Mutasynthesis, chemobiosynthesis, and back to semi-synthesis: combining synthetic chemistry and biosynthetic engineering for diversifying natural products
Jonathan
Kennedy
*
Environmental Research Institute, University College Cork, Lee Road, Cork, Ireland. E-mail: jonathan.kennedy@ucc.ie; Fax: +353 (0)21 490 1932; Tel: +353 (0)21 490 1971
First published on 4th September 2007
Abstract
Covering: up to 2007
The combination of biological and chemical approaches for the generation of new and diverse natural products holds much promise. While mutasynthesis based approaches are still very relevant, more recent approaches have utilised genetic and metabolic engineering to generate key intermediates for chemical syntheses. This new semi-synthetic approach exploits the ability of biological systems to efficiently generate complex chiral molecules and of synthetic chemistry to elaborate these into new, or difficult to source, molecules.
 Jonathan Kennedy Jonathan Kennedy | Jonathan Kennedy studied biology at the University of Sheffield, UK, where he received his PhD degree for his work on the ACV synthase and penicillin biosynthesis in Aspergillus nidulans under the guidance of Prof. Geoffrey Turner. As a post-doctoral researcher at the University of Wisconsin-Madison, WI, USA, with Prof C. Richard Hutchinson he then studied the biosynthesis of the polyketide natural products lovastatin and daunorubicin. He then moved to industry, joining Kosan Biosciences, Hayward, CA, USA, where he remained until 2006. He is currently a Marie-Curie Transfer of Knowledge Fellow at the Environmental Research Institute, University College Cork, Ireland, where he is developing metagenomic approaches for accessing marine sponge metabolites. |
Introduction
Natural products remain an important source of lead compounds for drug development,1 however, while these compounds often exhibit great potency when tested against specific targets, other properties of these compounds, such as solubility, bioavailability, exposure, stability, and metabolism, can require improvement. To diversify natural products a number of approaches have been used.The application of synthetic chemistry to modify natural product drug leads has long been used to generate compounds with improved pharmacological properties. For example β-lactam antibiotics produced by semi-synthesis can have a broader spectrum (e.g.amoxicillin) and greater resistance to penicillinase (e.g.methicillin).2 Similarly with the cholesterol-lowering statin drugs, the semisynthetic simvastatin32 is more potent than the fungal polyketide natural product lovastatin31.3 While this semi-synthetic approach has been widely used to improve natural product drug candidates the structural complexity of many natural products can make even relatively simple chemical modifications challenging. Total chemical synthesis has been achieved for many important natural products and this can lead to the synthesis of a wide range of analogues. Subsequent advances in the understanding of structure–activity relationships (SAR) can lead to pharmacologically improved molecules but the complexity of the syntheses can make these processes difficult to scale-up for all but the earliest stages of the drug development process. Exceptions to this occur when the SAR allows the synthesis of simplified analogues that retain potency or where the promise of the drug candidate is such that an extraordinary effort is made to synthesise sufficient material for early stage clinical trials as was the case for discodermolide43.4–8
Recently a more complete understanding of the genetics and biochemistry underlying natural product biosynthesis has led to the development of combinatorial biosynthesis technologies for the generation of new natural products.9 These methods have huge potential but are generally slow and unreliable compared to chemical methods, with each attempt at modification of the natural product scaffold requiring a new genetic construct and, often, a loss of biosynthetic efficiency.
Semi-synthesis, total chemical synthesis and combinatorial biosynthesis have succeeded in introducing greater diversity into natural products. However, combining chemical and biological approaches has great promise by utilising the strengths of each approach; biology generating stereochemically complex structures cheaply and in bulk, by fermentation; and chemistry to rapidly generate analogues and to introduce non-biological functional groups.
Precursor-directed biosynthesis
Precursor-directed biosynthesis is the earliest example of combining chemical and biological approaches for the generation of novel natural products.10 In its simplest form this technique involves the feeding of a precursor analogue to the producing organism with this unnatural precursor becoming incorporated into a new product (Fig. 1). This approach can be effective at generating new analogues of complex natural products; for example the orally active β-lactam antibiotic, penicillin V (phenoxymethylpenicillin) 1, is produced by adding phenoxyacetic acid to fermentations of Penicillium chrysogenum.2 However, there are drawbacks to precursor-directed biosynthesis; a mixture of natural and unnatural products with similar physical properties are often produced, leading to complex downstream purification procedures; high concentrations of synthetic precursor are often required to compete with the preferred natural precursor and; a limited range of intermediates are efficiently incorporated into the final product.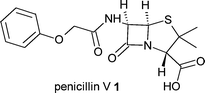
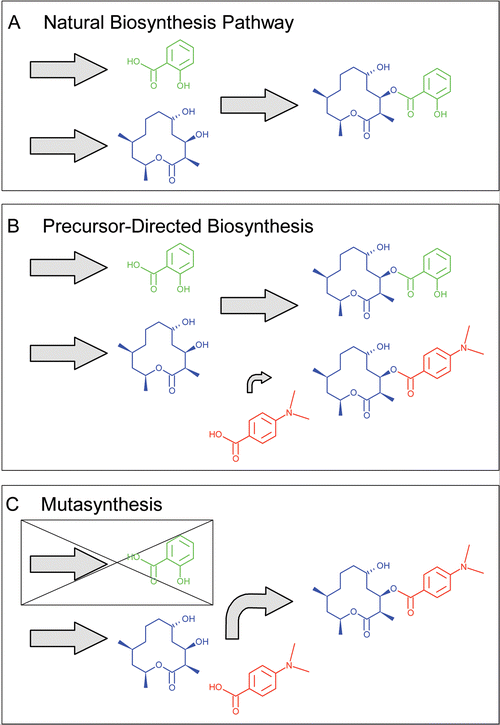 | ||
| Fig. 1 Precursor-directed biosynthesis and mutasynthesis. A. A hypothetical microbial natural product is made from a polyketide -derived 12 membered macrolide (in blue) and salicylic acid (in green). B. Precursor-directed biosynthesis. By adding the unnatural precursor 4-(dimethylamino)benzoic acid (in red) to the fermentation broth, this can be incorporated into the final product resulting in a mixture of the natural product and the precursor-directed product. C. Mutasynthesis. A strain which lacks the biosynthetic pathway for the intermediate salicylic acid is used. In this case the unnatural precursor 4-(dimethylamino)benzoic acid can be incorporated into the final product with no competition from the natural precursor, resulting in a single product. | ||
Mutasynthesis
Mutasynthesis is built upon precursor-directed biosynthesis but utilises mutant micro-organisms deficient in a key aspect of the biosynthetic pathway (Fig. 1). Typically mutant strains used for this approach are only able to synthesise the natural antibiotic when supplemented with a particular precursor and substitution of the natural precursor with a precursor analogue can lead to the production of a new natural product. This elegant approach was first described by Birch,11 although the term mutasynthesis was first used by Rinehart.12 Mutasynthesis overcomes some of the drawbacks of precursor-directed biosynthesis primarily due to the elimination of a competing natural precursor. Consequently, a less complex mixture of metabolites is produced, downstream purification is simpler, yields are higher and the incorporation of more diverse precursor analogues is possible.The development of mutasynthesis initially involved random mutagenesis followed by the isolation and characterisation of strains deficient in antibiotic biosynthesis. One of the earliest examples of mutasynthesis was in the production of new analogues of the aminoglycoside-aminocyclitol antibiotic neomycin B 2 (Fig. 2).13 The producing strain, Streptomyces fradiae, was subject to random mutagenesis and a mutant was selected that was only able to produce neomycin B 2 when supplemented with the aminocyclitol precursor deoxystreptamine3. When this mutant strain was supplemented with the aminocyclitols streptamine4 and epistreptamine 5 these unnatural precursors were utilised by the biosynthetic machinery to produce the novel neomycin analogues, hybrimycin A1 6 and hybrimycin B2 7 respectively.13 The success of mutasynthesis for the production of active neomycin analogues led to the application of this method to generate a whole range of aminoglycoside-aminocyclitol antibiotics using mutasynthesis.14 Mutasynthesis was subsequently successfully applied to many different classes of natural products including 14- and 16-membered macrolide antibiotics such as the erythromycins and platenolides, the aminocoumarin antibiotic novomycin, and β-lactam antibiotics.14
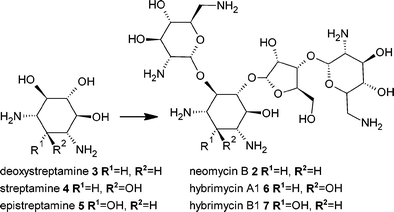 | ||
| Fig. 2 Mutasynthesis of neomycin analogues. | ||
The impact of genetics on mutasynthesis
As secondary metabolite biosynthesis pathways have become more fully understood at a biochemical and genetic level, new approaches have been developed for the diversification of natural products. Foremost amongst these approaches is combinatorial biosynthesis in which genes from different but related biosynthesis pathways are combined to generated novel compounds.9 However, the deeper understanding of the biochemistry and genetics of these pathways has also had an immediate impact on the mutasynthesis approach. Genetic and biochemical analyses can reveal the complete biochemical pathways from simple precursors to complex natural products, and all intermediates in between. Targeted disruption of discrete steps in the biosynthesis pathways has widened the scope for mutasynthesis allowing many more approaches to be evaluated. Several examples from different natural product classes are given below.Aminocoumarin antibiotics
The aminocoumarin antibiotic chlorobiocin8 consists of three components; an aminocoumarin moiety; a noviose sugar , and; a 3-dimethylallyl-4-hydroxybenzoic acid (DMAHB) 9 moiety. A key step in the biosynthesis of DMAHB is catalysed by CloQ, a dimethylallyl transferase. In order to produce analogues of 8, a strain of the producing organism, Streptomyces roseochromogenes, was constructed in which the cloQ gene was inactivated. This strain was unable to make chlorobiocins unless supplemented with DMAHB. Substitution of DMAHB with synthetic analogues allowed the incorporation of these into new and active chlorobiocin analogues (Fig. 3).15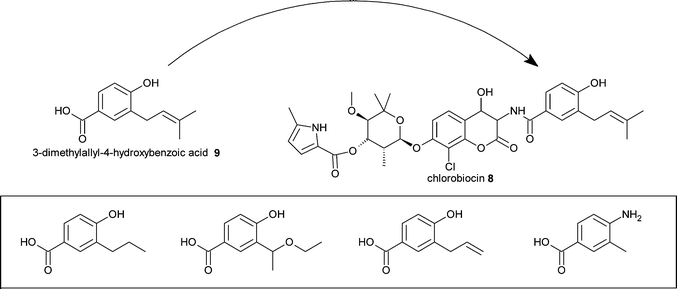 | ||
| Fig. 3 Mutasynthesis of chlorobiocin analogues. A selection of DMAHB analogues that were successfully incorporated into novel chlorobiocins are shown boxed. | ||
Non-ribosomal peptides
Precursor-directed biosynthesis approaches have long been used, with some success, to generate new analogues of non-ribosomal peptide derived compounds such as the immunosuppressant drug and fungal natural product cyclosporin A10.16 These approaches have often taken advantage of the apparently natural loose substrate specificity of certain domains of the non-ribosomal peptide synthetase (NRPS) enzymatic machinery. More recently a mutasynthesis approach was developed for the generation of novel vancomycin-type antibiotics (Fig. 4). The nonproteinogenic amino acid 3,5,-dihydroxyphenylglycine (DPG) 11 is a component of the glycopeptides vancomycin12 and the related balhimycin13. The biosynthesis of this amino acid was eliminated by disruption of the dpgA gene in Amycolatopsis balhimycina.17 Subsequent feeding of the disrupted strains with DPG analogues 14 and 16 resulted in the production of the novel and active glycopeptide antibiotics 15 and 17. Similarly the lipopeptide calcium-dependent antibiotic (CDA) 18 from Streptomyces coelicolor incorporates the nonproteinogenic amino acid 4-hydroxyphenylglycine (HPG) 19. Elimination of the supply of this intermediate via disruption of hmaS, a 4-hydroxymandelate synthase gene, resulted in a strain only able to produce CDA when supplemented with HPG or other intermediates (Fig. 5). The substitution of HPG with analogues 20 and 21 resulted in the biosynthesis of the corresponding analogues of CDA, 22 and 23.18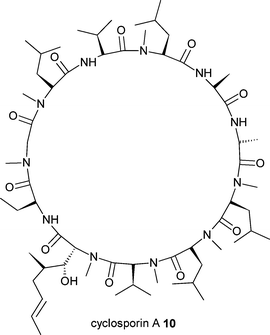
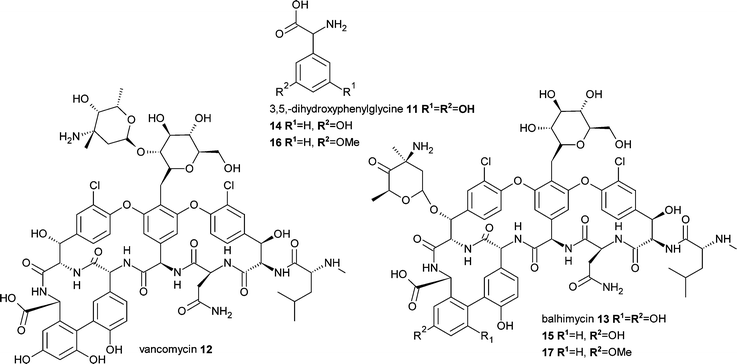 | ||
| Fig. 4 Mutasynthesis of glycopeptide antibiotics. | ||
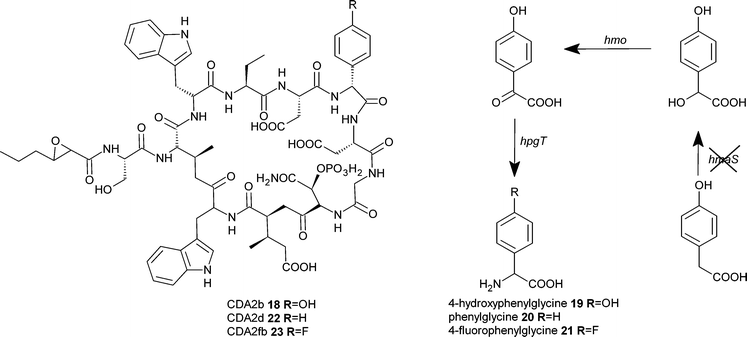 | ||
| Fig. 5 Mutasynthesis of calcium-dependent antibiotic. | ||
Polyketides
Perhaps the most notable examples of mutasynthesis of recent years have occurred in the polyketide class of natural products. The immunosuppressant polyketide rapamycin24 is synthesised from a shikimate-derived 4,5-dihydoxycyclohex-1-enecarboxylic acid (DHCHC) 25 which serves as a starter unit for the rapamycin PKS. As recently reviewed by Weissman,19 researchers at Biotica were able to generate a strain of Streptomyces hygroscopicus that was unable to make the DHCHC starter unit. Feeding this strain with analogues of DHCHC resulted in the efficient incorporation of several of these analogues into the rapamycin macrolide. A similar approach was also successful with the angiogenesis inhibitor borrelidin26.20 In this case, analogues of the natural starter unit trans-cyclopentane-(1R,2R)-dicarboxylic acid 27 were successfully incorporated to generate novel borrelidin analogues.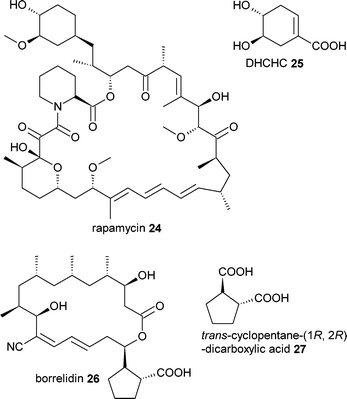
Mutasynthesis is also used for the commercial synthesis of the antiparasitic veterinary drug doramectin28.21 A mutant strain of the avermectin29 producing organism Streptomyces avermitilis was generated that is unable to generate the usual branched chain fatty acid starter unit.22 Supplementation of the culture with cyclohexanecarboxylic acid resulted in this being efficiently utilised as a starter unit for the generation of doramectin28.
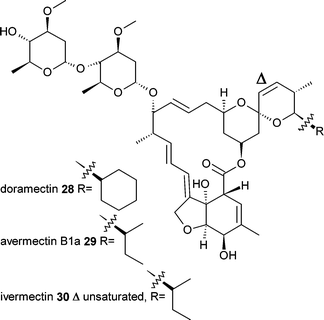
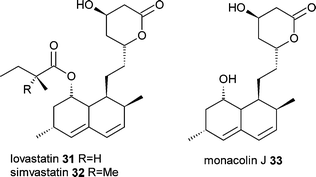
Another example of utilising biochemical and genetic knowledge for the generation of polyketide analogues is with lovastatin31. The more potent semisynthetic drug simvastatin32 differs from lovastatin by the addition of a single methyl group on the methylbutryate side-chain. Xie et al.23 found that the enzyme responsible for the addition of the side chain, LovD, was able to accept acyl-thioesters as substrates. Using an E. coli expression system for LovD and by feeding an N-acetylcysteamine thioester of dimethylbutyrate and the lovastatin intermediate monacolin J33, the authors were able to generate simvastatin32, with other analogues produced with alternative acyl-thiosters.24 It is easy to see this advance to a more straightforward mutasynthesis approach using a mutant of the lovastatin producer, Aspergillus terreus, that lacks side chain biosythesis and is able to take up and utilise the acyl-thioester intermediate.
Chemobiosynthesis
The examples above all demonstrate that genetic inactivation of a pathway for intermediate biosynthesis coupled with the feeding of a synthetic analogue of the intermediate can be effective for generating chemical diversity in natural products. However, the elucidation of the enzymology of modular polyketide synthase (PKS) systems has enabled the development of a new variant of mutasynthesis that has been called chemobiosynthesis (Fig. 6). A specific form of mutasynthesis, chemobiosynthesis, has been specifically used with multienzyme polyketide synthases in which the multienzyme carries a specific mutation disabling a particular enzymatic function. The feeding of a substrate analogue as a thioester then allows incorporation of this analogue into the final polyketide . This method was first used to generate new erythromycin analogues.256-Deoxyerythronolide B synthase (DEBS), the PKS responsible for the biosynthesis of the erythromycin macrolide, 6-deoxyerythronolide B (6-dEB) 34, consists of three large polypeptides, each of which contains two complete modules. In normal biosynthesis the growing polyketide chain is passed from module to module remaining bound to the PKSvia thioester linkages. By introducing a specific mutation in the PKS, changing the active-site cysteine of the ketosynthase of module 1 to an alanine, the PKS was inactivated. The normal substrate of module 2 is a diketide that is normally bound to the enzyme as a thioester. When this diketide was introduced into the strain as an N-acetylcysteamine thioester it was processed by the mutant DEBS PKS and elaborated into the usual product 6-dEB34. The authors then demonstrated the potential for this technique to generate new erythromycin analogues by feeding diketide analogues as thioesters to give new 6-dEB analogues including aromatic and ring expanded versions of 6-dEB, inaccessible by synthetic chemical methodology. Chemobiosynthesis has also been used with mutants deficient in the DEBS loading module.26 Here simple non-chiral acyl-thioesters were used to generate a range of 6-dEB analogues, and in addition defined alternative, simpler, and inexpensive thioester carriers that were utilised with equal efficiency to the originally used N-acetylcysteamine.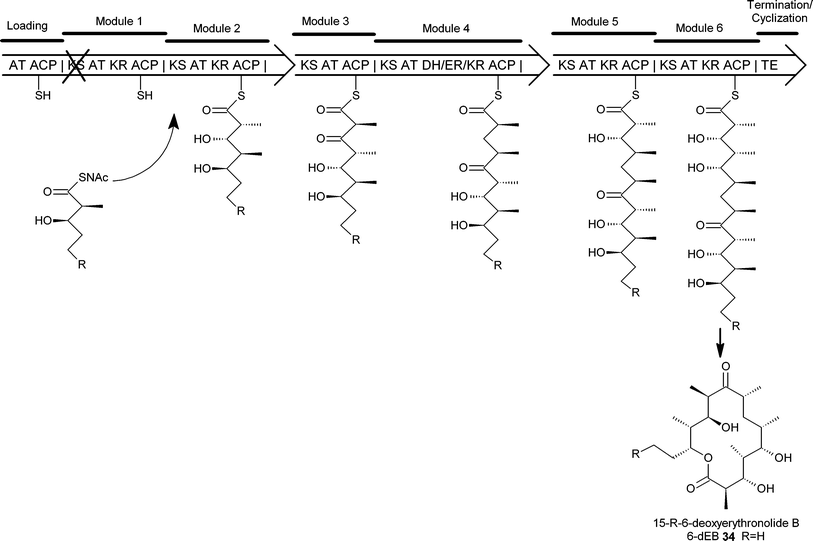 | ||
| Fig. 6 Chemobiosynthesis of 6-dEB analogues. A point mutation in the KS of module 1 renders the DEBS PKS inactive. By supplementing a strain expressing such a mutant PKS with an acyl-thioester similar to that normally accepted by module 2, activity can be restored and novel groups can be incorporated into the macrolide backbone (AT-acyltransferase, ACP-acyl carrier protein , KS-ketosynthase, KR-ketoreductase, DH-dehydratase, ER-enoylreductase, TE-thioesterase, SNAc-N-acetylcysteamine). | ||
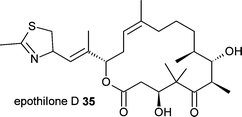
The chemobiosynthesis approach has also been used to generate the anti-cancer compound epothilone D 35 in the heterologous host Escherichia coli.27,28 This system also circumvents difficulties with genetics and chemobiosynthesis in the original producer, the myxobacterium Sorangium cellulosum, and has the potential to produce a range of epothilone analogues.
Modified semi-synthesis
Advancements in the understanding of natural product biosynthesis pathways and the development of the means to manipulate these pathways genetically have transformed the study of microbial secondary metabolism. However, the promise of combinatorial biosynthesis has yet to fulfil its potential. The use of genetics to generate new unnatural natural products has thus far been a relatively slow process, requiring a deep understanding of the underlying biosynthetic pathways (although recent advances are increasing the throughput of combinatorial biosynthesis technologies at least with PKS systems29–31). One area in which this biochemical and genetic knowledge has been applied to natural product biosynthesis is in developing direct fermentation processes for the production of compounds that were previously produced by semi-synthetic processes; for example metabolic engineering of microorganisms has led to direct fermentation processes for the anticancer drug epirubicin, and the antiparasitic drugs doramectin28 and ivermectin 30.32–34 These examples represent only a small fraction of the compounds that could theoretically be produced by direct fermentation processes, and while it is possible that in the future it will be possible to produce any natural product or natural product-like compound by fermentation, the technology for this is currently insufficiently robust.Similarly, total synthesis of complex natural products offers much potential (for a review of total synthesis of polyketides see Schetter and Mahrwald35) but the large-scale synthesis of complex natural products remains elusive, with low yields and multiple steps making chemical synthesis uneconomic.
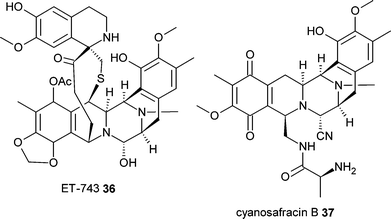
However the combination of current capabilities in synthetic chemistry with that of biochemistry and genetics offers much potential. One example of this approach is a method for the production of the antitumour compound ET-743 36, currently in clinical trials. This compound is a natural product, originally isolated from the marine tunicate Ecteinascidia turbinata.37 Wild harvest of the tunicate clearly could not supply a sustainable source of the compound to support clinical development and an alternative source was found. The bacterium Pseudomonas fluorescens produces the related safracin compounds and a semi-synthetic procedure based on conversion of microbially produced cyanosafracin B 37 has been used to supply kilogram quantities of ET-743 for clinical trials.38 The characterisation of the safracin biosynthesis pathway from P. fluorescens also promises to allow the biosynthesis and semi-synthesis of new analogues of ET-743 as well as offering a route to identifying the biosynthesis genes for ET-743 from E. turbinata microbial symbionts.39 This approach is similar to traditional semi-synthetic approaches, though in this case the semi-synthetic product is a difficult to source natural product.
Another example involves combining advances in metabolic engineering with synthetic chemistry to achieve synthesis of the sesquiterpene anti-malarial drug artemisinin42. Artemisinin-based therapies are currently one of the most effective treatments for malaria.40Artemisinin42 is currently extracted from the plant Artemisia annua and although a chemical synthetic route has been developed, neither of these routes can currently supply the demand for artemisinin at the low cost needed for large-scale treatment in the developing world. A potential solution to this supply problem is the reported production of the artemisinin precursor, artemisinic acid40, by an engineered strain of the yeast Saccharomyces cerevisiae (Fig. 7).41 This yeast strain was first engineered to increase farnesyl diphosphate38 production and also decrease the major sink for this intermediate, sterol biosynthesis. The authors then expressed the amorphadiene synthase gene from A. annua in this engineered yeast strain resulting in the conversion of farnesyl diphosphate38 to amorphadiene39. Finally, expression of a novel cytochrome P450 monooxygenase from A. annua, resulted in the conversion of the amorphadiene39 to artemisinic acid40 by a three-step oxidation. The microbially produced artemisinic acid can then be converted to artemisinin42 using a simple two step chemical procedure.42 This example could again be described as a semi-synthetic procedure to produce a natural drug, however in this case extensive metabolic engineering has been employed for the microbial production of the plant metabolite .
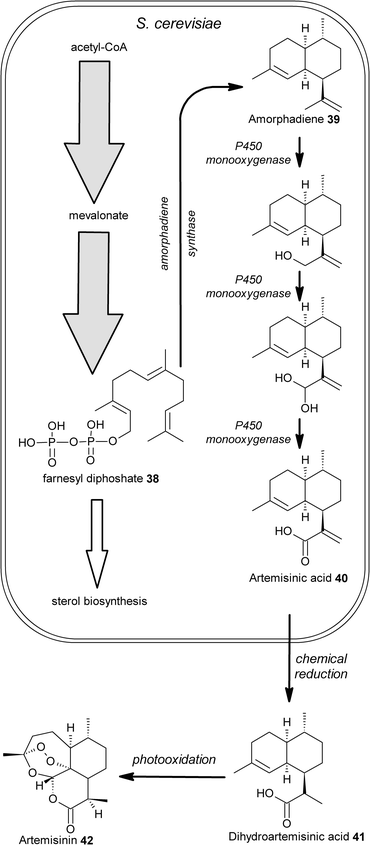 | ||
| Fig. 7 Production of the anti-malarial drug artemisinin42. Production of artemisinic acid40 was achieved in the yeast S. cerevisiae by; i) metabolic engineering to both increase production of farnesyl diphosphate38 and to downregulate the sterol biosynthesis pathway, a major sink for this intermediate; ii) expression of amorphadiene synthase for conversion of 38 to amorphadiene39; iii) expression of a cytochrome P450 monooxygenase for the three-step conversion of 39 to artemisinic acid40. The artemisinic acid produced by the yeast can then be chemically converted to artemisinin in a 2-step procedure involving reduction of 40 to dihydroartemisinic acid 41 followed by photooxidation to give artemisinin42. | ||
An interesting recent application of biotechnology in this area is to complement total synthesis strategies. One of the major challenges involved in the total syntheses of polyketide and other natural products is the presence of multiple chiral centers in the final product. A strategy used in total synthesis to overcome this is the synthesis of relatively small fragments, which contain many of the chiral centers of the final molecule. These fragments can then be combined to form the final structure. This fragment-based approach for the synthesis of polyketides has recently been proposed for use in combination with combinatorial biosynthesis.43
The search for natural products from diverse environmental niches has resulted in a very rich source of novel and promising drug-leads. However in many cases the development of these compounds is hindered by the lack of a sustainable supply of the compounds for clinical development.44 The sponge-derived compound discodermolide43 is a good example of a complex polyketide natural product that cannot be isolated from natural sources in sufficient quantities to allow clinical development.36 It is also a compound with enough promise to warrant an extraordinary total synthesis effort to advance the compound into clinical trials.4–8Discodermolide43 for clinical studies was prepared by total synthesis; a total of 60 g of 43 was made in a 39 step synthesis with an overall yield of 0.65%. While improvements to this synthesis have been made,45 a route for commercial total synthesis of 43 remains elusive.46Discodermolide43 has also become a target for alternative methods of production such as a direct biotechnological route, including, for example, attempts to isolate the biosynthesis genes from the metagenome of the sponge Discodermia dissoluta.47 Some progress has been made towards the synthesis of 43via a combined biotechnological and chemical approach (Fig. 8). The use of polyketide fragments as bio-intermediates was proposed in the patent literature and the first reported step in this direction was the production of bio-intermediates via chemobiosynthesis.48 Genetically engineered Streptomyces strains expressing a mutated and truncated DEBS PKS were fed diketide thioesters 44 and 45 that were processed into the respective triketide lactones 46 and 47 at high titers (Fig. 8). These triketide lactones can serve as intermediates in the synthesis of the common precursor 48 for discodermolide synthesis.49 In another published example a triketide lactone polyketide 49 that can be produced directly by fermentation by microbes expressing a truncated DEBS or oleandomycinPKS was converted to another key intermediate for discodermolide synthesis 50 and then on to fragment 51.49,50
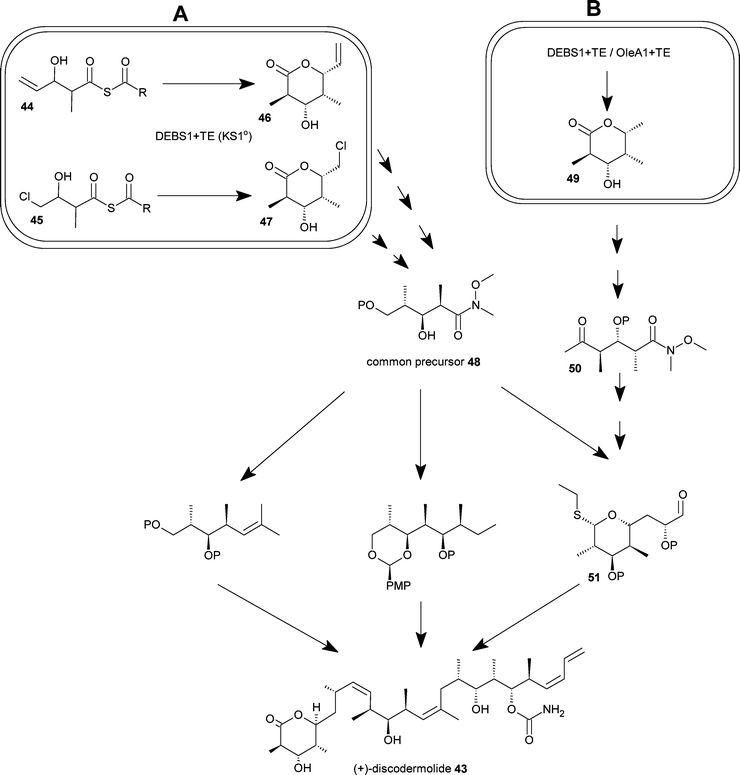 | ||
| Fig. 8 Use of bio-intermediates for discodermolide synthesis. The total synthesis of the complex natural product discodermolide is achieved using a fragment-based approach. In the examples shown here; A-the common precursor 48 can be synthesised from the triketide lactones 46 and 47 produced by chemobiosynthesis using a genetically modified Streptomycete; B-the triketide lactone 49 is produced by direct fermentation of an engineered Streptomycete, 49 can be chemically converted to the intermediate 50, and subsequently on to 51. These bio-intermediates can be produced in highly scalable procedures offering inexpensive access to large quantities of complex chiral molecules. | ||
The use of biotechnologically derived polyketide fragments will not only be of use for the synthesis of difficult to source natural products but will also allow access to analogues of polyketides that can currently only be produced by total chemical synthesis. For example, the anti-cancer epothilone compounds that are currently in clinical trials are produced by microbial fermentation and semi-synthesis. However, certain promising and potent analogues of epothilone have currently only been produced by total chemical synthesis,51 and the strategy of using biologically-derived polyketide fragments as biointermediates has the potential to make the syntheses of these and other polyketide analogues much simpler and more cost-effective.43
The strategy of using polyketide fragments as biointermediates for a wide range of potential syntheses gains credence from recent notable advances in the combinatorial biosynthesis of polyketides in what has been termed the Lego-isation of polyketides .52 Libraries of polyketide modules have been constructed and combined to generate multiple hybrid PKSs.29–31 These have then been assayed for the production of the predicted metabolite . Using this approach particular PKS modules have been identified that are more promiscuous in their processing of incoming substrates. This approach has now been extended to generate trimodular constucts to generate predicted tetraketides with multiple chiral centers.30
The future of chemical diversification of natural products
The combination of synthetic chemistry with the biosynthetic potential of micro-organisms has a long history in natural product research and development. The chemical modification of isolated natural products, semi-synthesis, has long been used for the generation of novel compounds with improved physical or biological properties. The possibilities of this hybrid approach were further extended with the advent of mutasynthesis. A deeper understanding of the underlying biosynthetic pathways and the application of molecular microbiological techniques allowed mutasynthesis (or precursor-directed biosynthesis) to be applied to a wide range of biosynthetic pathways and micro-organisms. The targeted inactivation of specific steps in the biosynthetic pathways of secondary metabolite precursors has allowed the incorporation of relatively simple chemically modified precursors into the natural product. In the case of polyketide derived natural products, the understanding of the enzymology of these systems has allowed the generation of mutant PKS systems deficient in starter unit loading (chemobiosynthesis). Feeding of synthetic starter unit substrate analogues, usually as thioesters, allows the incorporation of novel and unusual starter units directly into the polyketide chain.The precursor-directed biosynthesis or chemobiosynthesis approach described above involves the chemical synthesis of a biosynthetic precursor followed by feeding to a genetically modified microbe. Recently there has been a return to a modified semi-synthetic approach, with the addition of molecular genetics to the mix. This new semi-synthetic approach has evolved to address particular challenges in the economical production of drug candidates derived from difficult to obtain natural products. The artemisinin example highlights the possibility of using metabolic engineering of microbes to achieve high-level production of a complex precursor, artemisinic acid, that can be readily converted to the final product by chemical procedures. With polyketide -derived metabolites, the use of genetically engineered microbes to produce complex intermediates for chemical synthesis has also been demostrated. What links these final approaches is the acceptance of the current practical limitations of both total synthesis and biosynthetic engineering. While it may be possible to use total synthesis to produce enough material for initial clinical trials of complex natural products (e.g.discodermolide43) the cost of goods requires a more economical and scaleable synthesis. Similarly, while advances in the field of combinatorial biosynthesis, especially of polyketides , are impressive, constructing a PKS capable of synthesising a complex molecule such as discodermolide remains theoretical. Using the combinatorial biosynthesis approach to produce relatively small chiral molecules as a feedstock for chemical synthesis has the potential to dramatically increase the availability of such complex organic molecules both for screening and subsequent clinical development.
Acknowledgements
The author is in receipt of a Marie Curie Transfer of Knowledge Fellowship (grant no. MTKD-CT-2006-042062).References
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2007, 70, 461 CrossRef CAS.
- A. L. Demain and E. P. Elander, Antonie van Leeuwenhoek, 1999, 75, 5 CrossRef CAS.
- A. W. Alberts, Cardiology, 1990, 77(Suppl 4), 14 Search PubMed.
- S. J. Mickel, G. H. Sedelmeier, D. Niederer, R. Daeffler, A. Osmani, K. Schreiner, M. Seeger-Weibel, B. Berod, K. Schaer, R. Gamboni, S. Chen, W. Chen, C. T. Jagoe, F. R. Kinder, M. Loo, K. Prasad, O. Repic, W.-C. Shieh, R.-M. Wang, L. Waykole, D. D. Xu and S. Xue, Org. Process Res. Dev., 2004, 8, 92 Search PubMed.
- S. J. Mickel, G. H. Sedelmeier, D. Niederer, F. Schuerch, D. Grimler, G. Koch, R. Daeffler, A. Osmani, A. Hirni, K. Schaer, R. Gamboni, A. Bach, A. Chaudhary, S. Chen, W. Chen, B. Hu, C. T. Jagoe, H.-Y. Kim, F. R. Kinder, Y. Liu, Y. Lu, J. McKenna, M. Prashad, T. M. Ramsey, O. Repic, L. Rogers, W.-C. Shieh, R.-M. Wang and L. Waykole, Org. Process Res. Dev., 2004, 8, 101 Search PubMed.
- S. J. Mickel, G. H. Sedelmeier, D. Niederer, F. Schuerch, G. Koch, E. Kuesters, R. Daeffler, A. Osmani, M. Seeger-Weibel, E. Schmid, A. Hirni, K. Schaer, R. Gamboni, A. Bach, S. Chen, W. Chen, P. Geng, C. T. Jagoe, F. R. Kinder, G. T. Lee, J. McKenna, T. M. Ramsey, O. Repic, L. Rogers, L. W.-C. Shieh, R.-M. Wang and L. Waykole, Org. Process Res. Dev., 2004, 8, 107 Search PubMed.
- S. J. Mickel, G. H. Sedelmeier, D. Niederer, F. Schuerch, M. Seger, K. Schreiner, R. Daeffler, A. Osmani, D. Bixel, O. Loiseleur, J. Cercus, H. Stettler, K. Schaer, R. Gamboni, A. Bach, G.-P. Chen, W. Chen, P. Geng, G. T. Lee, E. Loeser, J. McKenna, F. R. Kinder, K. Konigsberger, K. Prasad, T. M. Ramsey, N. Reel, O. Repic, L. Rogers, W.-C. Shieh, R.-M. Wang, L. Waykole, S. Xue, G. Florence and I. Paterson, I., Org. Process Res. Dev., 2004, 8, 113 Search PubMed.
- S. J. Mickel, D. Niederer, R. Daeffler, A. Osmani, E. Kuesters, E. Schmid, K. Schaer, R. Gamboni, W. Chen, E. Loeser, F. R. Kinder, K. Konigsberger, K. Prasad, T. M. Ramsey, O. Repic, R.-M. Wang, G. Florence, I. Lyothier and I. Paterson, I., Org. Process Res. Dev., 2004, 8, 122 Search PubMed.
- C. D. Reeves, Crit. Rev. Biotechnol., 2003, 23, 95 CrossRef CAS.
- R. Thiericke and J. Rohr, Nat. Prod. Rep., 1993, 10, 265 RSC.
- A. J. Birch, Pure Appl. Chem., 1963, 7, 527 CrossRef.
- K. L. Rinehart, Jr., Pure Appl. Chem., 1977, 49, 1361 CrossRef CAS.
- W. T. Shier, K. L. Rinehart, Jr and D. Gottlieb, Proc. Natl. Acad. Sci. U. S. A., 1969, 63, 198 CrossRef CAS.
- S. J. Daum and J. R. Lemke, Annu. Rev. Microbiol., 1979, 33, 241 CrossRef CAS.
- U. Galm, S. Heller, S. Shapiro, M. Page, M. S. M. Li and L. Heide, Antimicrob. Agents Chemother., 2004, 48, 1307 CrossRef CAS.
- R. Traber, H. Hofmann and H. Kobel, J. Antibiot., 1989, 42, 591 CAS.
- S. Weist, C. Kittel, D. Bischoff, B. Bister, V. Pfeifer, G. J. Nicholson, W. Wohlleben and R. D. Sussmuth, J. Am. Chem. Soc., 2004, 126, 5942 CrossRef CAS.
- Z. Hojati, C. Milne, B. Harvey, L. Gordon, M. Borg, F. Flett, B. Wilkinson, P. J. Sidebottom, B. A. Rudd, M. A. Hayes, C. P. Smith and J. Micklefield, Chem. Biol., 2002, 9, 1175 CrossRef CAS.
- K. J. Weissman, Trends Biotechnol., 2007, 25, 139 CrossRef CAS.
- C. Olano, B. Wilkinson, C. Sanchez, S. J. Moss, R. Sheridan, V. Math, A. J. Weston, A. F. Brana, C. J. Martin, M. Oliynyk, C. Mendez, P. F. Leadlay and J. A. Salas, Chem. Biol., 2004, 11, 87 CAS.
- C. D. Denoya, R. W. Fedechko, E. W. Hafner, H. A. McArthur, M. R. Morgenstern, D. D. Skinner, K. Stutzman-Engwall, R. G. Wax and W. C. Wernau, J. Bacteriol., 1995, 177, 3504 CAS.
- H. A. I. McArthur, in Developments in industrial microbiology–BMP ′97, ed. C. R. Hutchinson and J. McAlpine, pp. 43–48, Society for Industrial Microbiology, Fairfax, VA, 1998 Search PubMed.
- X. Xie, K. Watanabe, W. A. Wojcicki, C. C. Wang and Y. Tang, Chem. Biol., 2006, 13, 1161 CrossRef CAS.
- X. Xie and Y. Tang, Appl. Environ. Microbiol., 2007, 73, 2054 CrossRef CAS.
- J. R. Jacobsen, C. R. Hutchinson, D. E. Cane and K. Khosla, Science, 1997, 277, 367 CrossRef CAS.
- S. Murli, K. S. MacMillan, Z. Hu, G. W. Ashley, S. D. Dong, J. T. Kealey, C. D. Reeves and J. Kennedy, Appl. Environ. Microbiol., 2005, 71, 4503 CrossRef CAS.
- C. N. Boddy, K. Hotta, M. L. Tse, R. E. Watts and C. Khosla, J. Am. Chem. Soc., 2004, 126, 7436 CrossRef CAS.
- S. C. Mutka, J. R. Carney, Y. Liu and J. Kennedy, Biochemistry, 2006, 45, 1321 CrossRef CAS.
- H. G. Menzella, R. Reid, J. R. Carney, S. S. Chandran, S. J. Reisinger, K. G. Patel, D. A. Hopwood and D. V. Santi, Nat. Biotechnol., 2005, 23, 1171 CrossRef CAS.
- H. G. Menzella, J. R. Carney and D. V. Santi, Chem. Biol., 2007, 14, 143 CrossRef CAS.
- S. S. Chandran, H. G. Menzella, J. R. Carney and D. V. Santi, Chem. Biol., 2006, 13, 469 CrossRef CAS.
- K. Madduri, J. Kennedy, G. Rivola, A. Inventi-Solari, S. Filippini, G. Zanuso, A. L. Colombo, K. M. Gewain, J. L. Occi, D. J. MacNeil and C. R. Hutchinson, Nat. Biotechnol., 1998, 16, 69 CrossRef CAS.
- T. A. Cropp, D. J. Wilson and K. A. Reynolds, Nat. Biotechnol., 2000, 18, 980 CrossRef CAS.
- X. Zhang, Z. Chen, M. Li, Y. Wen, Y. Songand and J. Li, Appl. Microbiol. Biotechnol., 2006, 72, 986 CrossRef CAS.
- B. Schetter and R. Mahrwald, Angew. Chem., Int. Ed., 2006, 45, 7506 CrossRef CAS.
- S. P. Gunasekera, M. Gunasekera, R. E. Longley and G. K. Schulte, J. Org. Chem., 1990, 55, 4912 CrossRef CAS.
- A. E. Wright, D. A. Forleo, G. P. Gunawardana, S. P. Gunasekera, F. E. Koehn and O. J. McConnell, J. Org. Chem., 1990, 55, 4508 CrossRef CAS.
- C. Cuevas, M. Perez, M. J. Martin, J. L. Chicharro, C. Fernandez-Rivas, M. Flores, A. Francesch, P. Gallego, M. Zarzuelo, F. de la Calle, J. Garcia, C. Polanco, I. Rodriguez and I. Manzanares, Org. Lett., 2000, 2, 2545 CrossRef CAS.
- A. Velasco, P. Acebo, A. Gomez, C. Schleissner, P. Rodriguez, T. Aparicio, S. Conde, R. Munoz, F. de la Calle, J. L. Garcia and J. M. Sanchez-Puelles, Mol. Microbiol., 2005, 56, 144 CrossRef CAS.
- M. Enserink, Science, 2005, 307, 33 CrossRef CAS.
- D. K. Ro, E. M. Paradise, M. Ouellet, K. J. Fisher, K. L. Newman, J. M. Ndungu, K. A. Ho, R. A. Eachus, T. S. Ham, J. Kirby, M. C. Chang, S. T. Withers, Y. Shiba, R. Sarpong and J. D. Keasling, Nature, 2006, 440, 940 CrossRef CAS.
- R. J. Roth and N. Acton, J. Nat. Prod., 1989, 52, 1183 CAS.
- D. V. Santi, G. Ashley and D. C. Myles, United States Patent Application, 2004 Search PubMed , Pub. No. US 2004/0018598 A1.
- J. L. Fortman and D. H. Sherman, ChemBioChem, 2005, 6, 960 CrossRef CAS.
- A. B. Smith III, B. S. Freeze, M. Xian and T. Hirose, Org. Lett., 2005, 7, 1825 CrossRef CAS.
- S. J. Mickel, Curr. Opin. Drug Discovery Dev., 2004, 7, 869 CAS.
- A. Schirmer, R. Gadkari, C. D. Reeves, F. Ibrahim, E. F. DeLong and C. R. Hutchinson, Appl. Environ. Microbiol., 2005, 71, 4840 CrossRef CAS.
- R. Regentin, J. Kennedy, N. Wu, J. R. Carney, P. Licari, J. Galazzo and R. Desai, Biotechnol. Prog., 2004, 20, 122 CrossRef CAS.
- A. B. Smith, Y. Qiu, D. R. Jones and K. Kobayashi, J. Am. Chem. Soc., 1995, 117, 12011 CrossRef.
- S. J. Shaw, D. Zhang, K. F. Sundermann and D. C. Myles, Synth. Commun., 2006, 36, 1735 CrossRef CAS.
- A. Rivkin, F. Yoshimura, A. E. Gabarda, T. C. Chou, H. Dong, W. P. Tong and S. J. Danishefsky, J. Am. Chem. Soc., 2003, 125, 2899 CrossRef CAS.
- D. H. Sherman, Nat. Biotechnol., 23, 1083 Search PubMed.
| This journal is © The Royal Society of Chemistry 2008 |