CAS.
- H. Takahata and M. Ichinose, Heterocycles, 2006, 67, 407 CrossRef CAS.
- M. A. Wijdeven, P. N. M. Botman, R. Wijtmans, H. E. Schoemaker, F. P. J. T. Rutjes and R. H. Blaauw, Org. Lett., 2005, 7, 4005 CrossRef CAS.
- P.-Q. Huang, Z.-Q. Guo and Y.-P. Ruan, Org. Lett., 2006, 8, 1435 CrossRef CAS.
- L. S. Santos and R. A. Pilli, Tetrahedron Lett., 2001, 42, 6999 CrossRef CAS.
- D. R. Artis, I.-S. Cho, S. Jaime-Figueroa and J. M. Muchowski, J. Org. Chem., 1994, 59, 2456 CrossRef CAS.
- D. J. Hart and Y.-M. Tsai, J. Org. Chem., 1982, 47, 4403 CrossRef CAS.
- A. B. Smith, III and D.-S. Kim, Org. Lett., 2004, 6, 1493 CrossRef.
- A. B. Smith, III and D.-S. Kim, J. Org. Chem., 2006, 71, 2547 CrossRef CAS.
- E. Conchon, Y. Gelas-Mialhe and R. Remuson, Tetrahedron: Asymmetry, 2006, 17, 1253 CrossRef CAS.
- N. Toyooka, Z. Dejun, H. Nemoto, H. M. Garraffo, T. F. Spande and J. W. Daly, Tetrahedron Lett., 2006, 47, 577 CrossRef CAS.
- N. Toyooka, Z. Dejun, H. Nemoto, H. M. Garraffo, T. F. Spande and J. W. Daly, Tetrahedron Lett., 2006, 47, 581 CrossRef CAS.
- S. Yu, W. Zhu and D. Ma, J. Org. Chem., 2005, 70, 7364 CrossRef CAS.
- G. Cai, W. Zhu and D. Ma, Tetrahedron, 2006, 62, 5697 CrossRef CAS.
- K. M. Maloney and R. L. Danheiser, Org. Lett., 2005, 7, 3115 CrossRef CAS.
- A. B. Smith, III and D.-S. Kim, Org. Lett., 2005, 7, 3247 CrossRef.
- N. Toyooka, M. Kawasaki, H. Nemoto, S. Awale, Y. Tezuka and S. Kadota, Synlett, 2005, 3109 CrossRef CAS.
- M. Movassaghi and A. E. Ondrus, Org. Lett., 2005, 7, 4423 CrossRef CAS.
- B. Sayah, N. Pelloux-Léon and Y. Vallée, J. Org. Chem., 2000, 65, 2824 CrossRef.
- A. E. Ondrus and M. Movassaghi, Tetrahedron, 2006, 62, 5287 CrossRef CAS.
- H. Takahata, H. Ouchi, M. Ichinose and H. Nemoto, Org. Lett., 2002, 4, 3459 CrossRef CAS.
- H. Takahata, Y. Saito and M. Ichinose, Org. Biomol. Chem., 2006, 4, 1587 RSC.
- A. Dinsmore, K. Mandy and J. P. Michael, Org. Biomol. Chem., 2006, 4, 1032 RSC.
- M. I. Choudhary, S. A. Nawaz, Zaheer-ul-Haq, M. K. Azim, M. N. Ghayur, M. A. Lodhi, S. Jalil, A. Khalid, A. Ahmed, B. M. Rode, Atta-ur-Rahman, A.-ul-H. Gilani and V. U. Ahmad, Biochem. Biophys. Res. Commun., 2005, 332, 1171 CrossRef CAS.
- D. L. Comins and H. Hong, J. Am. Chem. Soc., 1993, 115, 8851 CrossRef CAS.
- D. Stærk, K. B. Nezhad, J. Asili, S. A. Emami, A. Ahi, M. Sairafianpour and J. W. Jaroszewski, Biochem. Syst. Ecol., 2005, 33, 957 CrossRef CAS.
- A. G. Damu, P.-C. Kuo, L.-S. Shi, C.-Y. Li, C.-S. Kuoh, P.-L. Wu and T.-S. Wu, J. Nat. Prod., 2005, 68, 1071 CrossRef CAS.
- C.-W. Yang, W.-L. Chen, P.-L. Wu, H.-Y. Tseng and S.-J. Lee, Mol. Pharmacol., 2006, 69, 749 CAS.
- T.-H. Chuang, S.-J. Lee, C.-W. Yang and P.-L. Wu, Org. Biomol. Chem., 2006, 4, 860 RSC.
- M. G. Banwell, A. Bezos, C. Burns, I. Kruszelnicki, C. R. Parish, S. Su and M. O. Sydnes, Bioorg. Med. Chem. Lett., 2006, 16, 181 CrossRef CAS.
- M. G. Banwell and M. O. Sydnes, Aust. J. Chem., 2004, 57, 537 CrossRef CAS.
- F. A. Davis and M. Santhanaraman, J. Org. Chem., 2006, 71, 4222 CrossRef CAS.
- J. Barluenga, F. Aznar, C. Ribas and C. Valdés, J. Org. Chem., 1999, 64, 3736 CrossRef CAS.
- M. Katoh, H. Mizutani and T. Honda, Tetrahedron Lett., 2005, 46, 5161 CrossRef CAS.
- H. Matsuda, K. Yoshida, K. Miyagawa, Y. Nemoto, Y. Asao and M. Yoshikawa, Bioorg. Med. Chem. Lett., 2006, 16, 1567 CrossRef CAS.
- F. Zavala-Chávez, R. García-Mateos, M. Soto-Hernández and G. Kite, Z. Naturforsch., C, 2006, 61, 155 CAS.
- A. Martins, M. Wink, A. Tei, M. Brum-Bousquet, F. Tillequin and A.-P. Rauter, Phytochem. Anal., 2005, 16, 264 CrossRef CAS.
- M. B. Selenu, F. Carrus and L. Bonsignore, Boll. Chim. Farm., 2005, 144, 66 Search PubMed; Chem. Abstr., 2006, 145, 331626 Search PubMed.
- P.-L. Ding, Z.-X. Liao, H. Huang, P. Zhou and D.-F. Chen, Bioorg. Med. Chem. Lett., 2006, 16, 1231 CrossRef CAS.
- I. S. Movsumov, E. A. Garaev and M. I. Isaev, Chem. Nat. Compd., 2006, 42, 116 CrossRef CAS.
- P.-L. Ding, Y.-Q. Yu and D.-F. Chen, Phytochem. Anal., 2005, 16, 257 CrossRef CAS.
- T.-T. Jong, M.-R. Lee, Y.-C. Chiang and S.-T. Chiang, J. Pharm. Biomed. Anal., 2006, 40, 472 CrossRef CAS.
- H. Reinhard, H. Rupp, F. Sager, M. Streule and O. Zoller, J. Chromatogr., A, 2006, 1112, 353 CrossRef CAS.
- V. D. C. Shields, E. J. Rodgers, N. S. Arnold and D. Williams, Naturwissenschaften, 2006, 93, 127 CrossRef CAS.
- G. J. Sharam and R. Turkington, Can. J. Bot., 2005, 83, 1345 CrossRef CAS.
- S.-N. Choi, S.-Y. Kim, H.-W. Ryu and Y.-M. Lee, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2005, 61, m504 CrossRef.
- A. Johansson, E. Wingstrand and M. Håkansson, J. Organomet. Chem., 2005, 690, 3846 CrossRef CAS.
- Y. Reyes-Ortega, J. L. Alcántara-Flores, M. del C. Hernández-Galindo, R. Gutiérrez-Pérez, D. Ramírez-Rosales, S. Bernès, B. M. Cabrera-Vivas, A. Durán-Hernández and R. Zamorano-Ulloa, J. Mol. Struct., 2006, 788, 145 CrossRef CAS.
- B. Jasiewicz, W. Boczoń, B. Warżajtis, U. Rychlewska and T. Rafałowicz, J. Mol. Struct., 2005, 753, 45 CrossRef CAS.
- S. Jana and D. C. Sherrington, Angew. Chem., Int. Ed., 2005, 44, 4804 CrossRef CAS.
- B. Jasiewicz and W. Boczoń, J. Mol. Struct., 2005, 752, 115 CrossRef CAS.
- K. Yoshizawa, Y. In, T. Ishida and T. Shioiri, Heterocycles, 2005, 66, 667 CrossRef CAS.
- C. Strohmann, S. Dilsky and K. Strohfeldt, Organometallics, 2006, 25, 41 CrossRef CAS.
- E. G. Ijpeij and G.-J. M. Gruter, Synthesis, 2006, 1408 CrossRef CAS.
- R. Otte, R. Fröhlich, B. Wibbeling and D. Hoppe, Angew. Chem., Int. Ed., 2005, 44, 5492 CrossRef CAS.
- C. Strohmann, C. Däschlein and D. Auer, J. Am. Chem. Soc., 2006, 128, 704 CrossRef.
- T. Borowiak, I. Wolska, W. Wysocka and T. Brukwicki, J. Mol. Struct., 2005, 753, 27 CrossRef CAS.
- T. Brukwicki and W. Wysocka, J. Mol. Struct., 2006, 785, 225 CrossRef CAS.
- V. Galasso, F. Asaro, F. Berti, I. Habuš, B. Kovač and C. De Risi, Chemical Physics, 2004, 301, 33 Search PubMed.
- V. Galasso, F. Asaro, F. Berti, A. K. Przybył, J. Włodarczak, W. Wysocka, I. Habuš and B. Kovač, Chem. Phys., 2005, 314, 25 CrossRef CAS.
- V. Galasso, A. K. Przybył, V. Christov, B. Kovač, F. Asaro and E. Zangrando, Chem. Phys., 2006, 325, 365 CrossRef CAS.
- Atta-ur-Rahman, A Pervin, M. I. Choudhary, N. Hasan and B. Sener, J. Nat. Prod., 1991, 54, 929.
- J. E. Rode, E. D. Raczyńska, E. Górnicka and J. C. Dobrowolski, J. Mol. Struct., 2005, 749, 51 CrossRef CAS.
- A. D. McElhinney and S. P. Marsden, Synlett, 2005, 2528 CAS.
- G. Bélanger, R. Larouche-Gauthier, F. Ménard, M. Nantel and F. Barabé, J. Org. Chem., 2006, 71, 704 CrossRef CAS.
- C. Botuha, C. M. S. Galley and T. Gallagher, Org. Biomol. Chem., 2004, 2, 1825 RSC.
- D. Gray and T. Gallagher, Angew. Chem., Int. Ed., 2006, 45, 2419 CrossRef CAS.
- D. Stead, P. O'Brien and A. J. Sanderson, Org. Lett., 2005, 7, 4459 CrossRef CAS.
- P. R. Blakemore, C. Kilner, N. R. Norcross and P. C. Astles, Org. Lett., 2005, 7, 4721 CrossRef CAS.
- B. Danieli, G. Lesma, D. Passarella, A. Sacchetti and A. Silvani, Tetrahedron Lett., 2005, 46, 7121 CrossRef CAS.
- B. Danieli, G. Lesma, D. Passarella, A. Sacchetti, A. Silvani and A. Virdis, Org. Lett., 2004, 6, 493 CrossRef CAS.
- R. T. Tlegenov and A. Aitmambetov, Russ. J. Bioorg. Chem., 2005, 31, 495 Search PubMed.
- A. M. Gazaliev, O. A. Nurkenov, I. V. Kulakov, A. A. Ainabaev and D. V. Bessonov, Russ. J. Appl. Chem., 2006, 79, 508 CrossRef CAS.
- T. S. Zhivotova, A. M. Gazaliev, S. D. Fazylov, A. B. Karimova, D. M. Turdybekov and K. M. Turdybekov, Russ. J. Org. Chem., 2005, 41, 898 CrossRef CAS.
- O. V. Bakbardina, N. Z. Rakhimzhanova, M. A. Gazalieva, S. D. Fazylov and E. Z. Baimagambetov, Russ. J. Appl. Chem., 2006, 79, 504 CrossRef CAS.
- O. A. Nurkenov, G. G. Baikenova, D. M. Turdybekov, M. K. Ibraev, A. M. Gazaliev and K. M. Turdybekov, Russ. J. Gen. Chem., 2006, 76, 129 CrossRef CAS.
- I. P. Dubovik, M. M. Garazd, V. I. Vinogradova and V. P. Khilya, Chem. Nat. Compd., 2006, 42, 133 CrossRef CAS.
- M. V. Veselovskaya, M. M. Garazd, V. I. Vinogradova and V. P. Khilya, Chem. Nat. Compd., 2006, 42, 277 CrossRef CAS.
- S. K. Chellappan, Y. Xiao, W. Tuekmantel, K. J. Kellar and A. P. Kozikowski, J. Med. Chem., 2006, 49, 2673 CrossRef CAS.
- J. A. Abin-Carriquiry, M. H. Voutilainen, J. Barik, B. K. Cassels, P. Iturriaga-Vásquez, I. Bermudez, C. Durand, F. Dajas and S. Wonnacott, Eur. J. Pharmacol., 2006, 536, 1 CrossRef CAS.
- D. M. Hodgson, C. D. Bray and P. G. Humphreys, Synlett, 2006, 1 CrossRef CAS.
- B. Goldfuss, Synthesis, 2005, 2271 CrossRef CAS.
- M. S. Sigman and D. R. Jensen, Acc. Chem. Res., 2006, 39, 221 CrossRef CAS.
- J. A. Mueller, A. Cowell, B. D. Chandler and M. S. Sigman, J. Am. Chem. Soc., 2005, 127, 14817 CrossRef CAS.
- J. M. Keith, R. J. Nielsen, J. Oxgaard and W. A. Goddard, III, J. Am. Chem. Soc., 2005, 127, 13172 CrossRef CAS.
- D. L. J. Clive, M. Yu, J. Wang, V. S. C. Yeh and S. Kang, Chem. Rev., 2005, 105, 4483 CrossRef CAS.
- R. B. Andrade and S. F. Martin, Org. Lett., 2005, 7, 5733 CrossRef CAS.
- G. A. Wallace and C. H. Heathcock, J. Org. Chem., 2001, 66, 450 CrossRef CAS.
- Y. Matsumura, S. Aoyagi and C. Kibayashi, Org. Lett., 2004, 6, 965 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2008 |