The role of ligand-containing loops at copper sites in proteins
Christopher
Dennison
*
Institute for Cell and Molecular Biosciences, Medical School, Newcastle University, Newcastle upon Tyne, UK NE2 4HH. E-mail: christopher.dennison@ncl.ac.uk; Fax: +44 (0)191 222 7424; Tel: +44 (0)191 222 7127
First published on 8th October 2007
Abstract
Covering: primarily 2003–2007
Many approaches are being used to engineer metalloproteins, with most of these informed by, and aiming to further elucidate, the basic structural requirements for biological metal centers. Cupredoxins are type 1 (T1) copper-containing electron transfer (ET) proteins with a β-barrel fold that is thought to constrain metal site structure. The T1 copper ion is bound by ligands mainly originating from a single active site loop whose length and structure varies. This Highlight article will focus on protein engineering studies which have investigated the role of the metal-binding loop for active site integrity and functionality. Scaffold differences are present within the cupredoxin family and their influence has also been assessed. Given the widespread occurrence of β-barrel domains in nature, and the array of metal sites in proteins composed of loop regions, the studies described on this model system have implications for a variety of metalloproteins.
 Christopher Dennison Christopher Dennison | Chris Dennison received his PhD in Inorganic Chemistry in 1994 from the University of Newcastle upon Tyne with Prof. A. G. Sykes FRS and Prof. W. McFarlane. He was a postdoctoral fellow with Prof. G. W. Canters at Leiden University until 1996 and then moved to University College Dublin as a lecturer. In 1999 he returned to the Department of Chemistry at the University of Newcastle upon Tyne, and in 2004 moved to the Institute for Cell and Molecular Biosciences (Medical School, Newcastle University) |
1 Type 1 copper sites and cupredoxins
The cupredoxins (see Fig. 1) are a family of proteins possessing a mononuclear type 1 (T1) copper site that function as electron shuttles in a range of organisms.1–3 Cupredoxin domains are found in a number of copper-containing enzymes including nitrite reductases (NIRs),4,5 multi-copper oxidases6–10 and cytochrome c oxidase.11,12 A T1 copper site has three strong equatorial ligands provided by the thiolate sulfur of a Cys and the imidazole nitrogens of two His residues (see Fig. 1B). There is usually a distant axial interaction provided by a Met approximately 2.6 to 3.3 Å from the copper ion (see Fig. 1B) resulting in a distorted tetrahedral geometry. Azurin (AZ) is slightly unusual in that a second weak axial interaction is provided by the backbone carbonyl oxygen of Gly45 giving a distorted trigonal bipyramidal copper site (see Fig. 1B).13–15 The axial Met is not conserved at T1 copper sites and in some proteins a short bond to the Oε1 of a Gln is found in this position,16–18 whilst in other cases a non-coordinating side-chain resides here giving a three-coordinate site.2,7–10,19 The T1 copper site structure, and the nature of the coordinating atoms, are not ideal for either the cuprous or cupric ion, and the cupredoxin scaffold seems to provide a sub-optimal active site arrangement for both oxidation states of this metal which changes little upon redox inter-conversion.15,20–22 The reorganization energy (λ) is therefore small, with the inner sphere contribution (λi) thought to be as little as ∼0.1–0.3 eV, facilitating rapid electron transfer (ET).23–25 The importance of the rigid cupredoxin scaffold for function has been the subject of considerable debate.21,22,24–31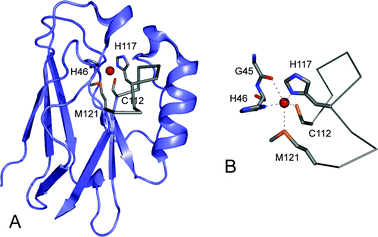 | ||
| Fig. 1 The structure of the cupredoxin azurin (AZ, 4AZU14) from Pseudomonas aeruginosa prepared using Pymol. (A) The overall β-barrel fold of the protein is shown in slate with the C-terminal ligand-containing loop as a Cα trace in grey. The side-chains of the Cys112, His117 and Met121 ligands on this loop, and also of the coordinating His46 are shown as stick models in grey with the copper ion in red. (B) The copper site of AZ demonstrating the distorted trigonal bipyramidal geometry with a second weak axial interaction provided by the backbone carbonyl oxygen of Gly45. | ||
Understanding the relationship between the distinctive T1 copper site spectroscopy, including the intense blue color (proteins possessing such sites are therefore referred to as blue copper proteins ) and unique electron paramagnetic resonance (EPR ) spectra, and structure has attracted considerable attention over the past four decades (some representative studies can be found in references 32–48). This level of interest is partly due to the fact that other classes of biological copper sites have more mundane spectroscopic features which are usually like those exhibited by Cu(II) complexes.44,48 An exception to this is the dinuclear CuA centre, also found in a cupredoxin domain that is the major component of subunit II of cytochrome c oxidase,11,12,49 and is also present in nitrous oxide reductase.50,51 The Cu2µ2-S(Cys)2 core of CuA with terminal His ligands at each metal ion gives rise to a mixed valence, fully delocalized oxidized form.52–54 Some successful attempts at synthesizing Cu(II) complexes with structural and spectroscopic properties similar to those of T1 copper and CuA sites have been reported,55,56 although this approach has not proved as successful as biomimetic studies with other metalloproteins.
The band at ∼600 nm (ε ≈ 2000–6000 M−1 cm−1) in the UV–vis spectrum which gives rise to the intense blue color of a T1 copper site has been assigned to a S(Cys)π → Cu(II) dx2−y2 ligand-to-metal charge transfer (LMCT) transition.47,48 The unusually small hyperfine coupling in the gz region of the EPR spectra of T1 copper sites has been attributed to the highly covalent nature of the Cu–S(Cys) bond.47,48 The Cys ligand is therefore a critical feature of T1 copper sites which has been confirmed by site-directed mutagenesis studies of the ligating residues.57–61 Another characteristic and extremely important feature of T1 copper sites is the reduction potential (Em value) which dictates the driving force for their ET reactions. All T1 copper sites have Em values higher than that of the Cu(II)/Cu(I) aqua couple (150 mV), and range from ∼200 to 1000 mV.21,48 In cupredoxins the variation is less with most members having Em values between 200 and 400 mV.3
Cupredoxins are made up of a β-barrel scaffold which is composed of two β-sheets consisting usually of eight β-strands.13–18,62–69 The surface loop, which is the focus of this review, contains the Cys, one of the His and usually the Met ligands (as mentioned above the Met can be replaced by a Gln or a non-coordinating residue), and links the two C-terminal β-strands (see Fig. 1). The second His ligand is located on a loop between β3 and β4 which is more in the core of the protein . Many of the residues in the main metal-binding loop are exposed on the surface of cupredoxins and contribute to a key area for associations with partners,70–72 which surrounds the C-terminal His ligand, and is usually hydrophobic. Furthermore, residues in this loop contribute to an array of interactions, including hydrogen-bonding contacts, which are thought to be critical for active site properties. The length, sequence and structure of this loop varies in cupredoxins (see Fig. 2) and could therefore provide a way to modify the copper site environment and tune important properties, including the Em value and the key recognition site for interactions, without compromising metal binding. The importance of this region has therefore been investigated by loop-directed mutagenesis.
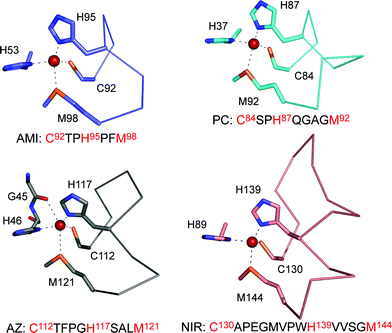 | ||
| Fig. 2 The T1 copper site structures of Paracoccus denitrificans amicyanin (AMI, 1AAC64), spinach plastocyanin (PC, 1AG668), P. aeruginosa azurin (AZ, 4AZU,14 chain A) and Alcaligenes xylosoxidansnitrite reductase (NIR, 1OE14) drawn with Pymol, highlighting differences in their ligand-containing loops which are shown as Cα traces. | ||
2 Loop-directed mutagenesis: the CuA experiment
Initial loop-directed mutagenesis experiments involving a cupredoxin domain reported the introduction of both T1 and CuA centers into subunit II of a quinol oxidase, which naturally lacks a copper site.73 The same CuA loop (CAEICGPGHSGM) was inserted in place of the T1 copper sequence of AMI resulting in an authentic dinuclear CuA centre.74 Similar experiments were subsequently performed with AZ75 and the crystal structure of this variant demonstrates the structural homology of the engineered CuA centers to those found in the native enzymes.76 The CuA centre promotes a single ET reaction like the mononuclear T1 copper site. It has been suggested that a CuA site is required in cytochrome c oxidase and nitrous oxide reductase, rather than a T1 copper site, to either maintain a low λi (lower than for a T1 copper site), to provide a site with the correct Em which is less susceptible to large changes caused by amino acid alterations in its vicinity, or to facilitate electronic coupling.77–79 Whatever the reason for having a dinuclear CuA over a mononuclear T1 copper centre (a similar situation exists for Fe–S ET proteins where even higher nuclearity is found), the C-terminal ligand-containing loop dictates the type of site formed.3 Loop-directed mutagenesis: the structural influence of interchanging native T1 copper site sequences
Loop-directed mutagenesis has been used to change the C-terminal ligand-containing loops of various cupredoxins with other known T1 copper binding sequences.80–89 The initial work involved the introduction of a series of longer loops into AMI, which has the shortest naturally occurring T1 copper-binding sequence (see Fig. 2).80–82 All of the loop variants have authentic T1 copper sites (as judged spectroscopically) with Em values higher, and ET capabilities lower, than those of AMI. Unfortunately, structures are not available for any of these AMI chimeras and thus a detailed analysis of the atomic features underlying the observed effects is not possible. With this in mind, and in order to systematically assess the importance of metal binding loops, detailed studies have been undertaken in this area using the cupredoxin AZ due to its stability and ease of crystallization.The AZ studies initially concentrated on shortening the ten residue (including the Cys and Met ligands) active site loop (see Fig. 2),83–87 and two chimeras were prepared by introducing the PC (9 residue)87 and AMI (7 amino acid)84,86 sequences as shown in Fig. 3. The loops of AZ, PC and AMI, as well as being different in length, have dissimilar structures. Both of the resulting loop-contraction variants: AZPC (AZ with the PC loop) and AZAMI (AZ with the AMI loop) have redox-active T1 copper sites and crystal structures have been determined. The Cα traces of these two chimeras are compared with that of AZ in Fig. 4 and 5. It is apparent from these superimpositions that shortening the C-terminal ligand-containing loop of AZ has little effect on the overall structure of the protein .
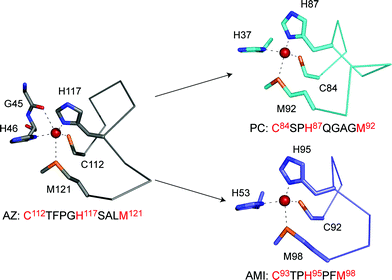 | ||
| Fig. 3 The active site structures of AZ (4AZU,14 chain A), PC (1AG668) and AMI (1AAC64) drawn with Pymol. The arrows indicate two loop contraction experiments that have been performed in AZ resulting in AZPC (top, giving the active site loop C112SPH115QGAGM120) and AZAMI (bottom, giving the loop sequence C112TPH115PFM118). The conformations that these loops adopt when grafted onto the AZ scaffold are shown in Fig. 6 and 7. | ||
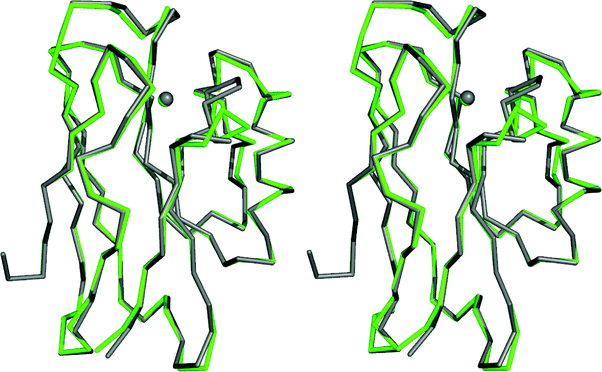 | ||
| Fig. 4 Overlay (prepared with Pymol) of the Cα traces, and the copper ions, of AZ (4AZU,14 chain A, grey) and AZPC (2HX7,87 chain A, green). | ||
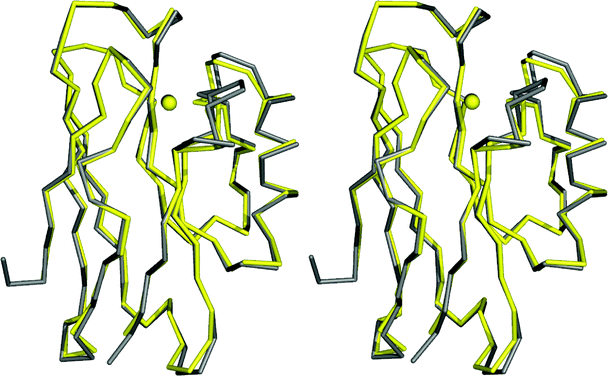 | ||
| Fig. 5 Overlay (prepared with Pymol) of the Cα traces, and the copper ions, of AZ (4AZU,14 chain A, grey) and AZAMI (2FTA,86 chain A, yellow). | ||
The Cu(II) site structures of AZPC and AZAMI are compared in Fig. 6 and 7 respectively to that of AZ, and also of the protein whose loop has been introduced.86,87 The coordination geometry of the cupric ion is very similar to AZ in both of these chimeras (a change in the conformation of the axial Met ligand occurs in one of the two molecules in the asymmetric unit of AZPC,87 which matches the arrangement found in PC68). Significantly, the active site loops of AMI and PC adopt structures almost identical to those found in the proteins from which the loop originates, when grafted onto the AZ scaffold (see Fig. 6 and 7).64,68,86,87 In the case of AZAMI the homology of the loop backbone to that in AMI is maybe not so surprising considering the highly constrained nature of this region resulting from the presence of Pro residues either side of the central His ligand. However, the side-chains of the Phe and Thr residues on the loop adopt conformations in AZAMI as found in AMI, and the hydrogen bonding contacts made by the hydroxyl group of the latter are replicated. In AZPC, the structural similarity of the introduced loop to that in PC is more remarkable, especially considering the inherently flexible GlnGlyAlaGly sequence linking the His and Met ligands. The Gln side chain is found in a different conformation in AZPC than in PC which is associated with alterations between the two scaffold proteins .62,68,87 In both AZAMI and AZPC a key active site hydrogen bond to the thiolate sulfur of the Cys ligand, provided by the backbone amide of Phe114 in AZ, is removed,86,87 as a Pro residue is introduced in this position. Analysis of the Phe114Pro AZ variant highlights this change as the main cause of altered spectroscopic properties in the AZ loop variants.90 In AZPC there is very little change at the copper site upon reduction at neutral pH (the structure of Cu(I) AZAMI has not been determined).87
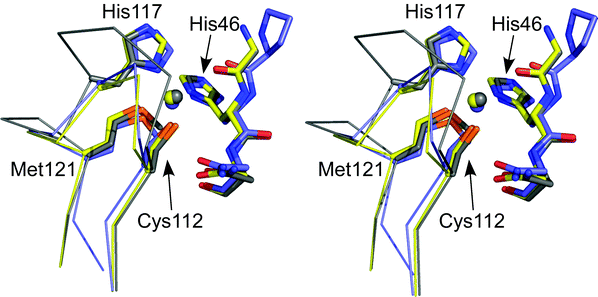 | ||
| Fig. 6 Stereoview (prepared with Pymol) showing an overlay of the active sites of Cu(II) AZAMI (2FTA,86 chain A, yellow), AZ (4AZU,14 chain A, gray) and AMI (1AAC,64 slate). The side chains of the coordinating residues (labeled as in AZ) and the amino acid on either side of the N-terminal His ligand are shown as stick models, copper atoms as spheres, and the backbone of the C-terminal ligand-containing loops as Cα traces. Reprinted with permission from reference 87; copyright (2007) American Chemical Society. | ||
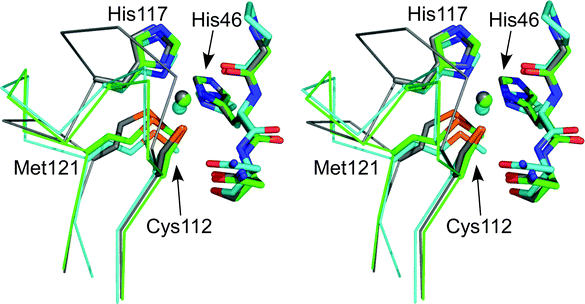 | ||
| Fig. 7 Stereoview (prepared with Pymol) showing an overlay of the active sites of Cu(II) AZPC (2HX7,87 chain A, green), AZ (4AZU,14 chain A, gray) and PC (1AG6,68 cyan). The side chains of the coordinating residues (labeled as in AZ) and the amino acid on either side of the N-terminal His ligand are shown as stick models, copper atoms as spheres, and the backbone of the C-terminal ligand-containing loops as Cα traces. Reprinted with permission from reference 87; copyright (2007) American Chemical Society. | ||
The T1 copper binding loop of AMI has also been introduced into the PC and PAZ scaffolds as well as that of AZ.83,84 The influence on the spectroscopic properties is limited in all cases, but is largest in AZAMI and PAZAMI. As discussed above, in AZAMI this is mainly due to the removal of one of the hydrogen bonds to the Cys ligand90 which is not a factor for PAZAMI (the PAZ loop sequence is C78TPH81YGMGM86i.e. PAZ has a Pro two residues after the Cys ligand and therefore does not possess this hydrogen bond). More significant changes are found at the active site of PAZAMI with the major effects being a shorter axial Cu–S(Met) bond and a more tetragonal copper site geometry as compared to PAZ. These differences give rise to the spectroscopic alterations, and scaffold-loop interactions seem to be a major contributing factor.88 Additionally, the T1 copper-binding loop of NIR, which is the longest known (15 residues), has been replaced with that of AMI (see Fig. 2). This represents a much more challenging loop-contraction, not only due to the dramatic decrease in loop length, but also because NIR is a trimeric protein with the T1 copper-binding loop at the monomer–monomer interface.4,5 Furthermore, NIR also possesses a catalytic (type 2) copper site which is situated at the inter-subunit interface and is coordinated by two His residues from one monomer and a third from the adjacent monomer. Nevertheless, the NIRAMI loop contraction variant has an authentic T1 Cu(II) site which is similar to that of WT NIR.89 The NIRAMI mutant has a number of interesting properties which will be discussed in a subsequent article along with the structure of this loop-contraction variant.
4 Basic requirements of a T1 copper-binding loop
As described above the effect of contracting the ligand-containing loop of AZ (and those of other cupredoxins and cupredoxin domains) to that of AMI, which is the shortest naturally occurring sequence found for a T1 copper site, has been investigated.83–89 The active site structure and spectroscopic properties of the AZAMI variant (with the ligand-containing loop sequence CTPHPFM) are very similar to those of the WT protein .84,86 The next question addressed was whether the copper-binding loop of a cupredoxin could be shortened further. Variants of AZ with loop sequences CPHPFM (AZAMI-T) and CTPHPM (AZAMI-F) were therefore constructed.86 The AZAMI-T variant was isolated but did not fold correctly, could not bind copper and had a tendency to form disulfide-linked multimers in solution. On the other hand, AZAMI-F bound copper giving a site with T1 properties. The Cu(II) site structure of AZAMI-F is very similar to that of AZ as shown in Fig. 8 (the Cu(II)–O(Gly45) distance is slightly elongated) and its spectroscopic properties are almost identical to those of AZAMI (the second hydrogen bond to the Cys ligand is also absent86,90). At slightly alkaline pH the active site structure of AZAMI-F changes very little upon reduction. The Em of AZAMI-F is similar to that of AZ demonstrating that the native loop is not required to give the particular value that AZ requires for its physiological function. However, the surface hydrophobic patch has been significantly altered in AZAMI-F which influences intermolecular ET reactivity (vide infra). These studies demonstrate that the T1 copper-binding loop can be shortened further than that which naturally occurs whilst still allowing the correct type of biological metal site to form. The instability of AZAMI-T, as compared to AZAMI-F, is probably related to the hydrogen bonding pattern around the active site, as the Thr residue on the loop makes a number of key interactions. Removal of both the Thr and Phe residues on the loop has also been attempted by introducing the sequence CPHPM but this variant does not fold correctly and cannot bind copper.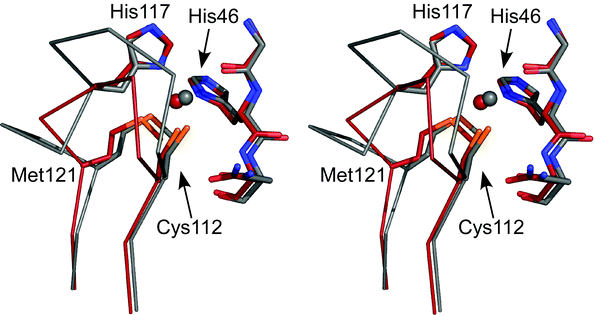 | ||
| Fig. 8 Stereoview (prepared with Pymol) showing an overlay of the active sites of Cu(II) AZAMI-F (2FT6,86 brick red) and AZ (4AZU,14 chain A, gray). The side chains of the coordinating residues (labeled as in AZ) and the amino acids on either side of the N-terminal His ligand are shown as stick models, copper atoms as spheres, and the backbone of the C-terminal ligand-containing loops as Cα traces. | ||
5 The influence of loop mutations on Em values
The introduction of the AMI and PC loops into AZ has an intriguing influence on the Em value (see Fig. 9).84,87 In AZAMI Em is lowered by ∼35 mV to match that found in AMI whilst in AZPC the value is raised by ∼70 mV giving an Em which is almost identical to that of PC. The Em values of PAZAMI and PCAMI are also lower than those of the WT proteins by ∼50 and 60 mV respectively, but do not match that of AMI,83,84 whilst those of AZAMI-F and AZ are almost the same.86 The Em effects seen are not due, in most cases, to large active site changes resulting from these loop mutations as the structures described above demonstrate. The exceptions to this are PAZAMI, where the more tetragonal active site structure as compared to PAZ results in the observed decrease in Em,88 and AZAMI-F where an increased Cu to O(Gly45) distance (see Fig. 8) may have an influence on Em.86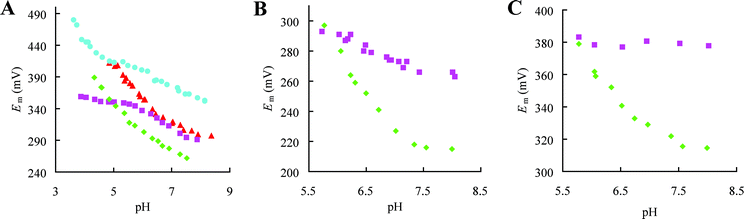 | ||
| Fig. 9 Dependence on pH of the reduction potential (Em) of various loop contraction variants. (A) AZ (■), AZAMI (✦), AZAMI-F (▲) and AZPC (●). (B) PAZ (■) and PAZAMI (✦). (C) PC (■) and PCAMI (✦). As the pH is lowered some of the proteins exhibit a marked increase in Em with a slope of about −60 mV per pH unit. This is due to protonation of the C-terminal His ligand in the Cu(I) protein , and the data can be fit (not shown) giving pKa values of 6.7, 6.7, 5.9, 5.5 and 4.3 for Cu(I) PAZAMI, PCAMI, AZAMI-F, AZAMI and AZPC respectively.83,84,86,87 The assessment of the influence of a particular loop mutation on Em is made from comparisons at pH 7.5–8.0 where the values are generally less dependent on pH. | ||
The thermodynamics of reduction of T1 copper sites has been found to be dominated by solvent reorganization effects.91 The solvent structure near the active site of AZPC, AZAMI and AZAMI-F are all alike and are different to those seen for AZ and PC14,68,86,87 and is reminiscent of that found for AMI64 (the arrangement in PAZAMI is similar to this and is different from that in PAZ67,88). These changes can be mainly attributed to the replacement of the bulky phenyl group at position 114 with a Pro.90 Solvent accessibility does not seem to be a major determinant of the Em trends as the active sites of AZAMI and AZPC, which have Em values over 100 mV apart, are both significantly more solvent exposed, and to a similar extent, than in AZ.14,86,87 The increases in Em in all of the loop elongation variants of AMI, which indicates stabilisation of the Cu(I) over the Cu(II) protein , have been attributed to mainly entropic effects thought to originate from reduction-induced increases in flexibility of the introduced loop, due to poor packing of the non-native loops against the AMI scaffold.81,92 The structures of the AZ loop-contraction variants show that in these chimeras the introduced loops pack well against this cupredoxin scaffold.86,87 In PAZAMI the interactions between the scaffold and the shortened loop seems to be sub-optimal which may contribute to the influence of this mutation on Em.88
The hydrogen bonding pattern and the influence of protein -derived dipoles in the vicinity of the copper ion are further features which can influence the Em value of a T1 copper site.93–96 The second hydrogen bond to the Cys ligand present at certain T1 copper centres including that in AZ, but missing in PC, AMI and PAZ, has been suggested as having a major influence on Em.96 The elimination of this interaction in the Phe114Pro AZ variant results in an 85 mV decrease in Em90 whilst its introduction in various PAZ and AMI mutants gives rise to 115–180 mV increases.97,98 The removal of this hydrogen bond in the AZ loop-contraction variants has a range of effects and it therefore appears unlikely that this single interaction is the sole determinant of Em. Theoretical studies have shown that numerous permanent protein dipoles around a T1 copper site have a major influence on Em and tune down the value for PC compared to that found for the solvated active site.95 Similar calculations have not been performed for AZ but it appears that the most likely cause of the alterations in the Em values in the loop variants, particularly those of AZAMI and AZPC, is the homology of the structures of the introduced loops to those found in AMI and PC respectively, with contributions from dipoles in this region being a major factor.87
6 The influence of loop mutations on ET reactivity
The intrinsic ET reactivity of all of the loop-contraction variants has been analysed using the electron self exchange (ESE) reaction, which involves the two oxidation states of a redox couple swapping an electron.3,99,100 It is known that AZ molecules interact via their hydrophobic patches when self exchanging, with ET involving the solvent exposed His ligand in the centre of this region.101–103 Surprisingly, the intrinsic ET reactivity of AZAMI is very similar to that of AZ.84 The introduction of the AMI loop into PC and PAZ has a more significant effect (2- and 8-fold decreases respectively) on the ESE rate constant (kese).83,84 The AZPC variant has a kese value 30-fold smaller than that of AZ,87 whereas in AZAMI-F the decrease is 150-fold.86 The very small active site changes upon redox inter-conversion for AZPC and AZAMI-F86,87 indicate that these variations are probably not due to different λi values (which are most likely comparable to that for AZ in all cases). The solvent reorganization energy (λo) could have been affected by the mutations but the magnitude of any alterations is not easily determined, and there does not seem to be a simple reliance on loop length. The structural data indicate that the most likely cause of the altered kese values are changes in the hydrophobic interaction surface. In the crystal structure of AZ adjacent molecules pack via these areas with the interface composed of 79% non-polar atoms,14,103 with a significant contribution from hydrophobic residues in the loop. The AZ loop variants possess fewer such side chains and generally have less non-polar atoms at this interface,86,87 with the removal of Phe114 being a significant factor, which disfavours homo-dimer formation and decreases ESE reactivity.86,90 In AZAMI the introduced Phe117 compensates for the loss of hydrophobic residues.84,86 In PAZAMI, whose active site structure also changes little upon reduction, the hydrophobicity around the exposed His ligand is increased and in this case the nature of the side chains present in this region may be more important88 (there is currently no structural information available for PCAMI and so the minor decrease in kese in this variant cannot be rationalised). The ligand-binding loop makes a significant contribution to the hydrophobic patch of cupredoxins which allows the properties of this area to be modified to facilitate complex formation conducive to fast ET with a particular partner.7 The influence of loop mutations on His ligand protonation and dissociation
The dissociation and protonation of the His ligand in the centre of the C-terminal loop is a well-studied characteristic of various reduced cupredoxins80,104–112 and may act as a switch to regulate ET reactivity.110,113,114 AZ does not exhibit this behavior in the accessible pH range, as demonstrated by the absence of a significant influence of pH on Em (see Fig. 9A), and a very low pKa has been estimated.115 His115 protonates in Cu(I) AZAMI-F86 (see Fig. 9A) and a similar effect is observed in Cu(I) AZAMI84 and AZPC87 with pKa values of 5.5 and 4.3 respectively. In AZAMI-F and AZPC this has been verified by crystallographic studies of the Cu(I) protein at lower pH values (see Fig. 10 for the AZAMI-F structures demonstrating His ligand protonation).86,87 The pKa of the His ligand on the loop in AZPC is very similar to the value of 4.9 for PC.100,104,105,112 Values almost identical to the pKa of ∼6.7 for the His96 ligand of AMI80,92,107,112 are obtained in PAZAMI (in PAZ the His ligand has a pKa of 4.9108) and PCAMI (in AZAMI the pKa is slightly lower).83,84The introduction of the AMI and PC loops provides a pKa value for the His ligand either identical or very similar to that of the protein from which the loop originates. Loop elongation in AMI always results in a decrease in the pKa for the His ligand, except when the AZ loop is introduced, when there is little effect, which is also the case in the PCAZ variant.87,92 A feature of the loop, which influences the pKa of the coordinating His, seems not to be reproduced in variants in which the AZ loop is introduced. Detailed analysis of the thermodynamics of dissociation and protonation of the AMI loop elongation variants has highlighted that the decreases in pKa are related to an unidentified entropic feature of the loop92 (thermodynamic studies of the AZ variants have been performed and highlight varying enthalpic and entropic contributions to the observed effects116). From all of these studies it appears that the pKa of the His ligand is, in most cases, related to loop length, with a shorter sequence facilitating dissociation and protonation.108 It should not be overlooked that all of the loop mutations may have influenced rotational barriers involved in the dissociation and protonation of the C-terminal His ligand which could have a significant effect on the pKa value.117 The structural features which control the pKa of the C-terminal His ligand at a T1 copper site remain to be identified.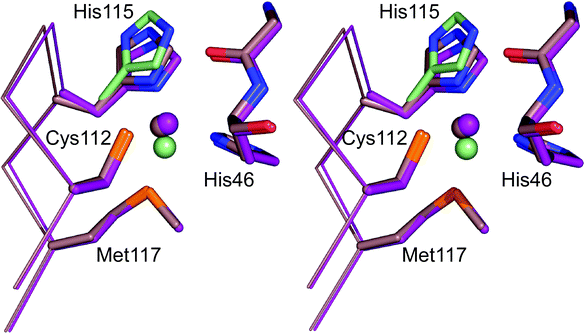 | ||
| Fig. 10 Overlay (prepared in Pymol) of the active site structures of the Cu(I) form of AZAMI-F at pH 8 (2FT8,86 pale pink) and pH 6 (2FT7,86 purple). The second conformations of the copper ion and His115 at pH 6 are colored pale green. | ||
8 The second coordination sphere
The T1 copper site loop mutations described usually have little effect on active site structure (the ligands are not changed) but have a significant influence on the protein environment close to the metal ion that is not involved in direct ligation (the second coordination sphere).86–89 This feature is known to be of paramount importance in various metalloenzymes yet its influence on ET sites, such as those found in cupredoxins, have only started to be investigated in detail.90,97,98,118–122 The metal-binding loop provides a convenient way to tune many second coordination sphere features without compromising metal binding, and the overall fold and stability of the protein and thus allows many key active site properties to be adjusted.9 Conclusions
The studies highlighted here demonstrate that metal-binding loops in proteins are tolerant to sequence modifications. The loop mutations described give rise to mainly subtle effects on structure and spectroscopy (except for the CuA loop mutations), as the copper binding residues are not altered. Many of the key physiological properties of cupredoxins and cupredoxin domains are controlled to some extent by the sequence and structure of the C-terminal ligand-containing loop. Thus the allowed variability in this region enables these attributes to be fine-tuned to ensure efficient reactivity in a particular ET pathway. These principles could apply to tuning the properties of a range of metal sites in proteins , not only those based on the Greek-key β-barrel scaffold.10 Acknowledgements
BBSRC are thanked for funding (grant BB/C504519). The contributions of group members to the studies described are greatly appreciated. This work has been supported by very fruitful collaborations with Mark Banfield and Albrecht Messerschmidt. Mark Banfield prepared all of the protein structure figures for this review.11 References
- E. T. Adman, Adv. Protein Chem., 1991, 42, 144–197.
- A. M. Nersissian and E. L. Shipp, Adv. Protein Chem., 2002, 60, 271–340 CAS.
- C. Dennison, Coord. Chem. Rev., 2005, 249, 3025–3054 CrossRef CAS.
- M. J. Ellis, F. E. Dodd, G. Sawers, R. R. Eady and S. S. Hasnain, J. Mol. Biol., 2003, 328, 429–438 CrossRef CAS.
- S. V. Antonyuk, R. W. Strange, G. Sawers, R. R. Eady and S. S. Hasnain, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12041–12046 CrossRef CAS.
- A. Messerschmidt, R. Ladenstein, R. Huber, M. Bolognesi, L. Avigliano, R. Petruzzelli, A. Rossi and A. Finazzi-Agró, J. Mol. Biol., 1992, 224, 179–205 CrossRef CAS.
- I. Zaitseva, V. Zaitsev, G. Card, K. Moshkov, B. Bax, A. Ralph and P. Lindley, J. Biol. Inorg. Chem., 1996, 1, 15–23 CrossRef CAS.
- T. Bertrand, C. Jolivalt, T. Briozzo, E. Caminade, N. Joly, C. Madzak and C. Mougin, Biochemistry, 2002, 41, 7325–7333 CrossRef CAS.
- N. Hakulinen, L. L. Kiiskinen, K. Kruus, M. Saloheimo, A. Paananen, A. Koivula and J. Rouvinen, Nat. Struct. Biol., 2002, 9, 601–605 CAS.
- A. B. Taylor, C. S. Stoj, L. Ziegler, D. J. Kosman and P. J. Hart, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15459–15464 CrossRef CAS.
- S. Iwata, C. Ostermeier, B. Ludwig and H. Michel, Nature, 1995, 376, 660–669 CrossRef CAS.
- T. Tsukihara, H. Aoyama, E. Yamashita, T. Tomizaki, H. Yamaguchi, K. Shinzawa-Itoh, R. Nakashima, R. Yaono and S. Yoshikawa, Science, 1995, 269, 1069–1074 CrossRef CAS.
- E. N. Baker, J. Mol. Biol., 1988, 203, 1071–1095 CAS.
- H. Nar, A. Messerschmidt, R. Huber, M. van de Kamp and G. W. Canters, J. Mol. Biol., 1991, 221, 765–772 CrossRef CAS.
- B. R. Crane, A. J. Di Bilio, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2001, 123, 11623–11631 CrossRef CAS.
- P. J. Hart, A. M. Nersissian, R. G. Herrmann, R. M. Nalbandyan, J. S. Valentine and D. Eisenberg, Protein Sci., 1996, 5, 2175–2183 CrossRef CAS.
- M. Koch, M. Velarde, M. D. Harrison, S. Echt, M. Fischer, A. Messerschmidt and C. Dennison, J. Am. Chem. Soc., 2005, 127, 158–166 CrossRef CAS.
- Y. Xie, T. Inoue, Y. Miyamoto, H. Matsumura, K. Kataoka, K. Yamaguchi, M. Nojini, S. Suzuki and Y. Kai, J. Biochem., 2005, 137, 455–461 CrossRef CAS.
- S. Kim, J. C. Mollet, J. Dong, K. Zhang, S. Y. Park and E. M. Lord, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 16125–16130 CrossRef CAS.
- W. E. B. Shepard, B. F. Anderson, D. A. Lewandoski, G. E. Norris and E. N. Baker, J. Am. Chem. Soc., 1990, 112, 7817–7819 CrossRef CAS.
- H. B. Gray, B. G. Malmström and R. J. P. Williams, J. Biol. Inorg. Chem., 2000, 5, 551–559 CrossRef CAS.
- U. Ryde, M. H. M. Olsson, K. Pierloot and B. O. Roos, J. Mol. Biol., 1996, 261, 586–596 CrossRef CAS.
- M. A. Webb, C. M. Kwong and G. R. Loppnow, J. Phys. Chem. B, 1997, 101, 5062–5069 CrossRef CAS.
- U. Ryde and M. H. M. Olsson, Int. J. Quantum Chem., 2001, 81, 335–347 CrossRef CAS.
- M. Cascella, A. Magistrato, I. Tavernelli, P. Carloni and U. Rothlisberger, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19641–19646 CrossRef CAS.
- B. G. Malmström, Eur. J. Biochem., 1994, 223, 711–718 CAS.
- R. J. P. Williams, Eur. J. Biochem., 1995, 234, 363–381 CrossRef CAS.
- J. R. Winkler, P. Wittung-Stafshede, J. Leckner, B. G. Malmström and H. B. Gray, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 4246–4249 CrossRef CAS.
- U. Ryde, M. H. M. Olsson, B. O. Roos, J. O. A. De Kerpel and K. Pierloot, J. Biol. Inorg. Chem., 2000, 5, 565–574 CrossRef CAS.
- U. Ryde and M. H. M. Olsson, Int. J. Quantum Chem., 2001, 81, 335–347 CrossRef CAS.
- C. Zong, C. J. Wilson, T. Shen, P. Wittung-Stafshede, S. L. Mayo and P. G. Wolynes, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 3159–3164 CrossRef CAS.
- J. Peisach, W. G. Levine and W. E. Blumberg, J. Biol. Chem., 1967, 242, 2847–2858 CAS.
- K. G. Paul and T. Stigbrand, Biochim. Biophys. Acta, 1970, 221, 255–263 CAS.
- B. G. Malmström, B. Reinhammar and T. Vänngård, Biochim. Biophys. Acta, 1970, 205, 48–57 CAS.
- D. R. McMillin, R. A. Holwerda and H. B. Gray, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 1339–1341 CrossRef CAS.
- D. R. McMillin, R. C. Rosenberg and H. B. Gray, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 4760–4762 CrossRef CAS.
- E. I. Solomon, J. Rawlings, D. R. McMillin, P. J. Stephens and H. B. Gray, J. Am. Chem. Soc., 1976, 98, 8046–8048 CrossRef CAS.
- D. M. Dooley, J. Rawlings, J. H. Dawson, P. J. Stephens, L. E. Andreasson, B. G. Malmström and H. B. Gray, J. Am. Chem. Soc., 1979, 101, 5038–5046 CrossRef CAS.
- J. H. Dawson, D. M. Dooley, R. Clark, P. J. Stephens and H. B. Gray, J. Am. Chem. Soc., 1979, 101, 5046–5053 CrossRef CAS.
- E. I. Solomon, J. W. Hare, D. M. Dooley, J. H. Dawson, P. J. Stephens and H. B. Gray, J. Am. Chem. Soc., 1980, 102, 168–178 CrossRef CAS.
- K. W. Penfield, R. R. Gay, R. S. Himmelwright, N. C. Eickman, V. A. Norris, H. C. Freeman and E. I. Solomon, J. Am. Chem. Soc., 1981, 103, 4382–4388 CrossRef CAS.
- J. E. Roberts, J. F. Cline, V. Lum, H. Freeman, H. B. Gray, J. Peisach, B. Reinhammar and B. M. Hoffman, J. Am. Chem. Soc., 1984, 106, 5324–5330 CrossRef CAS.
- K. W. Penfield, A. A. Gewirth and E. I. Solomon, J. Am. Chem. Soc., 1985, 107, 4519–4529 CrossRef CAS.
- E. I. Solomon and M. D. Lowery, Science, 1993, 259, 1575–1581 CAS.
- C. R. Andrews, H. Yeom, J. S. Valentine, B. G. Karlsson, N. Bonander, G. van Pouderoyen, G. W. Canters, T. M. Loehr and J. Sanders-Loehr, J. Am. Chem. Soc., 1994, 116, 11489–11498 CrossRef CAS.
- A. P. Kalverda, J. Salgado, C. Dennison and G. W. Canters, Biochemistry, 1996, 35, 3085–3092 CrossRef CAS.
- E. I. Solomon, K. W. Penfield, A. A. Gewirth, M. D. Lowery, S. E. Shadle, J. A. Guckert and L. B. LaCroix, Inorg. Chim. Acta, 1996, 243, 67–78 CrossRef.
- E. I. Solomon, R. K. Szilagyi, S. DeBeer George and L. Basumallick, Chem. Rev., 2004, 104, 419–458 CrossRef CAS.
- M. Wilmanns, P. Lappalainen, M. Kelly, E. Sauer-Eriksson and M. Saraste, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 11955–11959 CAS.
- K. Brown, M. Tegoni, M. Prudêncio, A. S. Pereira, S. Besson, J. J. Moura, I. Moura and C. Cambillau, Nat. Struct. Biol., 2000, 7, 191–195 CrossRef CAS.
- K. Brown, K. Djinovic-Carugo, T. Haltia, I. Cabrito, M. Saraste, M. Prudêncio, J. J. G. Moura, I. Moura, M. Tegoni and C. Cambillau, J. Biol. Chem., 2000, 275, 41133–41136 CrossRef CAS.
- P. M. H. Kroneck, W. A. Antholine, J. Riester and W. G. Zumft, FEBS Lett., 1988, 242, 70–74 CrossRef CAS.
- P. M. H. Kroneck, W. A. Antholine, D. H. W. Kastrau, G. Buse, G. C. M. Steffens and W. G. Zumft, FEBS Lett., 1990, 268, 274–276 CrossRef CAS.
- P. Lappalainen, R. Aasa, B. G. Malmström and M. Saraste, J. Biol. Chem., 1993, 268, 26416–26421 CAS.
- N. Kitajima, K. Fujisawa and Y. Moro-oka, J. Am. Chem. Soc., 1990, 112, 3210–3212 CrossRef CAS.
- W. B. Tolman, J. Biol. Inorg. Chem., 2006, 11, 261–271 CrossRef CAS.
- T. J. Mizoguchi, A. J. Di Bilio, H. B. Gray and J. H. Richards, J. Am. Chem. Soc., 1992, 114, 10076–10078 CrossRef CAS.
- B. G. Karlsson, M. Nordling, T. Pascher, L. C. Tsai, L. Sjölin and L. G. Lundberg, Protein Eng., 1991, 4, 343–349 CrossRef CAS.
- T. den Blaauwen, M. Van de Kamp and G. W. Canters, J. Am. Chem. Soc., 1991, 113, 5050–5052 CrossRef CAS.
- J. P. Germanas, A. J. Di Bilio, H. B. Gray and J. H. Richards, Biochemistry, 1993, 32, 7698–7702 CrossRef CAS.
- D. R. Casimiro, A. Toy-Palmer, R. C. Blake and H. J. Dyson, Biochemistry, 1995, 34, 6640–6648 CrossRef CAS.
- J. M. Guss and H. C. Freeman, J. Mol. Biol., 1983, 169, 521–563 CrossRef CAS.
- A. Romero, H. Nar, R. Huber, A. Messerschmidt, A. P. Kalverda, G. W. Canters, R. Durley and F. S. Mathews, J. Mol. Biol., 1994, 236, 1196–1211 CrossRef CAS.
- L. M. Cunane, Z. W. Chen, R. C. E. Durley and F. S. Mathews, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1996, 52, 676–686 CrossRef CAS.
- J. M. Guss, E. A. Merritt, R. P. Phizackerely and H. C. Freeman, J. Mol. Biol., 1996, 262, 686–705 CrossRef CAS.
- R. L. Walter, S. E. Ealick, A. M. Friedman, R. C. Blake, P. Proctor and M. Shoham, J. Mol. Biol., 1996, 263, 730–751 CrossRef CAS.
- T. Inoue, N. Nishio, S. Suzuki, K. Kataoka, T. Kohzuma and Y. Kai, J. Biol. Chem., 1999, 274, 17845–17852 CrossRef CAS.
- Y. Xhu, M. Ökvist, Ö. Hansson and S. Young, Protein Sci., 1998, 7, 2099–2105 CrossRef CAS.
- C. S. Bond, R. E. Blankenship, H. C. Freeman, J. M. Guss, M. J. Maher, F. M. Selvaraj, M. C. J. Wilce and K. M. Willingham, J. Mol. Biol., 2001, 306, 47–67 CrossRef CAS.
- L. Chen, R. Durley, B. J. Poliks, K. Hamada, Z. Chen, F. S. Mathews, V. L. Davidson, Y. Satow, E. Huizinga, F. M. D. Vellieux and W. G. J. Hol, Biochemistry, 1992, 31, 4959–4964 CrossRef CAS.
- M. Ubbink, M. Ejdebäck, B. G. Karlsson and D. S. Bendall, Structure, 1998, 6, 323–335 CrossRef CAS.
- A. Impagliazzo and M. Ubbink, J. Am. Chem. Soc., 2004, 126, 5658–5659 CrossRef CAS.
- J. von der Oost, P. Lappalainen, A. Musacchio, A. Warne, L. Lemieux, J. Rumbley, R. B. Gennis, R. Aasa, T. Pascher, B. G. Malmström and M. Saraste, EMBO J., 1992, 11, 3209–3217 CAS.
- C. Dennison, E. Vijgenboom, S. de Vries, J. van der Oost and G. W. Canters, FEBS Lett., 1995, 365, 92–94 CrossRef CAS.
- M. Hay, J. H. Richards and Y. Lu, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 461–464 CrossRef CAS.
- H. Robinson, M. C. Ang, Y. G. Gao, M. T. Hay, Y. Lu and A. H. J. Wang, Biochemistry, 1999, 38, 5677–5683 CrossRef CAS.
- S. De Beer George, M. Metz, R. K. Szilagyi, H. Wang, S. P. Cramer, Y. Lu, W. B. Tolman, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 2001, 123, 5757–5767 CrossRef CAS.
- M. H. M. Olsson and U. Ryde, J. Am. Chem. Soc., 2001, 123, 7866–7876 CrossRef CAS.
- H. J. Hwang, S. M. Berry, M. J. Nilges and Y. Lu, J. Am. Chem. Soc., 2005, 127, 7274–7275 CrossRef.
- C. Dennison, E. Vijgenboom, W. R. Hagen and G. W. Canters, J. Am. Chem. Soc., 1996, 118, 7406–7407 CrossRef CAS.
- C. Buning, G. W. Canters, P. Comba, C. Dennison, L. Jeuken, M. Melter and J. Sanders-Loehr, J. Am. Chem. Soc., 2000, 122, 204–211 CrossRef CAS.
- R. Remenyi, L. J. C. Jeuken, P. Comba and G. W. Canters, J. Biol. Inorg. Chem., 2001, 6, 23–26 CrossRef CAS.
- S. Yanagisawa and C. Dennison, J. Am. Chem. Soc., 2003, 125, 4974–4975 CrossRef CAS.
- S. Yanagisawa and C. Dennison, J. Am. Chem. Soc., 2004, 126, 15711–15719 CrossRef CAS.
- C. Dennison, Dalton Trans., 2005, 3436–3442 RSC.
- C. Li, S. Yanagisawa, B. M. Martins, A. Messerschmidt, M. J. Banfield and C. Dennison, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 7258–7263 CrossRef CAS.
- C. Li, M. J. Banfield and C. Dennison, J. Am. Chem. Soc., 2007, 129, 709–718 CrossRef CAS.
- M. Velarde, R. Huber, S. Yanagisawa, C. Dennison and A. Messerschmidt, Biochemistry, 2007, 46, 9981–9991 CrossRef CAS.
- K. Sato, S. J. Fairbank, C. Li, M. J. Banfield and C. Dennison, submitted.
- S. Yanagisawa, M. J. Banfield and C. Dennison, Biochemistry, 2006, 45, 8812–8822 CrossRef CAS.
- G. Battistuzzi, M. Bellei, M. Borsari, G. W. Canters, E. de Waal, L. J. C. Jeuken, A. Renieri and M. Sola, Biochemistry, 2003, 42, 9214–9220 CrossRef CAS.
- G. Battistuzzi, M. Borsari, G. W. Canters, G. di Rocco, E. de Waal, Y. Arendsen, A. Leonardi, A. Renieri and M. Sola, Biochemistry, 2005, 44, 9944–9949 CrossRef CAS.
- M. V. Botuyan, A. Toy-Palmer, J. Chung, R. C. Blake, P. Beroza, D. A. Case and H. J. Dyson, J. Mol. Biol., 1996, 263, 752–767 CrossRef CAS.
- M. H. M. Olsson and U. Ryde, J. Biol. Inorg. Chem., 1999, 4, 654–663 CrossRef CAS.
- M. H. M. Olsson, G. Hong and A. Warshel, J. Am. Chem. Soc., 2003, 125, 5025–5039 CrossRef CAS.
- H. Li, S. P. Webb, J. Ivanic and J. H. Jensen, J. Am. Chem. Soc., 2004, 126, 8010–8019 CrossRef CAS.
- C. A. P. Libeu, M. Kukimoto, M. Nishiyama, S. Horinouchi and E. T. Adman, Biochemistry, 1997, 36, 13160–13179 CrossRef CAS.
- M. C. Machczynski, H. B. Gray and J. H. Richards, J. Inorg. Biochem., 2002, 88, 375–380 CrossRef CAS.
- R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, 1985, 811, 265–322 CAS.
- K. Sato, T. Kohzuma and C. Dennison, J. Am. Chem. Soc., 2003, 125, 2101–2112 CrossRef CAS.
- M. van de Kamp, R. Floris, F. C. Hali and G. W. Canters, J. Am. Chem. Soc., 1990, 112, 907–908 CrossRef CAS.
- I. M. C. van Amsterdam, M. Ubbink, O. Einsle, A. Messerschmidt, A. Merli, D. Cavazzini, G. L. Rossi and G. W. Canters, Nat. Struct. Biol., 2002, 9, 48–52 CrossRef CAS.
- K. Sato, P. B. Crowley and C. Dennison, J. Biol. Chem., 2005, 280, 19281–19288 CrossRef CAS.
- J. L. Markley, E. L. Ulrich, S. P. Berg and D. W. Krogmann, Biochemistry, 1975, 14, 4428–4433 CrossRef CAS.
- F. A. Armstrong, H. A. O. Hill, B. N. Oliver and D. Whitford, J. Am. Chem. Soc., 1985, 107, 1473–1476 CrossRef CAS.
- J. M. Guss, P. R. Harrowell, M. Murata, V. A. Norris and H. C. Freeman, J. Mol. Biol., 1986, 192, 361–387 CrossRef CAS.
- A. Lommen and G. W. Canters, J. Biol. Chem., 1990, 265, 2768–2774 CAS.
- C. Dennison, T. Kohzuma, W. McFarlane, S. Suzuki and A. G. Sykes, J. Chem. Soc., Chem. Commun., 1994, 581–582 RSC.
- E. Vakoufari, K. S. Wilson and K. Petratos, FEBS Lett., 1994, 347, 203–206 CrossRef CAS.
- Z. Zhu, L. M. Cunane, Z. Chen, R. C. E. Durley, F. S. Mathews and V. L. Davidson, Biochemistry, 1998, 37, 17128–17136 CrossRef CAS.
- C. Dennison, A. T. P. Lawler and T. Kohzuma, Biochemistry, 2002, 41, 552–560 CrossRef CAS.
- G. Battistuzzi, M. Borsari, G. W. Canters, E. de Waal, A. Leonardi, A. Renieri and M. Sola, Biochemistry, 2002, 41, 14293–14298 CrossRef CAS.
- H. C. Freeman, in Coordination Chemistry-21, ed. J. L. Laurent, Pergamon Press, Oxford, 1981, pp 29–51 Search PubMed.
- A. J. Di Bilio, C. Dennison, H. B. Gray, B. E. Ramirez, A. G. Sykes and J. R. Winkler, J. Am. Chem. Soc., 1998, 120, 7551–7556 CrossRef CAS.
- L. J. C. Jeuken, P. van Vliet, M. P. Verbeet, R. Camba, J. P. McEvoy, F. A. Armstrong and G. W. Canters, J. Am. Chem. Soc., 2000, 122, 12186–12194 CrossRef CAS.
- C. Dennison, C. Li, A. Ranieri, M. Sola and S. Yanagisawa, manuscript in preparation.
- C. Buning and P. Comba, Eur. J. Inorg. Chem., 2000, 1267–1273 CrossRef CAS.
- R. Langen, G. M. Jensen, U. Jacob, P. J. Stephens and A. Warshel, J. Biol. Chem., 1992, 267, 25625–26627 CAS.
- M. K. Eidsness, A. E. Burden, K. A. Richie, D. M. Kurtz, R. A. Scott, E. T. Smith, T. Ichiye, B. Bean, T. Min and C. Kang, Biochemistry, 1999, 38, 14803–14809 CrossRef CAS.
- S. D. Zarić, D. M. Popović and E. W. Knapp, Chem.–Eur. J., 2000, 6, 3935–3942 CrossRef CAS.
- S. Yanagisawa, K. Sato, T. Kohzuma and C. Dennison, Biochemistry, 2003, 42, 6853–6862 CrossRef CAS.
- I. J. Lin, E. B. Gebel, T. E. Machonkin, W. M. Westler and J. L. Markley, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 14581–14586 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2008 |