Quinoline, quinazoline and acridone alkaloids
Joseph P.
Michael
*
Molecular Sciences Institute, School of Chemistry, University of the Witwatersrand, Wits, 2050, South Africa. E-mail: joseph.michael@wits.ac.za
First published on 17th December 2007
Abstract
Covering: July 2005 to June 2006. Previous review: Nat. Prod. Rep., 2007, 24, 223–246
This review covers the isolation, structure determination, synthesis and biological activity of quinoline, quinazoline and acridone alkaloids from plant, microbial and animal sources; 115 references are cited.
1 Quinoline alkaloids
1.1 Quinoline alkaloids from rutaceous plants: occurrence, characterisation and biological activity
New quinoline alkaloids isolated from plant species belonging to the Rutaceae are listed in Table 1;1–17 they will be described in greater detail in the text that follows. Also tabulated are plant sources from which known quinoline alkaloids have been obtained for the first time.| Species | Alkaloid a | Ref. |
|---|---|---|
| a Only new alkaloids and new records for a given species are listed in the table; in many cases, the presence of several previously isolated alkaloids was confirmed. Structures of known alkaloids, if not specifically numbered, may be found in previous reviews in this series. b New alkaloid. | ||
| Almeidea coerulea | Dictamnine 37 | 1 |
| Skimmianine 33 | ||
| Araliopsis tabouensis | Flindersiamine | 2 |
| (−)-Tabouensinium chlorideb16 | ||
| Boronella koniambiensis | (−)-cis-1,2-Dihydroxy-1,2-dihydromedicosmineb19 | 3 |
| Evolitrine | ||
| Medicosmine 18 | ||
| Brombya sp. nov. | Kokusaginine 24 | 4 |
| 5,6,7-Trimethoxydictamnineb25 | ||
| Esenbeckia pentaphylla | Dictamnine | 5 |
| Eduline | ||
| γ-Fagarine 31 | ||
| Flindersiamine | ||
| Kokusaginine | ||
| N-Methylflindersine | ||
| N-Methyl-4-methoxyquinolin-2-one 39 | ||
| Evodia fargesii | Dictamnine | 6 |
| Haplopine 32 | ||
| Robustine 22 | ||
| Haplophyllum sieversii | Anhydroevoxine 36 | 7 |
| Flindersine 34 | ||
| Haplamine 35 | ||
| Melicope bonwickii | Evellerine 27 | 8 |
| Kokusaginine | ||
| 7-(2,3-Epoxyprenyloxy)-4-methoxyfuro[2,3-b]quinoline 26 | ||
| 7-(2-Hydroxy-3-chloroprenyloxy)-4-methoxyfuro[2,3-b]quinolineb28 | ||
| Melicope semecarpifolia | (S)-(+)-Geibalansine | 9 |
| Philotheca deserti var. deserti | Evolitrine | 10 |
| γ-Fagarine | ||
| Maculosidine | ||
| Pteleine 29 | ||
| Robustine | ||
| Pilocarpus grandiflorus | Dictamnine | 11 |
| 4-Methoxyquinolin-2(1H)-one | ||
| Platydesmine 38 | ||
| Ruta chalepensis | Quinoline-4-carbaldehyde 1 | 12 |
| Spathelia excelsa | (−)-2-(10-Hydroxy-10-methyldodecyl)-3-methoxyquinolin-4(1H)-oneb3 | 13 |
| 2-(11-Hydroxy-11-methyldodecyl)-3-methoxyquinolin-4(1H)-oneb4 | ||
| (+)-2-(12-Hydroxytridecyl)-3-methoxyquinolin-4(1H)-oneb5 | ||
| 2-(12-Oxotridecyl)-3-methoxyquinolin-4(1H)-oneb2 | ||
| 2-(3-Hydroxy-3-methylbutyl)-6-hydroxyquinolin-4(1H)-oneb6 | ||
| 2-(3-Hydroxy-3-methylbutyl)-7-hydroxyquinolin-4(1H)-oneb7 | ||
| Spiranthera odoratissima | Dictamnine | 14 |
| Toddalia aculeata (= T. asiatica) | N-Methyl-3-(2,3-epoxy-3-methylbutyl)-4-hydroxy-7-methoxyquinolin-2-oneb14 | 15 |
| N-Methyl-3-(2,3-dihydroxy-3-methylbutyl)-4,7-dimethoxyquinolin-2-oneb15 | ||
| Zanthoxylum ailanthoides | Evolitrine | 16 |
| Pteleine | ||
| Zanthoxylum heitzii | Skimmianine | 17 |
The antimicrobial activity of an extract from the pharmacologically useful herb Ruta chalepensis has been traced by bioassay-guided fractionation to an unusual component, quinoline-4-carbaldehyde 1.12 While the authors make no mention of the fact, this compound has apparently never before been found in plants, although it has previously been isolated from the myxobacteriumArchangium gephyra.18 The compound strongly inhibited the growth of Clostridium perfringens, a noxious component of human gastrointestinal microflora, but its effect on Escherichia coli was weaker. It had little or no effect on the beneficial gastrointestinal bacteria Bifidobacterium bifidum, B. longum and Lactobacillus acidophilus, which perhaps explains the phytoprotective effects of the herbal remedy.
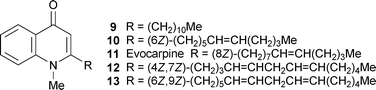
The isolation of the six new 2-alkylquinolin-4(1H)-one alkaloids 2–7 from the previously uninvestigated plant Spathelia (= Sohnreyia) excelsa has helped to cast light on the ambiguous taxonomic position of the genus within the Rutaceae.13 In particular, the occurrence of related alkaloids such as 8 in the genus Dictyoloma is seen as evidence for moving the subfamily Spathelioideae close to or within the Dictyolomatoideae. The structures of the alkaloids themselves were elucidated with the help of extensive spectroscopic methods, among which two-dimensional and long-range NMR experiments proved crucial in positioning the substituents on the quinolinone nucleus and the side chain. The more familiar 2-alkylquinolin-4-one alkaloids 9–13 isolated from unripe fruits of the Chinese herbal remedy Evodia rutaecarpa displayed promising antimycobacterial activities in in vitro tests with Mycobacterium fortuitum, M. smegmatis and M. phlei (minimum inhibitory concentrations 2–32 µg ml−1); the most abundant compound, evocarpine 11, was also the most active.19 The therapeutic potential of E. rutaecarpa and its alkaloidal metabolites for the treatment of mycobacterial diseases such as tuberculosis is thus intriguing, but remains to be explored.
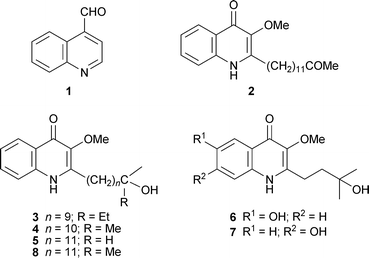
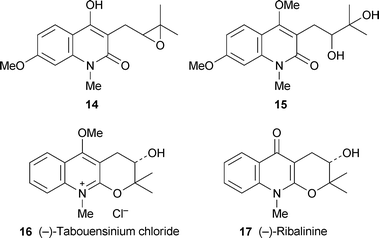
Hemiterpenoid quinoline alkaloids and their tricyclic (pyranoquinoline and furoquinoline) derivatives are among the most characteristic alkaloids of the Rutaceae, and novel variants of these structures continue to be identified. Two new functionalised 3-prenylquinolinones, isolated from the leaves and stems of the Indian medicinal plant Toddalia aculeata, were shown by a combination of spectroscopic techniques to have the gross structures 14 and 15.15 Their stereochemistries were not elucidated, and specific rotations were not reported. The unnamed alkaloids strongly inhibited the growth of the bacteria E. coli, Bacillus cereus and Lactobacillus lactis at millimolar concentrations. A cyclised analogue, the novel quaternary pyrano[2,3-b]quinoline alkaloid (−)-16, was isolated with chloride as counter-ion from the stem bark of the West African tree Araliopsis tabouensis, used in folk medicine for the treatment of venereal diseases.2 The compound, given the name tabouensinium chloride, was fully characterised by spectroscopic methods. Dequaternisation of 16 with anhydrous pyridine yielded the known alkaloid (−)-ribalinine 17, which was also identified in the plant extract together with another ten known alkaloids. Since (−)-17 is known have the 3S configuration, the absolute stereochemistry of 16 was assumed to be the same.
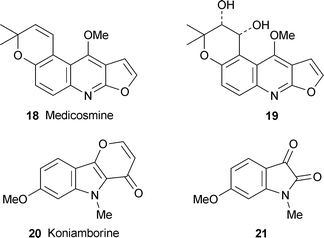
The rare alkaloid medicosmine 18 has been identified as a major metabolite in an extract of the aerial parts of Boronella koniambiensis, a rutaceous tree endemic to New Caledonia.3 Also isolated were the new diol analogue 19, the unprecedented pyrano[3,2-b]indole alkaloid koniamborine 20, and 6-methoxy-1-methylisatin 21 in its first isolation from a natural source. The cis relationship of the hydroxy groups in 19 was inferred from the size of the coupling constant between the attached hydrogen atoms (J = 5 Hz) and NOE interactions with the methyl substituents of the dihydropyran ring, both effects being supported by molecular mechanics calculations to determine the lowest energy conformer of the system. The absolute configuration of the new alkaloid was not determined. Both medicosmine and koniamborine were found to be cytotoxic towards the murine L1210 leukemia cell line (IC50 48.0 and 38.2 µM, respectively).
Reliable 1H and 13C NMR spectroscopic data have been reported for the simple furo[2,3-b]quinoline alkaloid robustine 22, isolated from the Australian shrub Philotheca deserti var. deserti.10 Also reliably assigned for the first time are the NMR spectroscopic data for maculine 23, which was extracted from a known source, Esenbeckia grandiflora.20 Kokusaginine 24 and a related furo[2,3-b]quinoline, the new alkaloid 5,6,7-trimethoxydictamnine 25, were isolated from the stem bark of an as yet undescribed species of Brombya found in the Gap Creek area of Australia.4 The rare 4-methoxy-7-prenyloxyfuro[2,3-b]quinoline motif has turned up in three alkaloids isolated from Melicope bonwickii, a small tree that is widely distributed in the Pacific regions from the Philippines through Malaysia to north-eastern Australia.8 They include the known epoxy derivative 26 and the diol evellerine 27, together with the novel chlorine-containing alkaloid 28, one of a growing number of chlorinated alkaloids from the Rutaceae. (Since chloroform was used in the extraction process, could 28 be an artefact, perhaps formed by cleavage of the epoxide 26?) The structures were ascertained principally by means of one- and two-dimensional NMR spectroscopic methods, but absolute configurations were not determined and optical rotations were not reported. Both 26 and 28 showed significant activity when tested against the HeLa (cervical cancer) cell line (IC50 6.0 and 11.4 µg ml−1, respectively). Antitumour activity was also displayed by two known alkaloids obtained from the root wood of Melicope semecarpifolia: pteleine 29 was moderately cytotoxic towards the P-388 cell line (ED50 8.94 µg ml−1), while both pteleine and (+)-7,8-dimethoxymyrtopsine 30 were active against the HT-29 cell line (ED50 15.2 and 41.6 µg ml−1, respectively).9
A study of the cytotoxicity of 14 known quinoline alkaloids isolated from several Uzbeki species of the genus Haplophyllum toward HeLa and HCT-116 cancer cells showed that γ-fagarine 31, haplopine 32, skimmianine 33, flindersine 34 and haplamine 35 had useful activity against the former cell line (IC50 <50 µM), while only haplamine was active against the latter (IC50 64.5 µM).21 Screening of extracts from Haplophyllum sieversii, a related species originating in Kazakhstan, for potentially useful antifungal activity against the plant pathogens Colletotrichum fragariae, C. gloeosporioides and C. aculatum also led to the isolation of flindersine and haplamine, as well as anhydroevoxine 36.7 Flindersine was especially effective in inhibiting the growth of the three fungi as well as that of Fusarium oxysporum and Phomopsis obscurans at micromolar concentrations, while haplamine showed the highest antifungal activity against Botrytis cinerea, although the effects of both alkaloids diminished with time. Haplamine was also selectively more toxic towards freshwater phytoplanktons such as Pseudanabaena sp. LW397 and the odour-producing cyanobacteriumOscillatoria perornata, which suggests a possible use as an algicide in commercial fish farming. The growth of another fungus, Leucoagaricus gongylophorus, which exists symbiotically with the insect pest Atta sexdens rubropilosa (a leaf-cutting ant), was inhibited by dictamnine 37 and platydesmine 38, two common alkaloids isolated for the first time from the rutaceous shrub Pilocarpus grandiflorus.11
In a study of the anti-HIV activity of 67 natural products (including 11 quinoline alkaloids) isolated from the root bark of Zanthoxylum ailanthoides, four alkaloids were able to inhibit viral replication in H9 lymphocyte cells at low concentrations (EC50 <1.34 µg ml−1) without significantly affecting the growth of uninfected H9 cells.16 The best therapeutic index (>231) was shown by γ-fagarine 31, while haplopine 32, (+)-platydesmine 38 and 4-methoxy-1-methylquinolin-2-one 39 were less effective. N-Methyl-3,3-diprenylquinoline-2,4-dione 40, obtained from Esenbeckia almawillia, showed antitumour activity towards five tumour cell lines; however, the synthetic derivatives 41–43 were found to be even more active.22
1.2 Quinoline alkaloids from rutaceous plants: synthesis
The simple Galipeaalkaloid 2-phenylquinoline 44 is often encountered among the products that result from novel approaches to the construction of the quinoline nucleus (Scheme 1). For example, it was formed in 84% yield by Sonogashira coupling of 2-iodoaniline 45 with 1-phenylprop-2-ynol 46 followed by ynol–enone isomerisation and cyclisation.23 Its formation by oxidative cyclisation of 2-aminobenzyl alcohol 47 with acetophenone (a modified Friedländer synthesis) has been accomplished in high yield with tris(triphenylphosphine)chlororhodium(I) (Wilkinson's catalyst) in dioxane,24 with bis(cyclooctadienyl)chloroiridium(I) dimer or iridium(III) chloride in the absence of solvent,25 or over palladium on carbon.26 The latter method was less successful (40% yield) for the synthesis of 2-pentylquinoline 48, another simple Galipeaalkaloid. Finally, the cycloisomerisation of the alkynylimine 49 to give 44 was effected in 91% yield on a one-gram scale with chloro(cyclopentadienyl)bis(triphenylphosphine)ruthenium and the additional ligand 2-(dicyclohexylphosphino)-2′,6′-dimethoxybiphenyl (SPhos).27![Reagents and conditions: i, PdCl2(PPh3)2 (2 mol%), CuI (4 mol%), aq. Bu4NOH (1 M), THF, 80 °C, 20 h; ii, PhCOMe (2 equiv.), RhCl(PPh3)3 (1 mol%), KOH (1 equiv.), dioxane, 80 °C, 24 h; iii, PhCOMe (2 equiv.), [IrCl(cod)2]2 or IrCl3 (2 mol%), PPh3 (4 mol%), KOH (0.2 equiv.), no solvent, 100 °C, 3 h; iv, ketone (2 equiv.), 5% Pd/C (0.5 mol%), KOH (3 equiv.), dioxane, 100 °C, 20 h; v, 49 (0.2 M in PhMe), CpRu(PPh3)2Cl (10 mol%), SPhos (10 mol%), NH4PF6 (1 equiv.), 105 °C.](/image/article/2008/NP/b612168n/b612168n-s1.gif) | ||
| Scheme 1 Reagents and conditions: i, PdCl2(PPh3)2 (2 mol%), CuI (4 mol%), aq. Bu4NOH (1 M), THF, 80 °C, 20 h; ii, PhCOMe (2 equiv.), RhCl(PPh3)3 (1 mol%), KOH (1 equiv.), dioxane, 80 °C, 24 h; iii, PhCOMe (2 equiv.), [IrCl(cod)2]2 or IrCl3 (2 mol%), PPh3 (4 mol%), KOH (0.2 equiv.), no solvent, 100 °C, 3 h; iv, ketone (2 equiv.), 5% Pd/C (0.5 mol%), KOH (3 equiv.), dioxane, 100 °C, 20 h; v, 49 (0.2 M in PhMe), CpRu(PPh3)2Cl (10 mol%), SPhos (10 mol%), NH4PF6 (1 equiv.), 105 °C. | ||
The reduction of substituted quinolines in the presence of chiral ligands is proving to be a useful method for the enantioselective synthesis of 1,2,3,4-tetrahydroquinoline alkaloids and unnatural analogues. For example, when 2-propylquinoline 50, 2-pentylquinoline 48 and the 3,4-dimethoxyphenylethyl analogue 51 were activated by N-acylation with benzyl chloroformate prior to hydrogenation under pressure in the presence of bis(cyclooctadienyl)chloroiridium(I) dimer and the ligand (S)-segphos 52, the (S)-products 53 were isolated in high yield with enantiomeric excesses (ee) of about 90% (Scheme 2).28Reduction with lithium aluminium hydride then afforded the N-methyl alkaloids (S)-(+)-54, (S)-(+)-angustureine 55 and (S)-(−)-cuspareine 56. A more unusual reduction of the quinolines 48, 51 and 57 was mediated by the dihydropyridone 58 as the hydrogen transfer reagent in the presence of the chiral Brønsted acid 59 to give the (R)-tetrahydroquinolines 60–62 in excellent yields, the enantiomeric excesses again being about 90%.29 These three compounds are also known natural products,30 although apparently not recognised as such by the authors. Subsequent N-methylation produced the alkaloids (R)-(−)-angustureine ent-55, (R)-(+)-cuspareine ent-56 and (R)-(+)-galipinine 63 in high yield and comparable enantiomeric excesses.
![Reagents and conditions: i, [IrCl(cod)2]2 (0.5 mol%), (S)-segphos 52 (1 mol%), Li2CO3 (1.2 equiv.), ClCO2Bn, THF, rt, 10 min, then H2 (600 psi), 12–15 h; ii, LiAlH4, Et2O; iii, 58 (2.4 equiv.), 59 (2 mol%), C6H6, 60 °C, 12 h; iv, CH2O, AcOH; v, NaBH4.](/image/article/2008/NP/b612168n/b612168n-s2.gif) | ||
| Scheme 2 Reagents and conditions: i, [IrCl(cod)2]2 (0.5 mol%), (S)-segphos 52 (1 mol%), Li2CO3 (1.2 equiv.), ClCO2Bn, THF, rt, 10 min, then H2 (600 psi), 12–15 h; ii, LiAlH4, Et2O; iii, 58 (2.4 equiv.), 59 (2 mol%), C6H6, 60 °C, 12 h; iv, CH2O, AcOH; v, NaBH4. | ||
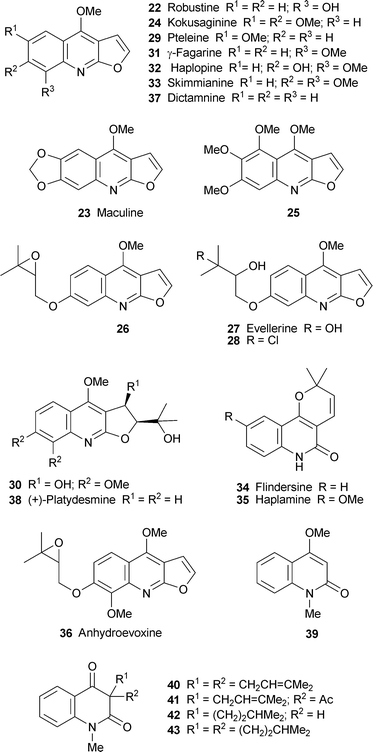
A quite different approach to (S)-(+)-angustureine 55 commenced with aniline, the methanesulfonyl derivative 64 of which was an efficient enough nucleophile for Mitsunobu reaction with (R)-(+)-oct-1-yn-3-ol 6531 (Scheme 3). The coupled product 66, with inverted configuration at the stereogenic centre, was obtained in quantitative yield. The key cyclisation step, an intramolecular hydroarylation, was accomplished with platinum(IV) chloride to give the dihydroquinoline 67 in 72% yield; much lower yields were obtained with gold(II) and mercury(II) catalysts. After hydrogenation of 67, cleavage of the methanesulfonamide yielded ent-60, methylation of which completed the synthesis of (S)-(+)-angustureine 55. The overall yield of this six-step synthesis from aniline was 55%.
 | ||
| Scheme 3 Reagents and conditions: i, DEAD, PPh3, THF, rt, 1 h; ii, PtCl4 (10 mol%), ClCH2CH2Cl, 70 °C, 2 h; iii, H2, 5% Pd/C, EtOH, rt, 3 h; iv, Red-Al, PhMe, 80 °C, 0.5 h; v, MeI, K2CO3, THF, reflux, 24 h. | ||
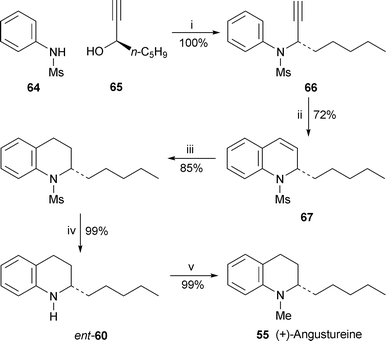
A one-step synthesis of 4-hydroxyquinolin-2(1H)-one derivatives entailed microwave heating of anilines 68 with diethyl malonate and p-toluenesulfonic acid for just a few minutes to give the products 69 in yields of 84% or higher32 (Scheme 4). The products could in turn be microwaved with dimethyl sulfate and potassium carbonate in N,N-dimethylformamide to give N,O-dimethyl derivatives, among them the alkaloids 39, 70, 71 and folimine 72, in yields of 65–70%. In another paper of interest, the O,O-dialkylation of 4-hydroxyquinolin-2(1H)-one, normally bedevilled by competing N-alkylation, was accomplished efficiently in the presence of silver(I) carbonate.33 Among the products prepared by this route was the simple alkaloid 2,4-dimethoxyquinoline 73 (montanine or monrutanine), which was obtained in 75% yield by stirring the reactants with iodomethane in benzene at room temperature for three days.
 | ||
| Scheme 4 Reagents and conditions: i, CH2(CO2Et)2, p-TsOH, microwave irradiation (320 W), 6–11 min; ii, Me2SO4, K2CO3, DMF, microwave irradiation, 3–6 min; iii, MeI (4 equiv.), Ag2CO3 (2 equiv.), C6H6, rt (dark), 3 d. | ||
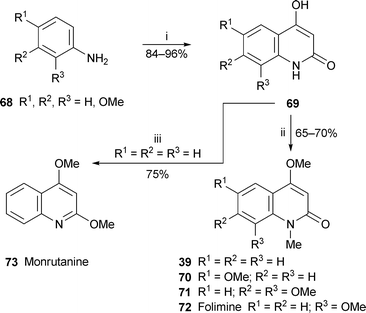
The concise synthesis of orixalone A 74 shown in Scheme 5 provides an illustration of a general new method for constructing 3-substituted 4-hydroxyquinolin-2-ones by tandem Michael–Dieckmann reaction.34 In the step of interest, conjugate addition of the anion of 2-isopropyl-1,3-dithiane to the N-arylacrylamide 75 generated an intermediate enolate ion that condensed with the strategically placed ester to produce the hydroxyquinolinone 76 in 65% yield. Methylation of the enolic hydroxy group followed by hydrolysis of the dithiane with silver nitrate completed the synthesis of the target alkaloid.
 | ||
Scheme 5
Reagents and conditions: i, COCl2 (20% in PhMe), dioxane, 0 °C to rt, 1 h; ii, NaH, DMF, 0 °C, 30 min, then MeI, rt, 18 h; iii, MeOH–DMF (1 : 15), DMAP (10 mol%), 50 °C, 18 h; iv, H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCOCl, EtNPri2, CH2Cl2, rt, 3 h; v, 2-isopropyl-1,3-dithiane, n-BuLi, THF, −78 °C to rt, 40 min; vi, Me2SO4, Cs2CO3, DMF, rt, 18 h; vii, aq. AgNO3 (3 M), EtOH, 50 °C, 2 h. CHCOCl, EtNPri2, CH2Cl2, rt, 3 h; v, 2-isopropyl-1,3-dithiane, n-BuLi, THF, −78 °C to rt, 40 min; vi, Me2SO4, Cs2CO3, DMF, rt, 18 h; vii, aq. AgNO3 (3 M), EtOH, 50 °C, 2 h. | ||
The first unassailable evidence of cis-dihydrodiol intermediate formation in the bacterial biotransformation of an alkaloid has been obtained by Boyd and co-workers, who studied the action of the B8/36 bacterial mutant strain of Sphingomonas yanoikuyae, a source of biphenyl dioxygenase, on dictamnine 37 and its synthetic 4-chloro analogue 7735 (Scheme 6). This bacterially mediated oxidation of dictamnine produced cis-diols (+)-78 and (−)-79 in yields of 20–29% and 0–3%, respectively, while a third isolable product, the quinolin-2-one (+)-80 (0–1%), resulted from oxidation of the furan ring. Similarly, the chloro analogues (+)-81 (10%), (+)-82 (30%) and (−)-83 were obtained from the biotransformation of 77. The (7S,8R) absolute configuration for 78 and 81 and (5R,6S) configuration for 79 and 82 were determined by 1H NMR analysis of the diastereomeric boronates formed from the diols with both (R)- and (S)-2-(1-methoxyethyl)phenylboronic acid. This spectroscopic method also showed that the enantiomeric excesses of the diols were better than 98%; however, the acyclic diols 80 and 83 had lower enantiomeric excesses (75% and 18%, respectively). The cis-diols 78 and 81 were then cleverly exploited as starting materials for the synthesis of several other furoquinoline alkaloids. The inspiration for their approach was a classic (1974) labelling study by Grundon and co-workers,36 in which dictamnine 37 had been revealed as the biosynthetic precursor of other ring A-oxygenated furoquinoline alkaloids, probably via an arene oxide intermediate. Boyd's team accomplished the synthesis of just such an intermediate, (+)-84, from the remarkably stable diol 78 in four steps as shown in Scheme 6. Thermal or acid-catalysed isomerisation of 84 then afforded robustine 22 as the sole regioisomer in almost quantitative yield; and methylation of robustine with diazomethane produced γ-fagarine 31. Both of these products could be further monohydroxylated in the presence of cytochrome P-450. An alternative biomimetic transformation was achieved by exposing the cis-diol 78 to whole cells of the recombinant bacterial strain E. coli nar B (a source of naphthalenecis-diol dehydrogenase), thereby producing the catechol 85 in very poor yield (<5%). Surprisingly, the chloro analogue 81 was a much better substrate for this enzymatic dehydrogenation, since it gave the catechol 86 in about 40% yield. Methylation of this product followed by replacement of the chlorine atom with methoxy on exposure to sodium methoxide completed a synthesis of skimmianine 33. Selective demethylation of skimmianine with boron tribromide was the method of choice for producing additional quantities of the catechol 85, which was then converted by standard means into the four additional alkaloids haplopine 32, 7-prenyloxy-γ-fagarine 87, the unnamed compound 88 and isohaplopine 3,3′-dimethylallyl ether 89. Finally, acid-catalysed dehydration of the 5,6-cis-diol 79 followed by methylation of the resulting phenol completed a synthesis of pteleine 29.
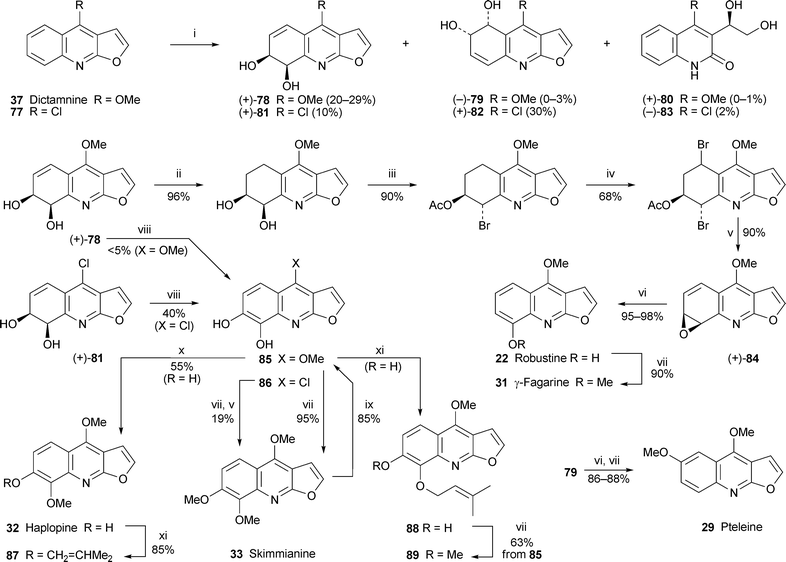 | ||
| Scheme 6
Reagents and conditions: i, Sphingomonas yanoikuyae B8/36, O2; ii, H2, Pd/C; iii, AcOCMe2COBr, MeCN; iv, NBS, CCl4; v, NaOMe; vi, TFA; vii; CH2N2; viii, Escherichia coli nar B; ix, BBr3; x, CH2N2, short time (60 s); xi, ClCH2CHCMe2, K2CO3. | ||
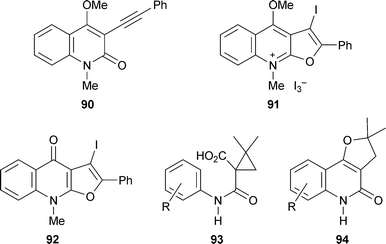
Two new methods for making furoquinolines are of interest. Cyclisation of the 3-alkynylquinolin-2-one 90 with iodine in dichloromethane afforded the quaternary salt 91 in 56% yield, while heating the product with sodium iodide in acetonitrile effected O-demethylation to give the corresponding quinolin-4-one 92 (85% from 90).37 Treatment of the cyclopropane-containing anilides 93 with polyphosphoric acid at 100–110 °C gave the angularly fused furoquinolinones 94 in moderate yield.38 Neither method has yet been applied to the synthesis of natural products, although possible applications can easily be envisaged.
1.3 Quinoline alkaloids from non-rutaceous plants
The new C27N3 pentacyclic Lycopodium alkaloid(−)-lycoperine A 95, obtained from the club moss L. hamiltonii (Lycopodiaceae), has an unprecedented structure in which two identical N-acetyloctahydroquinoline units are attached to a central 2,6-cis-disubstituted piperidine ring.39 Since the presence of amide rotamers in the parent alkaloid made interpretation of the NMR spectra difficult because of broadening of signals, the structural elucidation was performed on the bis(N-ethyl) derivative 96, obtained from 95 by reduction with lithium aluminium hydride. The relative stereochemistry of the piperidine ring was inferred after comparison of chemical shifts with those of known 2,6-disubstituted piperidine alkaloids, while that of the octahydroquinoline units was elucidated by NOESY correlations. However, since the number of 13C NMR signals for 96 showed that the compound was not symmetrical, the two octahydroquinolines had to have the same absolute configurations. Lycoperine A was found to be a reasonably good inhibitor of bovine erythrocyte acetylcholinesterase (IC50 60.9 µM).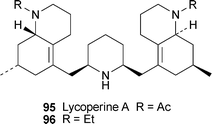
Improvements to the previously reported40 total synthesis of quinine 97 and quinidine 98 by Kobayashi and co-workers (cf. ref. 41a) entailed use of the Teoc (2-trimethylsilylethoxycarbonyl, Me3SiCH2CH2O2C) protecting group on the key piperidine intermediate 99, which obviated the need for protecting group exchanges that had lengthened the earlier route.42 Wadsworth–Emmons coupling of 99 with the quinoline-containing phosphonate 100 produced the alkene 101 in 95% yield, after which Sharpless asymmetric dihydroxylation with AD-mix-β afforded diol 102, converted in turn into the epoxide 103 as previously described (Scheme 7). The Teoc group was easily removed with caesium fluoride, the liberated secondary amine then undergoing spontaneous cyclisation to yield quinine 97 in 78% yield. A similar sequence of reactions commencing with the dihydroxylation of 101 with AD-mix-α was used in completing the synthesis of quinidine 98.
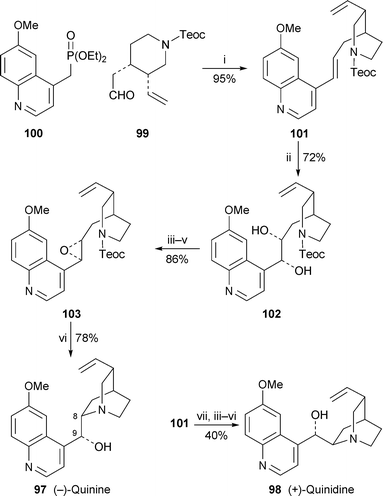 | ||
| Scheme 7 Reagents and conditions: i, NaH, THF; ii, AD-mix-β; iii, MeC(OMe)3, PPTS; iv, Me3SiCl; v, K2CO3, MeOH; vi, CsF, DMF–ButOH (9 : 1), 110 °C, 12 h; vii, AD-mix-α. | ||
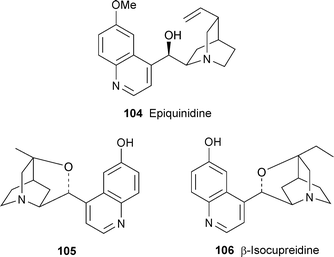
Various cyclised, rearranged and fluorinated products have been isolated from the reaction of quinidine, epiquinidine 104 and their acetates in the hydrofluoric acid–antimony pentafluoride superacidic medium.43 The chiral amine 105 has been prepared from (−)-quinine 97 by a lengthy reaction sequence.44 This product was designed as a pseudoenantiomer of β-isocupreidine 106, available in a single step from quinidine, which is a useful chiral catalyst for asymmetric Baylis–Hillman reactions of hexafluoroisopropyl acrylate. The new compound fulfilled expectations by giving products of opposite absolute configuration when used instead of 106 in Baylis–Hillman reactions.
1.4 Quinoline alkaloids from fungal and microbial sources
A review on intercellular communication in Pseudomonas aeruginosa bacterial colonies highlights the role played by 4-quinolone ‘signalling’ molecules, especially 2-heptyl-3-hydroxyquinolin-4(1H)-one 107, the major Pseudomonasquinolone signal (PQS).45 Included in the review is a synopsis of the biosynthesis of PQS that details the genes involved in the conversion of anthranilic acid into 2-heptylquinolin-4(1H)-one (HHQ) 108, and its subsequent export into adjacent cells and conversion into PQS by a putative mono-oxygenase. The mechanisms by which the 4-quinolones are formed are described, as are the processes by which ‘quorum sensing’ (cell-to-cell communication) takes place. It now seems certain that PQS is produced during infection by P. aeruginosa, and that it is a key compound in determining the virulence of the pathogen. Finally, there is some interesting speculation on the production of PQS by other bacteria, and the possibility of ‘cross-talk’ between bacteria belonging to different species or even genera.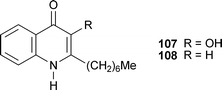
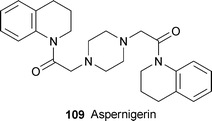
An extract from a culture of Aspergillus niger strain IFB-E003, an endophytic fungus isolated from healthy leaves of the grass Cynodon dactylon, yielded the novel cytotoxic alkaloid aspernigerin 109, which has an unprecedented structure containing two identical tetrahydroquinoline units attached by oxoethyl spacers to the nitrogen atoms of a central piperazine ring.46 The symmetrical structure was deduced principally from NMR spectroscopic data, which also suggested that the carbonyl groups of the amides were preferentially orientated towards the phenyl rings in solution. However, X-ray crystallographic analysis revealed that, in the solid state, the piperazine ring adopted a chair conformation, with the amide carbonyl groups pointing in opposite directions and orientated towards the reduced rings of the tetrahydroquinoline units. The structure was further confirmed by a straightforward synthesis from piperazine, bromoacetic acid and 1,2,3,4-tetrahydroquinoline (28% overall yield). Aspernigerin showed significant antitumour activity when tested against human nasopharyngeal epidermoid (KB), cervical carcinoma (HeLa) and colorectal carcinoma (SW1116) cell lines, giving IC50 values of 22, 46 and 35 µM, respectively. These values suggest that 109 might well be an interesting lead compound for the development of new anticancer drugs, especially in view of the ease with which analogues could be made.
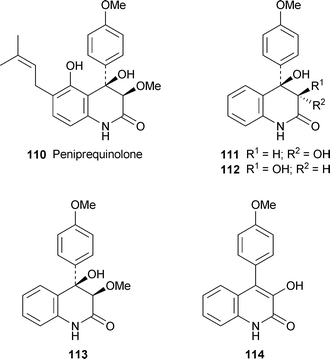
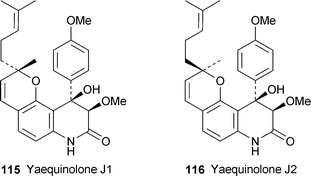
A growing family of highly oxygenated 4-aryl-3,4-dihydroquinolin-2-ones found in various Penicillium species and exemplified by peniprequinolone 110 has recently been augmented by the new diastereomeric compounds (−)-111 and (−)-112, which were isolated from cultures of the marine species P. janczewskii strain H-TW5/869 together with 110 and another known compound, (−)-113.47 The relative stereochemistries of the new metabolites were established by means of quantitative NOE data supported by conformational analysis by standard calculational methods. In addition, acid-induced dehydration of both 111 and 112 yielded the same product, the 3-hydroxy-2(1H)-quinolinone 114. The absolute configurations of the four compounds were not determined. Compounds 110–112, but not 113, showed moderate cytotoxicity towards eight human tumour cell lines, with 112 being especially active against the human ovarian adenocarcinoma (SKOV-3) cell line. Two related new alkaloids, (−)-yaequinolone J1 115 and (+)-yaequinolone J2 116, were isolated from a Japanese soil sample of Penicillium sp. FKI-2140.48 The unusual monoterpenoid structures were deduced from NMR spectroscopic data, with NOE correlations again proving key in establishing the relative stereochemistries. Both yaequinolones showed insecticidal activity in the brine shrimp assay (MIC 6.25 µg ml−1).
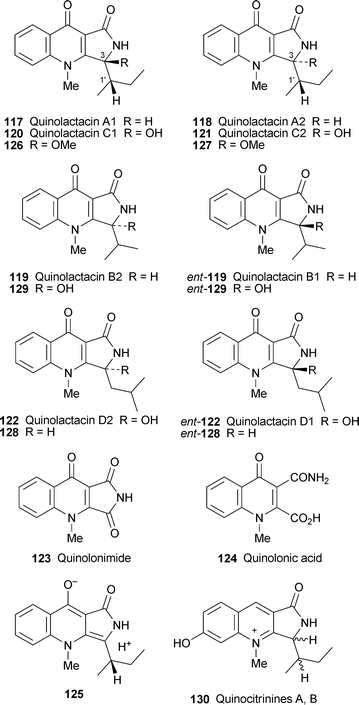
Another family of Penicillium metabolites whose numbers are growing are the quinolactacins, isolated from strains of P. citrinum. The absolute structures of quinolactacins A1 117, A2 118 and B2 (formerly quinolactacin B) 119 have recently been established by total synthesis49,50 (cf. ref. 41b,c). In the latest significant contribution to the literature, Capon and co-workers have identified several more members of the family from P. citrinum strain MST-F10130, obtained from an Australian soil sample.51 The new compounds include quinolactacin B1 ent-119 (as a racemate with its enantiomer, quinolactacin B2); the diastereomeric quinolactacins C1 120 and C2 121; and the enantiomeric quinolactacins D2 122 and D1 ent-122 (obtained as a racemic mixture as well). Also isolated were the new compounds quinolonimide 123 and quinolonic acid 124. The substituents at C-3 in the quinolactacins strongly imply the participation of the amino acidsL-isoleucine (for the A and C series), L-valine (for the B series) and L-leucine (for the D series) in the biosynthesis of the metabolites. Furthermore, on handling and storage, quinolactacins A1 and A2 interconverted by slow epimerisation at C-3, probably via an intermediate such as 125; and with time each accumulated small amounts of the oxidised products quinolactacins C1 and C2. Deliberate exposure of methanolic solutions of 117 and 118 to neutral, basic and acidic conditions led to observable interconversion as well as decomposition to the C series of compounds; in addition, heating the precursors with a catalytic amount of trifluoroacetic acid in methanol also yielded quinolonimide and two additional unidentified products, perhaps the methanolysis products 126 and 127. Quinolonic acid was also shown to be a hydrolysis product from quinolonimide. These results implied that the quinolactactin B1/B2 racemates probably resulted from handling and storage of the natural B2 derived from L-valine, while the D series might well be artefacts formed by oxidation of an as yet undetected natural product 128 and its artefactual enantiomer ent-128. In support of this hypothesis, controlled decomposition of quinolactacins B gave oxidised products with the putative structures 129 and ent-129. Whereas none of the quinolactacins displayed antimicrobial properties, quinolactacins C1, C2 and the D1/D2 racemate were significantly cytotoxic in the NS-1 bioassay (LD99 40, 40 and 7.5 µg ml−1, respectively). Related to the quinolactacins are the diastereomeric quinocitrinines A and B 130, the accumulation of which in P. citrinum strain VKM FW-800 cultures has been found to proceed simultaneously with culture growth, and to be stimulated in the presence of zinc ions.52 The addition of isoleucine during inoculation enhanced biomass accumulation, while tryptophan stimulated the biosynthesis of quinocitrinins and both leucine and isoleucine inhibited the biosynthesis when added during the stationary growth phase.53
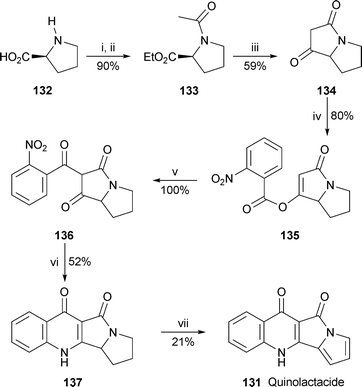
Yet another P. citrinum metabolite, quinolactacide 131, has been synthesised from L-proline 132 for biological evaluation54 (Scheme 8). Key steps included the Dieckmann-like cyclisation of N-acetylproline ethyl ester 133 to form the pyrrolizidinedione 134, esterification of the latter with 2-nitrobenzoyl chloride to give the enol ester 135, and efficient acyl migration catalysed by acetone cyanohydrin to produce the trione 136. Hydrogenation of the nitro group resulted in spontaneous cyclisation to give the tetracyclic product 137 in a modest yield of 52%. Disappointingly, the dehydrogenation of 137, eventually achieved with manganese dioxide, gave quinolactacide 131 in a maximum yield of 21%. However, the product proved to be an effective insecticide against the green peach aphid and diamondback moth (100% and 42% mortality, respectively, at 500 ppm), although it was inactive against three other insect pests and one mite.
 | ||
| Scheme 8 Reagents and conditions: i, SOCl2, EtOH, reflux, 4 h, then 20 °C, 12 h; ii, AcCl, py, THF, 0 °C, 1 h, then 20 °C, 2 h; iii, KOBut, THF–PhMe (1 : 1), reflux, 12 h; iv, 2-O2NC6H4COCl, NEt3, THF, 0 °C, 1 h, then 20 °C, 2 h; v, Me2C(OH)CN, NEt3, MeCN, 20 °C, 2 h; vi, H2 (1 atm), 10% Pd/C, MeOH, rt, 3 d; vii, MnO2, DMF–CHCl3 (1 : 3), reflux, 24 h. | ||
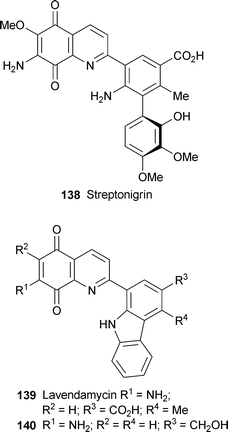
The well-known antitumour antibioticstreptonigrin 138 has been isolated from Kitasataspora sp. MJM383, a rare actinomycete strain isolated from a Chinese soil sample.55 Its close relative lavendamycin 139 provided the inspiration for a clutch of synthetic analogues that were subsequently evaluated as NADP(H):quinone oxidoreductase (NQO1)-directed antitumour agents.56 The best substrate, 140, exhibited the highest rate of reduction when metabolised by NQO1, and also showed the highest selective toxicity towards the NQO1-rich human colon adenocarcinoma (BE-NQ) cell line. Molecular docking studies supported a model in which substrate 140 participated in efficient hydrogen bonding to key residues of the active site, thereby promoting receptivity to hydride ion transfer.
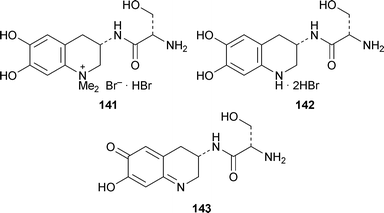
The tetrahydroquinoline chromophore 141 of the anachelins, complex siderophores isolated from the cyanobacterium Anabaena cylindrica, has been synthesis by Bethuel and Gademann57 in continuation of their investigations into the chemistry of these unique compounds (cf. ref. 41d). Wondering about the function of the unusual quaternary nitrogen in the natural product, they also made the bis-nor analogue 142 for comparative studies. They found that this catechol was rapidly oxidised in air at a physiologically relevant pH of 6.5, and more slowly at pH 4, to what is presumably an ortho-quinone or its tautomer such as 143. Almost instantaneous oxidation was observed upon addition of ferric salts, and oxidation could also be enzymatically induced with catechol oxidases. In stark contrast, the quaternary chromophore 141 was completely stable under all oxidative conditions, and in addition formed complexes characterisable by UV-visible spectroscopy when treated with ferric salts. The interesting conclusion is that the permanent quaternary ammonium group in the natural products has a bioprotective function, allowing them to complex with iron(III) without undergoing oxidative destruction.
The biosynthetic gene cluster responsible for the production of the complex thiodepsipeptide thiocoraline 144 in the actinomycete Micromonospora sp. has been isolated and sequenced, and the functions of many of these genes have been deduced.58 When a region of the relevant DNA embracing these genes was expressed heterologously in Streptomyces albus and S. lividans, these microorganisms were indeed found to produce thiocoraline. Of particular interest for this review was the identification of the genes involved in the biosynthesis of the unusual starter unit, 3-hydroxyquinaldic acid 145; the putative enzymes in the biosynthetic conversion of L-tryptophan 146 into 145 are shown in Scheme 9. Several genes in the sequential growth of the peptide chain from the 3-hydroxyquinaldic acid starter were also identified.
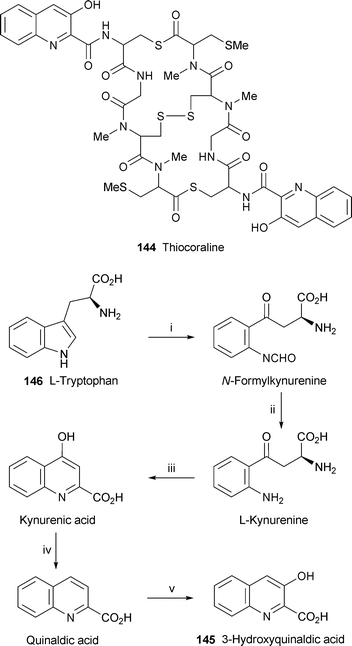 | ||
| Scheme 9 Enzymes in the putative biosynthesis of 3-hydroxyquinaldic acid : i, Trp-2,3-dioxygenase; ii, kynurenine formamidase; iii, kynurenine aminotransferase; iv, oxidoreductase; v, cytochrome P450. | ||
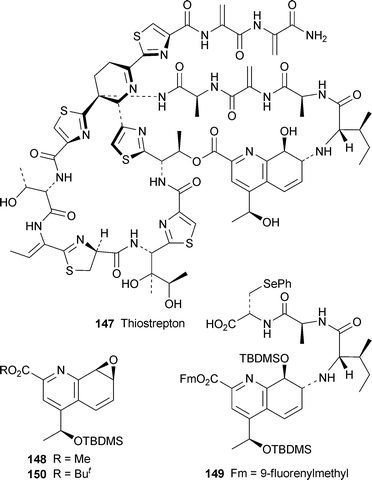
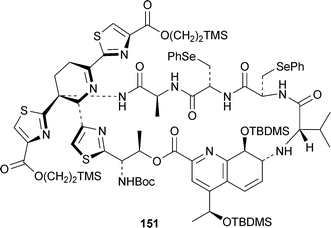
One of the most impressive achievements of the past few years has been the total synthesis of the dauntingly complex antitumour antibioticthiostrepton 147 by Nicolaou and co-workers. This work, communicated in a series of recent articles (cf. ref. 41e,f), has now been published in full in two articles that describe the construction of the building blocks (including the quinaldic acid derivatives 148 and 149)59 and their assembly to give the target,60 as well as several of the roadblocks that were encountered in the course of the extensive investigation. Also not to be overlooked are the continuing investigations of Hashimoto and co-workers (cf. ref. 41e), who have recently communicated syntheses of the dihydroquinoline building block 150,61 found not only in thiostrepton but also in siomycins A and C and thiopeptin A1b. They have also reported a synthesis of 151, the cyclic core of the siomycins.62
1.5 Quinoline alkaloids from animals
The known alkaloid jineol 152 and the novel natural product 153 (or, more probably, its quinolin-2-one tautomer) were isolated by bioassay-guided fractionation from the centipede Scolopendra subspinipes mutilans, used in traditional Chinese and Korean medicine for a variety of ailments.63 Both compounds were assayed for antioxidant activity by means of various tests involving low-density lipoprotein oxidation, the oxidised products of which are implicated in the development of atherosclerosis. While both showed antioxidant activities in the micromolar concentration range, jineol proved to be more active by more than an order of magnitude. Their mode of action appears to entail the trapping of free radicals, since both alkaloids functioned as scavengers for the 1,1-diphenyl-2-picrylhydrazyl radical. In addition, jineol formed complexes with copper(II), which has implications for suppressing the copper ion-induced formation of peroxy radicals in lipids.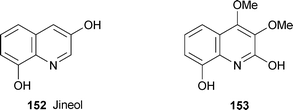
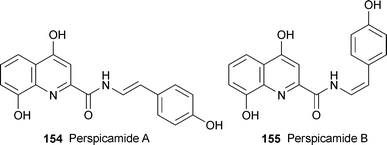
Extracts of freeze-dried samples of the Australian ascidian (sea squirt) Botrylloides perspicuum have yielded two new quinoline-2-carboxylic acid derivatives, perspicamide A 154 and its geometric isomer perspicamide B 155.64 The structures were determined from analysis of 2D NMR spectra. The main difference in the 1H NMR spectra of the isomeric compounds were the sizes of the coupling constant for the alkene protons (15 Hz for the trans isomer, and 9.4 Hz for the cis isomer), and the significant upfield shifts for the alkene and NH protons in 155. Perspicamide B was unstable on exposure to light, undergoing isomerisation to the trans isomer 154.
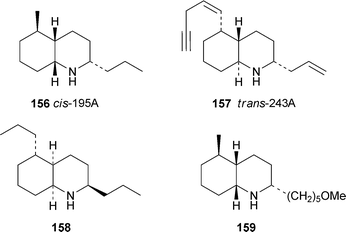
About 50 decahydroquinoline alkaloids (including stereoisomers) are listed in a comprehensive review detailing the more than 800 alkaloids detected in the skins of amphibians.65 These alkaloids are common in neotropical dendrobatid frogs; but, apart from the widely distributed cis-decahydroquinoline 195A (−)-156, occur rarely in mantellid frogs and bufonid toads. The alkaloids appear to be sequestered from myrmicine ants, a principal food source for the anurans. It has recently been shown that the presence of decahydroquinolines in amphibian skin might serve an antimicrobial function; trans-decahydroquinoline 243A (−)-157 and 158 (the synthetic epimer of cis-decahydroquinoline 223F) inhibited the growth of the Gram-positive bacterium Bacillus subtilis, while 158 and the racemic compound 159 were effective against the fungus Candida albicans.66
The lepadins, a group of 2,3,5-trisubstituted cis-fused decahydroquinoline alkaloids isolated from ascidians, remain attractive but challenging targets for total synthesis because of the variety of stereochemical arrangements in the natural products (e.g., lepadins B, D and G, 160–162). Mena and Bonjoch have now reported model studies in which the stereochemical consequences of a new synthetic approach to the alkaloids’ bicyclic core were investigated67 (Scheme 10). Lithium–halogen exchange of the protected 2-bromocyclohexenone 163 followed by treatment of the organometallic intermediate with either the (R)- or the (S)-epoxide 164 and subsequent functional group interconversions yielded the enones 165. These intermediates could in turn be transformed into the 3-butyl analogues 166. The pivotal transformation entailed hydrogenation of 165 and 166 over activated Pearlman's catalyst to give both cis- and trans-fused decahydroquinolines 167 and 168, all with the (8aR) absolute configuration. Unfortunately, the desired cis-fused products 167 were generally unfavoured. Since the actual sequence of reactions in the tandem hydrogenation process (reduction of the double bond, N-debenzylation and intramolecular reductive amination) was not established, the stereochemical outcome could not be rationalised; but it is intriguing that catalytic hydrogenation of the double bond in the precursors 166 (R = Bu) is able to produce a trans-relationship of the hydrogen atoms at C-4a and C-5.
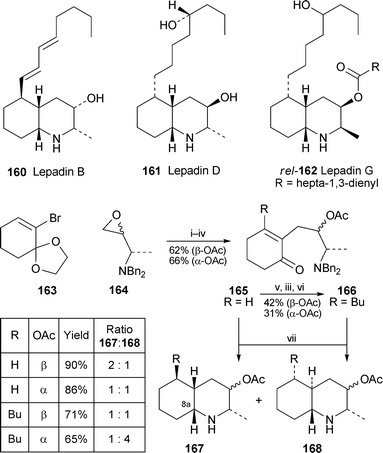 | ||
| Scheme 10 Reagents and conditions: i, 163 + n-BuLi, THF, −78 °C, 90 min; ii, add (R)- or (S)-164, BF3·Et2O, −78 °C, 2 h; iii, Ac2O, DMAP (cat.), py, rt, overnight; iv, aq. HCl–THF (1 : 1), rt, 1 h; v, n-BuLi, THF, −78 °C, 4 h, then aq. NH4Cl; vi, PCC, SiO2, CH2Cl2, rt, overnight; vii, H2, Pd(OH)2, EtOH, rt, overnight. | ||
2 Quinazoline alkaloids
2.1 Occurrence, characterisation and biological activity
The medicinally important alkaloid vasicine (peganine) 169 has been obtained in vitro for the first time from leaf- and petiole-derived callus cultures of Adhatoda zeylanica.68Chloride and oxalate salts of the related alkaloid deoxypeganine 170, obtained from Peganum harmala, have been characterised by X-ray crystallography.69 Other known quinazoline alkaloids isolated from new sources include peganol 171 from Nitraria schoberi;70rutaecarpine 172 from Phellodendron japonicum;71 and rutaecarpine, 1-hydroxyrutaecarpine 173 and evodiamine 174 from Spiranthera odoratissima.14 A far more unusual plant source of quinazoline alkaloids is the Asian medicinal plant Aconitum pseudo-laeve var. erectum (Ranunculaceae), which belongs to a genus notorious for producting highly toxic diterpene and norditerpene alkaloids. A methanolic extract from the roots of the plant yielded two norditerpene esters of 2-(2-methyl-4-oxoquinazolin-3(4H)-yl)benzoic acid, viz., the (+)-lycoctonine ester 175 and the (−)-14-O-acetyl-8-O-methylcammaconine ester 176.72 Also obtained was the simple methyl ester 177, isolated for the first time from a natural source. Could this compound perhaps be an artefact of the extraction procedure? The structures of all three compounds were elucidated by spectroscopic methods, one- and two-dimensional NMR spectroscopic techniques providing especially important information.Members of the fungal genus Aspergillus produce numerous piperazino[2,1-b]quinazoline derivatives formally derived from anthranilic acid and various amino acids. (−)-Janoxepin 178, isolated from a culture of A. janus, is an interesting new relative of these alkaloids in which the benzene ring has been oxidised to an oxepine.73 Full NMR spectroscopic characterisation included NOE experiments to establish the (Z)-configuration of the double bond. The absolute configuration at C-1 was determined to be (R)- by Marfey's method, suggesting that the isobutyl group is derived from the rare D-leucine instead of the common L-amino acid; but the structural diagram given in the article shows the (S)-configuration. Although janoxepin showed no antifungal or antibacterial activity, it had some antiplasmodial activity against the malaria parasite Plasmodium falciparum (IC50 approximately 28 mg ml−1, limited by precipitation in the test medium).
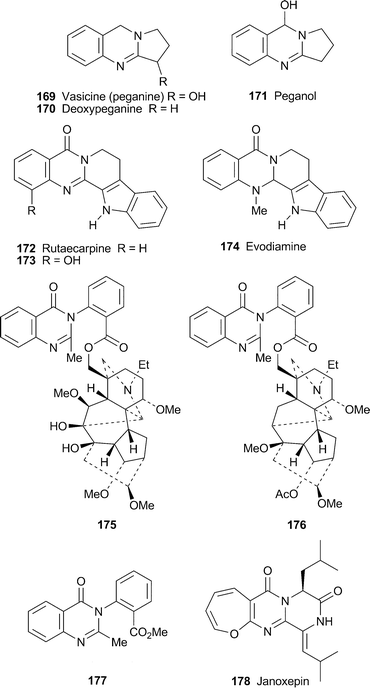
The potent antimalarial alkaloid febrifugine 179 is not used clinically because of unacceptable liver toxicity, ascribed to the ease of oxidation by cytochrome P-450 enzymes to give arene oxide 180, which can form covalent adducts with essential biomolecules. Some recent efforts to find less toxic analogues by Zhu et al. have concentrated on varying the quinazoline substituent to reduce the possibility of arene oxide formation.74 Out of a suite of 12 synthetic analogues, compounds 181–183 showed similar antimalarial activity to the parent alkaloid when tested against chloroquine-sensitive and chloroquine-resistant strains of the malaria parasite Plasmodium falciparum, but they were about a hundred times less cytoxic towards rat hepatocytes. Compounds 184 and 185, possessing electron-withdrawing substituents at the oxidisable position, also showed a similar diminution of cytotoxicity, but in addition were even better antimalarials than febrifugine itself. For comparison, four compounds bearing electron-donating methyl or methoxy substituents at C-7 or C-8 retained antimalarial activity comparable to that of febrifugine, but were about ten times as toxic.
Evaluation of the anti-inflammatory activity of rutaecarpine 172, evodiamine 174 and goshuyuamide II 186, the principal alkaloids of Evodia rutaecarpa fruits, showed that the first two were powerful inhibitors of prostaglandin E2 synthesis.75 While evodiamine also inhibited the induction of cyclooxygenase-2 (COX-2) and activation of NF-κB from various cancer cell lines, goshuyuamide II inhibited 5-lipoxygenase (5-LOX), resulting in the reduced synthesis of leukotrienes. None of the alkaloids affected the production of nitric oxide. The very different mechanisms of action of the three structurally similar alkaloids is surprising, and the phenomenon warrants further study. The oxidation of rutaecarpine by cytochrome P450 and the identification of the monohydroxylated metabolites continues to receive attention; and some of the human liver cytochrome P450 enzymes implicated in the oxidative metabolism of rutaecarpine have been characterised.76,77
2.2 Synthesis and other chemical studies
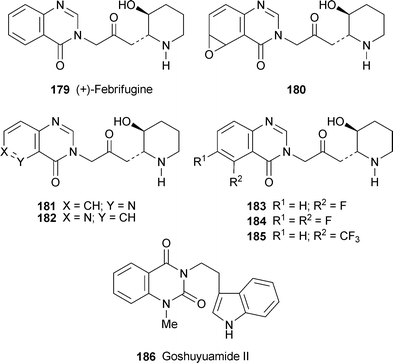
Two reviews devoted to the synthesis of quinazoline systems are of interest. The first, although not dealing specifically with alkaloids, presents a useful survey of methods for preparing quinazolines and quinazolinones monosubstituted or disubstituted in the heterocyclic ring.78 The second (by Eguchi, one of the major players in the field of quinazoline alkaloid synthesis) includes a survey of synthetic methodology for preparing quinazolines as well as a comprehensive review of recent total syntheses of quinazoline alkaloids belonging to all the major classes.79A previous synthesis of (+)-febrifugine by Takeuchi et al.80 (cf. ref. 41g) suffered from uncontrollable trans/cisisomerisation of the 2,3-disubstituted piperidine ring in the final step. This problem has now been solved by taking advantage of a trans-specific intramolecular boron trifluoride-catalysed conjugate addition of the ω-amido enone 187, prepared from the piperidine-2,3-diol 188 by a Wittig reaction81 (Scheme 11). The conjugate addition gave the adduct 189 in 75% yield when left for ten minutes at room temperature, but increasing amounts of the furan 190 and an acyclic 1,4-diketone were formed as the time and temperature were increased. As described in the earlier report, the synthesis was completed by reaction of the α-bromo ketone 191 derived from 189 with quinazolin-4(3H)-one 192 and removal of the Cbz protecting group to give racemic febrifugine (±)-179.
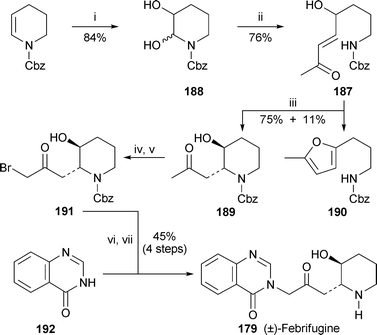 | ||
| Scheme 11
Reagents and conditions: i, oxone, K2CO3, Me2CO, H2O, rt, 2 h; ii, MeCOCHPPh3, MeCN, reflux, 1 h; iii, BF3·OEt2 (0.5 equiv.), MeCN, rt, 10 min; iv, 189 + TMSOTf, EtNPri2, CH2Cl2, rt, 20 min; v, add NBS, rt, 2 h; vi, add 192, rt, 4.5 h; vii, H2, 20% Pd(OH)2/C, MeOH–THF, rt, 3.5 h. | ||
In 2004, Honda and co-workers communicated a synthesis of (+)-febrifugine by a route in which the reductive deamination of an appropriately substituted proline ester with samarium diiodide and recyclisation of the intermediate amino ester produced the target's piperidine ring82 (cf. ref. 41h). Full details of this work have now been published in a paper that also includes a formal synthesis of (−)-isofebrifugine 19383 (Scheme 12). Commercially available (4R)-hydroxy-L-proline was converted into the protected ester derivative 194, which was oxidised to the lactam 195 with ruthenium tetroxide. Mild reduction to the aminal was followed by Horner–Emmons reaction with diethyl (N-methoxy-N-methylcarbamoylmethyl)phosphonate, the alkene intermediate undergoing intramolecular conjugate addition to give the 4,5-cis-disubstituted proline ester 196 (60%), accompanied by 5.5% of the 4,5-trans-isomer. Reduction of the Weinreb amide to the aldehyde and Wittig methylenation gave 197. Removal of the Boc protecting group with zinc bromide preceded the reductive deamination–recyclisation procedure, which afforded the piperidinone (+)-198 in 71% yield from 197. Since Takeuchi et al. have previously reported the conversion of ent-198 into (+)-isofebrifugine84 (cf.ref. 41i), the present route constitutes a formal synthesis of (−)-isofebrifugine 193.
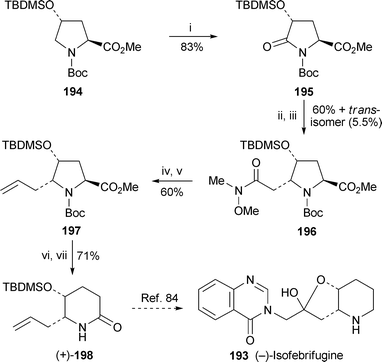 | ||
| Scheme 12
Reagents and conditions: i, RuO2·xH2O (cat.), NaIO4, H2O–EtOAc, rt, overnight; ii, LiBEt3H, THF, −78 °C, 30 min, then aq. H2O2 (30%), aq. NaHCO3, 0 °C, 20 min; iii, (EtO)2P(O)CH2CON(Me)OMe, NaH, THF, rt, overnight; iv, DIBAL, THF, −78 °C, 30 min; v, Ph3PCH2, THF, 0 °C to rt, then overnight; vi, ZnBr2, CH2Cl2, rt, overnight; vii, SmI2, THF–HMPA, MeOH, 0 °C to rt, then rt, 2 h. | ||
A simple synthesis of (±)-deoxyfebrifugine 199 (Scheme 13) entailed Eschenmoser coupling of the readily prepared quinazoline-containing bromoketone 200 with piperidine-2-thione 201 to produce the (Z)-enaminone 202 containing all of the target's skeletal elements.85Hydrogenation of the alkene in acidic methanol over Adams catalyst gave (±)-deoxyfebrifugine 199 in 58% yield. The versatility of this route was demonstrated by condensing 200 with a range of other thiolactams to produced enaminones 203 and 204 with substituents on the piperidine ring or with smaller or larger rings replacing piperidine. Reduction of the former produced N-substituted deoxyfebrifugine analogues 205.
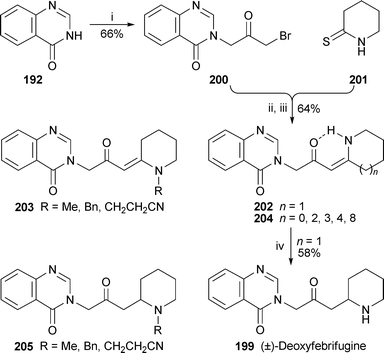 | ||
| Scheme 13 Reagents and conditions: i, BrCH2COCH2Br (3 equiv.), K2CO3, DMF, rt, 2 h; ii, THF, rt, 48 h; iii, N-methylpiperidine (2.5 equiv.), PPh3 (2.5 equiv.), MeCN, rt, 18 h; iv, H2 (3 atm), PtO2, MeOH–aq. HCl (8.5 M), rt, 6 h, then aq. K2CO3. | ||
The biological activity of many pyrrolo[2,1-b]quinazoline alkaloids makes them and their analogues popular targets for synthesis. In an alternative to the ‘Eguchi protocol’ (Staudinger reaction of an aryl azide with tributylphosphine followed by intramolecular aza-Wittig reaction with an amide or lactam), reductive cyclisation of the azides 206 with boron trifluoride etherate and ethanethiol yielded deoxyvasicinone 207 and derivatives 208 and 209 directly in yields of 96% and above86 (Scheme 14). A different approach by Liu et al. exploited the power of microwave heating in promoting one-pot domino reactions between anthranilic acids 210 and various N-Boc protected ω-amino acids 211.87 For example, microwave-enhanced heating of anthranilic acid itself with 211 (n = 1) in the presence of triethyl phosphite in pyridine at 200 °C for ten minutes produced deoxyvasicinone 207 in 89% yield. Similar reactions with appropriate reactants gave mackinazolinone 212 and 8-hydroxydeoxyvasicinone 213 in yields of 86% and 72%, respectively. In a related one-pot three-component coupling, the reaction of anthranilic acid 210 (R = H) with 211 (n = 1) as described above was followed by addition of the substituted benzaldehyde 214 and a further period of microwave heating at 230 °C to give a 79% yield of isaindigotone 215. These simple procedures permitted the preparation of many analogues of 207, 212 and 215, including the quinazolinone analogue 216 of linarinic acid (see below) and the mackinazolinone–isaindigotone hybrids 217, all of which were tested for cytotoxicity against six cancer cell lines. Although isaindigotone proved to be inactive, the hybrid structures 217 (R = OH, OMe; Ar = 3,5-dimethoxy-4-hydroxyphenyl) were surprisingly effective, prompting a follow-up investigation in which a library of 44 analogues was prepared and purified by automated procedures.88 Exhaustive testing of representative compounds revealed anticancer properties that were especially pronounced with the pendent 3,5-dimethoxy-4-hydroxyphenyl substituent as well as substituents at C-4. The active compounds were found to act as antimitotic agents by destabilising tubulin polymerisation in a dose-dependent fashion (IC50 0.3 to 11 µM). Mackinazolinone 212 can thus be regarded as a ‘privileged substructure’ for the synthesis of new drug candidates.
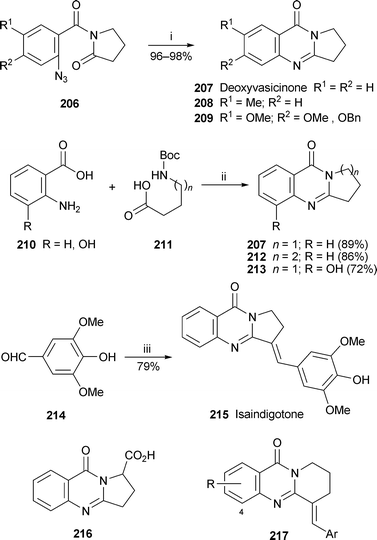 | ||
| Scheme 14 Reagents and conditions: i, BF3·OEt2 (10 equiv.), EtSH (20 equiv.), CH2Cl2, rt, 1.5–2 h; ii, P(OPh)3, py, microwave (200 °C), 10 min; iii, 207 (prepared in situ), P(OPh)3, py, microwave (230 °C), 12 min. | ||
When the pyrrolo[2,1-b]quinazoline alkaloid (−)-linarinic acid was first described, its absolute configuration could not be determined despite the availability of an X-ray crystal structure.89 The (1S) configuration shown in 218 has now been determined by asymmetric total synthesis supported by theoretical calculations90 (Scheme 15). Reductive amination of 2-nitrobenzaldehyde 219 with L-glutamic acid 220 afforded the product 221, which underwent cyclisation to the lactam 222 in boiling ethanol. Reduction of the nitro group with hydrazine and ferric chloride produced the target alkaloid, [α]18D −290 (c 0.01, MeOH), in 38% overall yield based on 219. The sign of the specific rotation agreed with that of the natural product ([α]18D −217). In addition, ab initio calculations by Hartree–Fock methods and density functional theory yielded a preferred conformation almost identical to that found by X-ray crystallography; the specific rotation for this low-energy conformer of the (S)-enantiomer was calculated as −263.
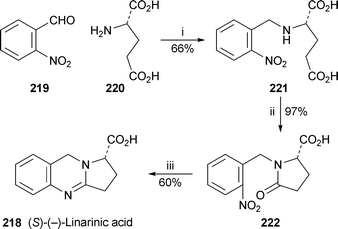 | ||
| Scheme 15 Reagents and conditions: i, 220 + aq. NaOH (2 M), then 219, EtOH, NaBH4, rt, 90 min and repeat, 45 min, then conc. HCl to pH 2–3; ii, EtOH, reflux, 3 h; iii, H2NNH2·H2O, FeCl3 (cat.), activated C, EtOH, reflux, 64 h. | ||
An important methodological study of the rhodium(I)-mediated coupling of unactivated alkenes to the C-2 site of 3,4-dihydroquinazolines by Wiedemann, Ellman and Bergman has found application in the total synthesis of (±)-vasicoline 22391 (Scheme 16). The parent heterocycle 224 was regioselectively alkylated on N-3 with 2-chlorocinnamyl bromide 225 to give 226 in 65% yield. An intramolecular variant of the coupling process was then induced with a catalytic quantity of [RhCl(cyclooctene)2]2 in the presence of the ligand Cy-[3.3.1]-Phoban 227 in boiling 1,2-dichlorobenzene to give the tricyclic product 228 in a fair yield of 60%. The failure of the corresponding 2-(N,N-dimethylamino)cinnamyl analogue of 226 to undergo cyclisation to give vasicoline directly was ascribed to trapping of the rhodium catalyst by the amine group, which prevented catalyst turnover. The transformation of the chlorine substituent in 228 to the requisite amine was not trivial; after considerable experimentation, Goldberg-type amination was accomplished with trifluoroacetamide and copper(I) iodide in the presence of trans-1,2-methylaminocyclohexane as ligand to give the aniline 229 in a modest yield of 37%. The final reductive methylation of the primary amine with formaldehyde was also tricky because of competing reactions with the heterocyclic amidine; it was eventually achieved under very mild conditions with KHFe(CO)4 under an atmosphere of carbon monoxide. The overall yield of vasicoline 223 was 10% based on commercially available precursors of 224 and 225.
![Reagents and conditions: i, K2CO3, DMF, 0 °C, 1 h, then 25 °C, overnight; ii, [RhCl(coe)2]2 (5 mol%), ligand 227 (15 mol%), 1,2-Cl2C6H4, 150 °C (sealed tube), 10 h; iii, F3CCONH2 (2 equiv.), K2CO3, CuI (25 mol%), trans-1,2-(NHMe)2cyclohexane (50 mol%), dioxane, 140 °C (sealed tube), 60 h; iv, add HCl (6 M), 75 °C, 9 h; v, KHFe(CO4) (prepared in situ), aq. H2CO (37%), EtOH, CO (5 atm), 105 °C, 48 h.](/image/article/2008/NP/b612168n/b612168n-s16.gif) | ||
| Scheme 16 Reagents and conditions: i, K2CO3, DMF, 0 °C, 1 h, then 25 °C, overnight; ii, [RhCl(coe)2]2 (5 mol%), ligand 227 (15 mol%), 1,2-Cl2C6H4, 150 °C (sealed tube), 10 h; iii, F3CCONH2 (2 equiv.), K2CO3, CuI (25 mol%), trans-1,2-(NHMe)2cyclohexane (50 mol%), dioxane, 140 °C (sealed tube), 60 h; iv, add HCl (6 M), 75 °C, 9 h; v, KHFe(CO4) (prepared in situ), aq. H2CO (37%), EtOH, CO (5 atm), 105 °C, 48 h. | ||
When the iminothioether 230, prepared in quantitative yield from the tetrahydro-β-carboline-1-thione 231, was heated at reflux with anthranilic acids 232 in dry acetic acid for an extended period, not only did the expected amidine formation take place, but tandem cyclodehydration resulted in formation of the quinazolin-4-one system, thereby completing one-pot syntheses of the bioactive alkaloids rutaecarpine 172, euxylophorine A 233 and the chloro analogue 234 in yields of 79–85%92 (Scheme 17). N-Methylrutaecarpine could be obtained in a similar manner from the indolyl N-methyl analogue of 230. However, when methyl anthranilate 235 was used instead of the acid, the reaction stopped at the amidine stage 236, necessitating further treatment with sodium hydride to effect cyclisation to rutaecarpine. The latter method proved to be suitable for the synthesis of rutaecarpine analogues 237 in which ring A was replaced by pyrrole, furan and thiophene units. In addition, the reaction of methyl anthranilate with 2-methylthio-Δ1-pyrroline 238 completed an efficient synthesis of deoxyvasicinone 207 under comparable conditions. A different approach to ring E-substituted derivatives of rutaecarpine entailed construction of the carboline section from the tricyclic ketone 239 and substituted phenylhydrazines by a Fischer indole synthesis.93 When the products, which included fluoro, chloro, bromo, methyl and methanesulfonyl analogues, were tested for inhibitory activity towards cyclooxygenases-1 and -2 (COX-1 and COX-2), those with 10-bromo and 10-methanesulfonyl substituents showed comparable activity to rutaecarpine itself against COX-2 (IC50 0.27, 0.35 and 0.28 µM, respectively).
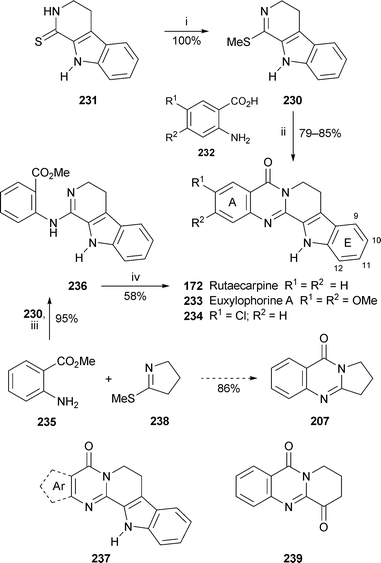 | ||
| Scheme 17 Reagents and conditions: i, MeI, aq. NaOH (2%), EtOH, rt, 24 h; ii, AcOH, reflux, 24 h; iii, 230 + 235, AcOH, reflux, 4 h; iv, NaH (3 equiv.), DMF, 0 °C to rt, 12 h. | ||
The considerable interest elicited by luotonin A 240 since its isolation from Peganum nigellastrum a decade ago94 is a result of its outstanding cytotoxicity towards murine leukemia P-388 cells at low concentrations and, to a lesser extent, its ability to inhibit topoisomerases I and II. A timely review by its discoverers covers the isolation, structure determination, total synthesis and biological activity of this topical alkaloid and some of its derivatives, including the interesting hybrid 14-azacamptothecin 241, which combines structural elements from 240 and the potent topoisomerase I inhibitorcamptothecin 242.95 In the interim, Curran and co-workers have reported yet another versatile synthesis of 240 in which 2-bromo-3-propargylquinazolin-4(3H)-one 243, prepared as shown in Scheme 18, was condensed with phenyl isocyanide in the presence of hexamethyldistannane under photochemical conditions to give the target in 47% yield.96 A small library of analogues 244 was also prepared by varying the propargylating reagent and the aryl isocyanide, but only 7-ethylluotonin (244, R1 = H; R2 = Et) was somewhat better as an inhibitor of topoisomerase I than its weakly active parent. Also of interest is a general synthesis of luotonin A analogues 245 and 246 by Jahng and co-workers, in which the products were readily formed by Friedländer condensation of tricyclic precursors 247 with 2-aminobenzaldehyde 248 (Y = CH) or 2-aminonicotinaldehyde 248 (Y = N).97 Although activity against topoisomerase I was weak, the aza analogue 246 (n = 1; Y = N) showed comparable cytotoxicity to luotonin A itself towards five human cancer cell lines.
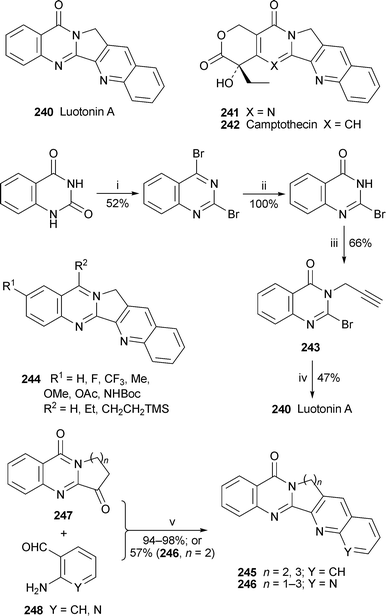 | ||
Scheme 18
Reagents and conditions: i, POBr3, PhNMe2, 105 °C, 4 h; ii, aq. NaOH (1 M), THF, rt, 2 h; iii, NaH, DMF, 0 °C, 10 min, then HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCH2Br, rt, 6.5 h; iv, PhNC, (Me3Sn)2, C6H6, W GE sunlamp (275 W), rt, 8 h; v, satd. aq. KOH, EtOH, reflux, 8 h. CCH2Br, rt, 6.5 h; iv, PhNC, (Me3Sn)2, C6H6, W GE sunlamp (275 W), rt, 8 h; v, satd. aq. KOH, EtOH, reflux, 8 h. | ||
Complementary routes to quinazolinobenzodiazepine alkaloids by Al-Said and co-workers are based on the reaction of isatoic anhydride 249 with amines, and entail creation of the quinazoline core at a late stage (Scheme 19).98 Sequential reaction of 249 with benzylamine and chloroacetyl chloride followed by base-induced cyclisation produced the benzodiazepinedione 250, which underwent acylation with 2-nitrobenzoyl chloride to give the readily hydrolysed imide 251 in moderate yield. Mild catalytic hydrogenation then afforded N-benzylsclerotigenin 252 in 77% yield. The synthesis of the more complex alkaloid asperlicin D 253 began with the reaction of 249 with the methyl ester of L-tryptophan, followed by N-acylation with 2-nitrobenzoyl chloride and reduction of the amine group to give the tripeptide-like cyclisation precursor 254 (R = NH2).99 Unfortunately, the Lewis acid-induced double cyclodehydration required to give the target proved to be inefficient, giving asperlicin D in yields of 30–40%. A change of route involved the use of 2-azidobenzoyl chloride instead of the nitro compound to give the azide 254 (R = N3) which, when subjected to the ‘Eguchi protocol’ (Staudinger reaction with tributylphosphine followed by intramolecular aza-Wittig reaction) afforded asperlicin D 253 in an improved yield of 62%.
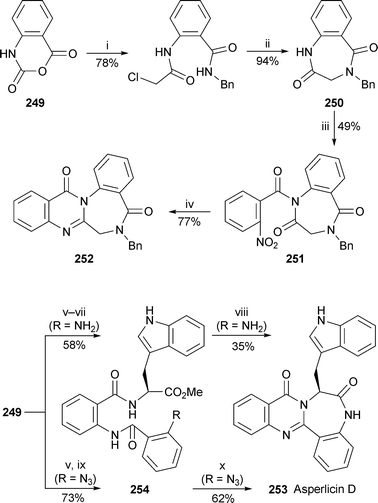 | ||
| Scheme 19 Reagents and conditions: i, BnNH2, MeCN, reflux, 3 h, then NEt3, ClCH2COCl, 0 °C, then rt, 24 h; ii, NaH, THF, rt, overnight; iii, 2-O2NC6H4COCl, NEt3, DMAP, CH2Cl2, rt, 2 h; iv, H2 (1 atm), 10% Pd/C, EtOAc, 50 °C; v, L-tryptophan methyl ester, NEt3, MeCN; vi, 2-O2NC6H4COCl, NEt3; vii, SnCl2, MeOH; viii, MgCl2, DMF, 130 °C, 36 h; ix, 2-N3C6H4COCl, NEt3; x, PBu3, mesitylene, 150 °C, 16 h, then PhSO3H (cat.), H2O–THF, 3 h. | ||
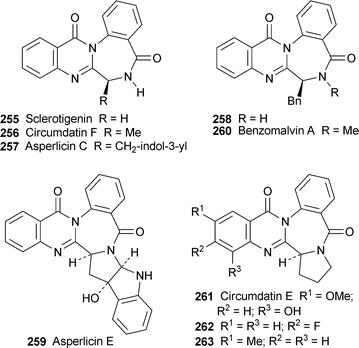
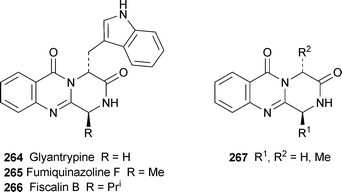
Liu et al. have achieved remarkably short syntheses of several quinazolinobenzodiazepine alkaloids by means of a one-pot domino procedure that owes its success to microwave heating100 (cf. Scheme 14 above). Simply heating two equivalents of anthranilic acid with N-Boc derivatives of α-amino acids and triphenyl phosphite in pyridine at 230 °C for 20 minutes in a sealed vessel under microwave irradiation afforded sclerotigenin 255, (±)-circumdatin F 256, (±)-asperlicin C 257 and the benzyl compound 258 in yields of 55%, 32%, 20% and 23%, respectively. The preparation of 257 also completes a formal synthesis of asperlicin E 259,101 while treatment of 258 with lithium hexamethyldisilazide followed by iodomethane gave a 70% yield of benzomalvin A 260. Sclerotigenin could also be prepared in 60% yield in a three-component one-pot procedure by the microwave heating of anthranilic acid and N-Boc-glycine with triphenyl phosphite in pyridine at 150 °C followed by further microwave heating at 250 °C with methyl anthranilate. When the glycine precursor was replaced by the N-Boc derivative of proline, the three-component method proved to be suitable for making two analogues of circumdatin F 261, namely 262 (34%) and 263 (29%). In related work, the three-component reaction of anthranilic acid with N-Boc glycine as described above followed by brief (1.5 minute) microwave heating with the methyl ester of D-tryptophan hydrochloride at 220 °C completed a syntheses of (−)-glyantrypine 264, in 55% yield and 70% ee, longer times resulting in enantiomeric erosion; but the optical purity could be raised to 100% by recrystallisation from methanol.102 With (S)-alanine and (S)-valine, on the other hand, the first stage required thermal heating at 55 °C, after which the brief microwave-induced reaction with D-tryptophan hydrochloride afforded fumiquinazoline F 265 and fiscalin B 266 in yields of 39% (ee 72%) and 20% (ee 50%), respectively. The tandem thermal–microwave method was also successfully applied to the preparation of model compounds 267 in yields of 56–64%.
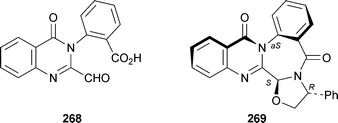
An interesting adaptation of the Meyers methodology for the diastereoselective synthesis of lactams was applied to the synthesis of an axially chiral quinazolinobenzodiazepine related to the asperlicins, benzomalvins and circumdatins. Reaction of the quinazolinone 268 with (R)-phenylglycinol in the presence of magnesium sulfate as dehydrating agent proceeded in boiling dichloromethane to give trans-(aS,R,S)-269 in 25% yield and a diastereomeric excess (de) of better than 95%.103 Alternatively, use of N-ethyl-2-fluoropyridinium tetrafluoroborate, an activating agent for carboxylic acids, in combination with diisopropylethylamine permitted the reaction to take place at ambient temperature, giving 269 in 85% yield but only 65% de.
3 Acridone alkaloids
New acridone alkaloids and their plant sources, all members of the Rutaceae, are listed in Table 2.5,104–108 Also included are known alkaloids isolated from new sources.| Species | Alkaloid a | Ref. |
|---|---|---|
| a Only new alkaloids and new records for a given species are listed in the table; in many cases, the presence of several previously isolated alkaloids was confirmed. Structures of known alkaloids, if not specifically numbered, may be found in previous reviews in this series. b New alkaloids. | ||
| Citrus maxima | Citracridone-III 272 | 104 |
| Citrusinine-I | ||
| Glycocitrine-I | ||
| Grandisine-I | ||
| 5-Hydroxynoracronycine 271 | ||
| 5-Hydroxynoracronycine alcoholb270 | ||
| Natsucitrine-II | ||
| Erthela bahiensis | Arborinine | 105 |
| Esenbeckia pentaphylla | 1-Hydroxy-3-methoxy-N-methylacridone | 5 |
| Oriciopsis glaberrima | Atalaphyllidine | 106 |
| (−)-Oriciacridone Ab273 | 107 | |
| (−)-Oriciacridone Bb274 | ||
| (+)-Oriciacridone Cb278 | 106 | |
| (+)-Oriciacridone Db279 | ||
| (+)-Oriciacridone Eb280 | ||
| (+)-Oriciacridone Fb282 | ||
| (+)-1,3,5-Trihydroxy-4-prenylacridoneb281 | ||
| Toddaliopsis bremekampii | Toddaliopsin Ab283 | 108 |
| Toddaliopsin Bb284 | ||
| Toddaliopsin Cb285 | ||
| Toddaliopsin Db286 |
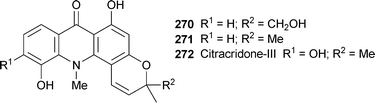
Among the known alkaloids isolated from the stem bark of Citrus maxima (the buntan shaddock) was the novel compound 270, which appears to be unique among acridone alkaloids in containing a hydroxymethyl group in the prenyl-derived pyran ring instead of the customary methyl group. The location of this unusual substituent was deduced from HMBC correlations.104 Compound 270 is thus a hydroxylated analogue of 5-hydroxynoracronycine 271, which was also found in the plant extract. Most of the alkaloids isolated in this work (see Table 2) showed cytotoxicity towards the HepG2 hepatoma and KB epidermoid cancer cell lines; 270 was most potent towards the KB cells (IC50 19.5 µM), while citracridone-III 272 was most effective against the HepG2 cell line (IC50 17.0 µM).
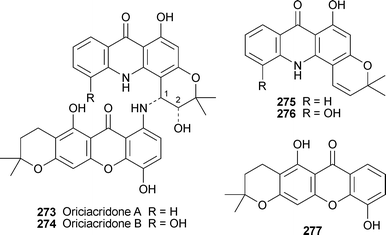
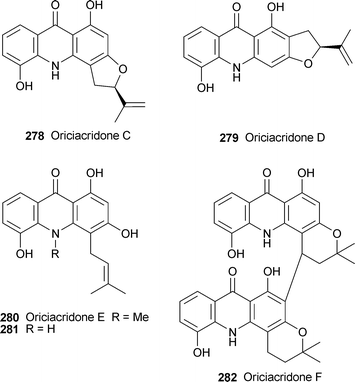
No fewer than seven new acridone alkaloids have been obtained from the stem bark of Oriciopsis glaberrima, a medicinal plant endemic to the rain forests of Cameroon. (−)-Oriciacridones A and B, 273 and 274, are unique acridone–xanthone dimers in which acridone moieties formally derived from N-desmethylnoracronycine 275 and its 5-hydroxy analogue 276, respectively, are linked by an amine group to dihydro-6-desoxyjacareulin 277.107 The structures were elucidated by means of detailed spectroscopic analyses; in particular, the linkage between the units was established with the aid of HMBC and NOESY experiments, while the cis-relationship of H-1 and H-2 was inferred from coupling constants of 3.7 and 3.9 Hz, respectively. Both alkaloids exhibited antimicrobial activity against a range of bacteria, fungi and microalgae. Two of the remaining new alkaloids, (−)-oriciacridone C 278 and (−)-oriciacridone D 279, are regioisomeric furoacridones. Their absolute configurations were inferred to be (R) on the basis of positive Cotton effects in their CD spectra; but it should be noted that the structural diagram given for oriciacridone D is of the (S)-enantiomer, as illustrated.106 Oriciacridone E 280 and its N-desmethyl analogue 281 are simple prenylated acridones; puzzlingly, large positive specific rotations are given for both compounds, though neither has any obvious chiral elements.106 Compound 281, although a new natural product, has previously been synthesised.109 The final new alkaloid, (+)-oriciacridone F 282, is a more conventional bisacridone involving dimerisation of two 5-hydroxy-N-desmethylnoracronycine units 276. The new alkaloids 273, 281 and 282 were reasonably effective inhibitors of Saccharomyces α-glucosidase at concentrations of 100 µM or less, while the same three alkaloids as well as 274 exhibited moderate radical scavenging ability towards 1,1-diphenyl-2-picrylhydrazyl, which implies some antioxidant activity.
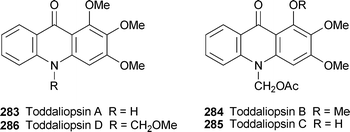
Toddaliopsins A–D, 283–286, are new acridone alkaloids isolated from the leaves of Toddaliopsis bremekampii, a small tree originating in the sand and dune forests of central southern Africa.108 The striking feature of these compounds is the functionalised N-methyl substituent in 284–286—the first time that substituted N-methyl groups have been found among the acridone alkaloids, although N-acetoxymethyl groups have previously been reported in quinolone alkaloids from other rutaceous genera. In particular, the N-methoxymethyl substituent in toddaliopsin D is unique; could the alkaloid perhaps be produced from toddaliopsin B as an artefact of the isolation procedure, which included an extraction with hot methanol? Also unusual is the 1-methoxy group in three of the four alkaloids, since the majority of naturally occurring acridones possess a 1-hydroxy substituent. From a chemotaxonomic viewpoint, the unusual N-substituents in the new alkaloids serve to distinguish the rare genus Toddaliopsis from the closely related Vepris, which contains conventional acridones as well as furoquinolines (not detected in the present study). Alkaloids 283–286 displayed moderate anti-inflammatory activity in a chemiluminescence assay (IC50 27.3, 48.3, 4.21 and 79.1 µg ml−1, respectively), the enhanced activity of toddaliopsin C perhaps relating to the presence of the C-1 hydroxy group.
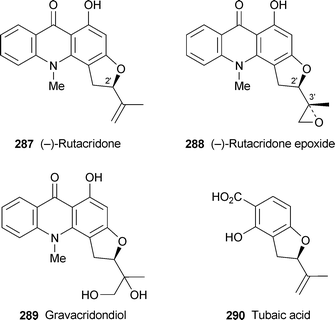
Bioassay-guided fractionation of root extracts from Ruta graveolens for algicidal activity towards the blue-green alga Oscillatoria perornata, a pest in commercial catfish farming, led to the isolation of the known alkaloids (−)-rutacridone 287, (−)-rutacridone epoxide 288 and gravacridondiol 289.110 Although the first two alkaloids have been known for some time, their absolute configurations have never been determined. This has now been remedied by comparison of the CD spectrum of 287 with that of (R)-(−)-tubaic acid 290; since both showed negative Cotton effects, (−)-rutacridone presumably also has the (2′R)-configuration. The (3′R)-configuration of the second stereogenic centre in rutacridone epoxide was inferred by comparing chemical shift differences for the epoxide ring's two methylene protons with those of related compounds with known absolute stereochemistry. Rutacridone epoxide proved to be a potent and selective algicide against O. perornata (IC50 9 × 10−3 µM); gravacridondiol was far less active in the bioassay, and the parent alkaloid rutacridone was inactive. The epoxide was also an effective fungicide against several agriculturally important fungal pathogens, but unfavourable mutagenic and cytotoxic effects are likely to preclude its use as an agrochemical.
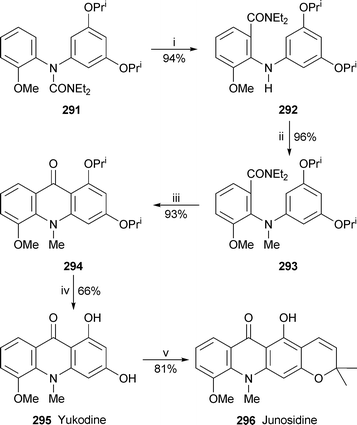
A new synthesis of two acridone alkaloids by Snieckus and co-workers employed an anionic aza-Fries rearrangement of an N,N-diarylurea as key step111 (Scheme 20). When the precursor 291, made by Buchwald C–N cross-coupling methodology, was treated with an excess of lithium tetramethylpiperidide, an efficient regioselective directed ortho-metallation of the methoxyaryl ring was followed by acyl migration to give the substituted anthranilamide 292 in 94% yield. Simply treating the subsequently formed N-methyl intermediate 293 with trifluoromethanesulfonic anhydride afforded the acridone 294 in 93% yield. Removal of the isopropyl protecting groups was achieved with boron trichloride to complete the first reported synthesis of yukodine 295. Finally, the reaction of 295 with senecialdehyde (prenal) and acetic acid in boiling toluene yielded the linearly fused pyranoacridone alkaloid junosidine 296 uncontaminated by its angularly fused regioisomer. The overall yield of junosidine was 33% based on a commercially available reactant, 1-chloro-3,5-dimethoxybenzene.
 | ||
| Scheme 20
Reagents and conditions: i, LiTMP (3 equiv.), THF, 0 °C, 2 h; ii, NaH, DMF, 0 °C, then rt, 2 h, then MeI; iii, Tf2O, CH2Cl2, 0 °C to rt, 0.5 h; iv, BCl3 (1.0 M in CH2Cl2), ClCH2CH2Cl, rt, 14 h; v, Me2CCHCHO, AcOH, PhMe, reflux, 24 h. | ||
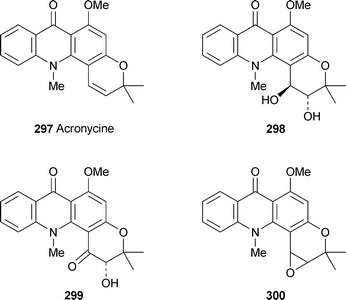
Tillequin and coworkers have continued to explore the chemistry and biological activity of the antitumour alkaloid acronycine 297 and its many synthetic analogues. A novel oxidation of the parent alkaloid with hydrogen peroxide was catalysed by a manganese–porphyrin complex in the presence of imidazole to give the racemic trans-diol 298, a valuable precursor for the preparation of bioactive ester derivatives, in 15% yield together with the hydroxyketone 299 (9%).112 The unstable epoxide intermediate 300, which is the active metabolite of acronycinein vivo, was not detected, although similar epoxides were isolated in parallel studies with chromenes. By contrast, oxidation of acronycine with potassium permanganate is known to give only the hydroxyketone.
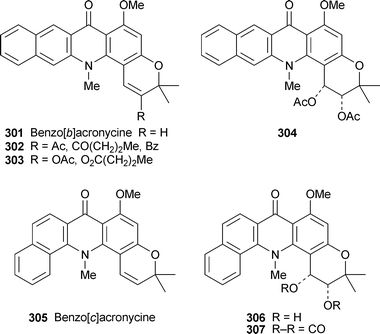
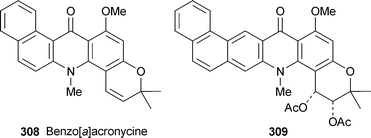
New derivatives of synthetic benzo[b]acronycine 301 include the enones 302, prepared by Friedel–Crafts acylation of 301, and the acylated enols 303.113 The cytotoxicities of these enol esters towards the murine L1210 leukemia cell line were comparable to that of the cis-diacetoxy analogue 304, which is now undergoing phase I clinical trials. Among many new angularly fused analogues of the benzoacronycines, benzo[c]acronycine 305, the cis-diol 306 and the cyclic carbonate 307 inhibited L1210 cell proliferation with approximately the same degree of effectiveness as acronycine itself.114 In general, however, benzo[c] ring fusion did not enhance cytotoxicity, probably because of the increased steric hindrance close to the reactive DNA-binding pyran ring region of the molecules. By contrast, most members of a suite of 22 derivatives of benzo[a]acronycine 308 exhibited submicromolar toxicity similar to that of 304 when tested against the L1210 leukemia and human epidermoid carcinoma (KB-3-1) cell lines, the activity correlating strongly with the ability of the compounds to bind covalently to DNA.115 Among the most active compounds was, once again, a cis-diacetoxy derivative 309 (IC50 0.7 and 0.15 µM, respectively), which was also active in vivo against C38 colon adenocarcinoma implanted into mice.
4 Acknowledgements
This material is based upon work supported by the National Research Foundation under grant number 2053652. Financial support from the University of the Witwatersrand is also gratefully acknowledged.5 References
- L. E. R. Cortez, D. A. G. Cortez, A. G. Ferreira, P. C. Vieira, M. F. das G. F. da Silva and J. B. Fernandes, Rev. Bras. Farm., 2006, 16, 164 ( Chem. Abstr. , 2007 , 146 , 202107 ) Search PubMed.
- H. K. Wabo, P. Tane, J. D. Connolly, C. C. Okunji, B. M. Schuster and M. M. Iwu, Nat. Prod. Res., 2005, 19, 591 CrossRef CAS.
- R. Grougnet, P. Magiatis, N. Fokialakis, S. Mitaku, A.-L. Skaltsounis, F. Tillequin, T. Sévenet and M. Litaudon, J. Nat. Prod., 2005, 68, 1083 CrossRef CAS.
- C. W. Halstead, P. I. Forster and P. G. Waterman, Nat. Prod. Commun., 2006, 1, 351 ( Chem. Abstr. , 2007 , 146 , 312823 ) CAS.
- D. S. Simpson and H. Jacobs, Biochem. Syst. Ecol., 2005, 33, 841 CrossRef CAS.
- Q.-W. Liu, C.-H. Tan, S.-J. Qu, X. Fan and D.-Y. Zhu, Zhongguo Tianran Yaowu, 2006, 4, 25 ( Chem. Abstr. , 2006 , 145 , 425429 ) Search PubMed.
- C. L. Cantrell, K. K. Schrader, L. K. Mamonov, G. T. Sitpaeva, T. S. Kustova, C. Dunbar and D. E. Wedge, J. Agric. Food Chem., 2005, 53, 7741 CrossRef CAS.
- I. Komala, M. Rahmani, M. A. Sukari, H. B. M. Ismail, G. E. C. Lian and A. Rahmat, Nat. Prod. Res., 2006, 20, 355 CrossRef CAS.
- H.-C. Chou, J.-J. Chen, C.-Y. Duh, T.-F. Huang and I.-S. Chen, Planta Med., 2005, 71, 1078 CrossRef CAS.
- A. Chlouchi, C. Girard, F. Tillequin, F. Bévalot, P. G. Waterman and F. Muyard, Biochem. Syst. Ecol., 2006, 34, 71 CrossRef CAS.
- R. C. de Souza, J. B. Fernandes, P. C. Vieira, M. F. das G. F. da Silva, M. F. P. Godoy, F. C. Pagnocca, O. C. Bueno, M. J. A. Hebling and J. R. Pirani, Z. Naturforsch., B, 2005, 60b, 787.
- J.-H. Cho, C.-H. Lee and H.-S. Lee, J. Microbiol. Biotechnol., 2005, 15, 646 CAS.
- M. da P. Lima, L. V. Rosas, M. F. das G. F. da Silva, A. G. Ferreira, J. B. Fernandes and P. C. Vieira, Phytochemistry, 2005, 66, 1560 CrossRef CAS.
- T. A. N. Ribeiro, E. A. da Silva Ndiaye, E. da S. Velozo, P. C. Vieira, J. Ellena and P. T. de Sousa Júnior, J. Braz. Chem. Soc., 2005, 16, 1347 CAS.
- S. C. Jain, M. K. Pandey, R. K. Upadhyay, R. Kumar, G. Hundal and M. S. Hundal, Phytochemistry, 2006, 67, 1005 CrossRef CAS.
- M.-J. Cheng, K.-H. Lee, I.-L. Tsai and I.-S. Chen, Bioorg. Med. Chem., 2005, 13, 5915 CrossRef CAS.
- J.-B. Bongui, A. Blanckaert, A. Elomri and E. Seguin, Biochem. Syst. Ecol., 2005, 33, 845 CrossRef CAS.
- B. Bohlendorf, E. Forche, N. Bedorf, K. Gerth, H. Irschik, R. Jansen, B. Kunze, W. Trowizsch-Kienast, H. Reichenbach and G. Hofle, Liebigs Ann., 1996, 49.
- M. Adams, A. A. Wube, F. Bucar, R. Bauer, O. Kunert and E. Haslinger, Int. J. Antimicrob. Agents, 2005, 26, 261 CrossRef.
- F. M. Nunes, B. A. Barros-Filho, M. C. F. de Oliveira, M. Andrade-Neto, M. C. de Mattos, J. Mafezoli and J. R. Pirani, Magn. Reson. Chem., 2005, 43, 864 CrossRef CAS.
- O. Jansen, V. Akhmedjanova, L. Angenot, G. Balansard, A. Chariot, E. Ollivier, M. Tits and M. Frédérich, J. Ethnopharmacol., 2006, 105, 241 CrossRef CAS.
- F. M. Nunes, B. A. Barros-Filho, M. C. F. de Oliveira, M. C. de Mattos, M. Andrade-Neto, F. G. Barbosa, J. Mafezoli, R. C. Montenegro, C. Pessoa, M. O. de Moraes, L. V. Costa-Lotufo, F. C. S. Galetti, C. L. Silva and A. O. de Souza, Nat. Prod. Commun., 2006, 1, 313 CAS.
- C. S. Cho, J. Organomet. Chem., 2005, 690, 4094 CrossRef CAS.
- C. S. Cho, H. J. Seok and S. C. Shim, J. Heterocycl. Chem., 2005, 42, 1219 CrossRef CAS.
- K. Taguchi, S. Sakaguchi and Y. Ishii, Tetrahedron Lett., 2005, 46, 4539 CrossRef CAS.
- C. S. Cho, W. X. Ren and S. C. Shim, Bull. Korean Chem. Soc., 2005, 26, 1286 CAS.
- M. Movassaghi and M. D. Hill, J. Am. Chem. Soc., 2006, 128, 4592 CrossRef CAS.
- S.-M. Lu, Y.-Q. Wang, X.-W. Han and Y.-G. Zhou, Angew. Chem., Int. Ed., 2006, 45, 2260 CrossRef CAS.
- M. Rueping, A. P. Antonchick and T. Theissmann, Angew. Chem., Int. Ed., 2006, 45, 3683 CrossRef.
- I. Jacquemond-Collet, J.-M. Bessière, S. Hannedouche, C. Bertrand, I. Fourasté and C. Moulis, Phytochem. Anal., 2001, 12, 312 CrossRef CAS.
- J.-S. Ryu, Bull. Korean Chem. Soc., 2006, 27, 631 CAS.
- V. Nadaraj, S. T. Selvi and R. Sasi, Arkivoc, 2006, x, 82.
- A. F. Morel, E. L. Larghi and M. M. Selvero, Synlett, 2005, 2755 CAS.
- G. D. Cuny, K. Lee and S. Choi, Lett. Org. Chem., 2006, 3, 68 Search PubMed.
- D. R. Boyd, N. D. Sharma, C. R. O'Dowd, J. G. Carroll, P. L. Loke and C. C. R. Allen, Chem. Commun., 2005, 3989 RSC.
- M. F. Grundon, D. M. Harrison and C. G. Sypropoulos, J. Chem. Soc., Perkin Trans. 1, 1974, 2181 RSC.
- I. Aillaud, E. Bossharth, D. Conreaux, P. Desbordes, N. Monteiro and G. Balme, Org. Lett., 2006, 8, 1113 CrossRef CAS.
- J. Su, J. Xiong, S. Liang, G. Qiu, X. Feng, H. Teng, L. Wu and X. Hu, Synth. Commun., 2006, 36, 693 CrossRef CAS.
- Y. Hirasawa, J. Kobayashi and H. Morita, Org. Lett., 2006, 8, 123 CrossRef CAS.
- J. Igarashi, M. Katsukawa, Y.-G. Wang, H. P. Acharya and Y. Kobayashi, Tetrahedron Lett., 2004, 45, 3783 CrossRef CAS.
- (a) J. P. Michael, Nat. Prod. Rep., 2005, 22, 631 Search PubMed; (b) J. P. Michael, Nat. Prod. Rep., 2004, 21, 657 Search PubMed; (c) J. P. Michael, Nat. Prod. Rep., 2007, 24, 231 Search PubMed; (d) J. P. Michael, Nat. Prod. Rep., 2007, 24, 233 Search PubMed; (e) J. P. Michael, Nat. Prod. Rep., 2003, 20, 476 RSC; (f) J. P. Michael, Nat. Prod. Rep., 2007, 24, 230 Search PubMed; (g) J. P. Michael, Nat. Prod. Rep., 2004, 21, 662 Search PubMed; (h) J. P. Michael, Nat. Prod. Rep., 2007, 24, 241 Search PubMed; (i) J. P. Michael, Nat. Prod. Rep., 2002, 19, 755 Search PubMed.
- J. Igarashi and Y. Kobayashi, Tetrahedron Lett., 2005, 46, 6381 CrossRef CAS.
- S. Debarge, B. Violeau, M.-P. Jouannetaud, J.-C. Jacquesy and A. Cousson, Tetrahedron, 2006, 62, 662 CrossRef CAS.
- A. Nakano, K. Takahashi, J. Ishihara and S. Hatakeyama, Heterocycles, 2005, 66, 371 CrossRef CAS.
- S. P. Diggle, P. Cornelis, P. Williams and M. Cámara, Int. J. Med. Microbiol., 2006, 296, 83 Search PubMed.
- L. Shen, Y.-H. Ye, X.-T. Wang, H.-L. Zhu, C. Xu, Y.-C. Song, H. Li and R.-X. Tan, Chem. Eur. J., 2006, 12, 4393 CrossRef CAS.
- J. He, U. Lion, I. Sattler, F. A. Gollmick, S. Grabley, J. Cai, M. Meiners, H. Schünke, K. Schaumann, U. Dechert and M. Krohn, J. Nat. Prod., 2005, 68, 1397 CrossRef CAS.
- R. Uchida, R. Imasato, K. Shiomi, H. Tomoda and S. Ōmura, Org. Lett., 2005, 7, 5701 CrossRef CAS.
- X. Zhang, W. Jiang and Z. Sui, J. Org. Chem., 2003, 68, 4523 CrossRef CAS.
- S.-J. Park, K.-N. Cho, W.-G. Kim and K.-I. Lee, Tetrahedron Lett., 2004, 45, 8793 CrossRef CAS.
- B. Clark, R. J. Capon, E. Lacey, S. Tennant and J. H. Gill, Org. Biomol. Chem., 2006, 4, 1512 RSC.
- A. G. Kozlovskii, V. P. Zhelifonova and T. V. Antipova, Appl. Biochem. Microbiol., 2005, 41, 499 ( Chem. Abstr. , 2006 , 144 , 83800 ) Search PubMed.
- A. G. Kozlovskii, V. P. Zhelifonova, T. V. Antipova and V. Y. Lysanskaya, Mikrobiologiya, 2006, 75, 279 Search PubMed.
- M. Abe, T. Imai, N. Ishii and M. Usui, Biosci., Biotechnol., Biochem., 2006, 70, 303 CrossRef CAS.
- Y.-Y. Jin, T.-M. Yoon, W.-K. Kim, K.-R. Kim, J.-K. Song, J.-G. Kim, J. Liu, Y.-Y. Yang, H.-J. Kwon and J.-W. Suh, J. Microbiol. Biotechnol., 2005, 15, 1140 CAS.
- M. Hassani, W. Cai, D. C. Holley, J. P. Lineswala, B. R. Maharjan, G. R. Ebrahimian, H. Seradj, M. G. Stocksdale, F. Mohammadi, C. C. Marvin, J. M. Gerdes, H. D. Beall and M. Behforouz, J. Med. Chem., 2005, 48, 7733 CrossRef CAS.
- Y. Bethuel and K. Gademann, J. Org. Chem., 2005, 70, 6258 CrossRef CAS.
- F. Lombó, A. Velasco, A. Castro, F. de la Calle, A. F. Braña, J. M. Sánchez-Puelles, C. Méndez and J. A. Salas, ChemBioChem, 2006, 7, 366 CrossRef CAS.
- K. C. Nicolaou, B. S. Safina, M. Zak, S. H. Lee, M. Nevalainen, M. Bella, A. A. Estrada, C. Funke, F. J. Zécri and S. Bulat, J. Am. Chem. Soc., 2005, 127, 11159 CrossRef CAS.
- K. C. Nicolaou, M. Zak, B. S. Safina, A. A. Estrada, S. H. Lee and M. Nevalainen, J. Am. Chem. Soc., 2005, 127, 11176 CrossRef CAS.
- T. Mori, Y. Satouchi, H. Tohmiya, S. Higashibayashi, K. Hashimoto and M. Nakata, Tetrahedron Lett., 2005, 46, 6417 CrossRef CAS.
- T. Mori, H. Tohmiya, Y. Satouchi, S. Higashibayashi, K. Hashimoto and M. Nakata, Tetrahedron Lett., 2005, 46, 6423 CrossRef CAS.
- M.-A. Yoon, T.-S. Jeong, D.-S. Park, M.-Z. Xu, H.-W. Oh, K.-B. Song, W. S. Lee and H.-Y. Park, Biol. Pharm. Bull., 2006, 29, 735 CrossRef CAS.
- M. J. McKay, A. R. Carroll and R. J. Quinn, J. Nat. Prod., 2005, 68, 1776 CrossRef CAS.
- J. W. Daly, T. F. Spande and H. M. Garraffo, J. Nat. Prod., 2005, 68, 1556 CrossRef CAS.
- C. Macfoy, D. Danosus, R. Sandit, T. H. Jones, H. M. Garraffo, T. F. Spande and J. W. Daly, Z. Naturforsch., C, 2006, 60, 932.
- M. Mena and J. Bonjoch, Tetrahedron, 2005, 61, 8264 CrossRef CAS.
- K. Jayapaul, P. B. K. Kishor and K. J. Reddy, In Vitro Cell. Dev. Biol.: Plant, 2005, 41, 682 Search PubMed.
- A. G. Tozhibaev, K. K. Turgunov, B. Tashkhodzhaev and K. M. Shakhidoyatov, Chem. Nat. Compd., 2006, 42, 340 CrossRef CAS.
- T. S. Tulyaganov and N. M. Kozimova, Chem. Nat. Compd., 2005, 41, 578 CrossRef CAS.
- C.-Y. Chiu, C.-Y. Li, C.-C. Chiu, M. Niwa, S. Kitanaka, A. G. Damu, E.-J. Lee and T.-S. Wu, Chem. Pharm. Bull., 2005, 53, 1118 CrossRef CAS.
- S. H. Shim, J. S. Kim, K. H. Son, K. H. Bae and S. S. Kang, J. Nat. Prod., 2006, 69, 400 CrossRef CAS.
- K. Sprogøe, S. Manniche, T. O. Larsen and C. Christophersen, Tetrahedron, 2005, 61, 8718 CrossRef CAS.
- S. Zhu, L. Meng, Q. Zhang and L. Wei, Bioorg. Med. Chem. Lett., 2006, 16, 1854 CrossRef CAS.
- Y. H. Choi, E. M. Shin, Y. S. Kim, X. F. Cai, J. J. Lee and H. P. Kim, Arch. Pharm. Res., 2006, 29, 293 Search PubMed.
- Y.-F. Ueng, M.-J. Don, W.-C. Jan, S.-Y. Wang, L.-K. Ho and C.-F. Chen, Drug Metab. Dispos., 2006, 34, 821 CrossRef CAS.
- S. K. Lee, J. H. Lee, H. H. Yoo, D. H. Kim, Y. Jahng and T. C. Jeong, J. Pharm. Biomed. Anal., 2006, 41, 304 CrossRef CAS.
- D. J. Connolly, D. Cusack, T. P. O'Sullivan and P. J. Guiry, Tetrahedron, 2005, 61, 10153 CrossRef CAS.
- S. Eguchi, Top. Heterocycl. Chem., 2006, 6, 113 Search PubMed.
- Y. Takeuchi, K. Azuma, M. Oshige, H. Abe, H. Nishioka, K. Sasaki and T. Harayama, Tetrahedron, 2003, 59, 1639 CrossRef CAS.
- Y. Takeuchi, M. Oshige, K. Azuma, H. Abe and T. Harayama, Chem. Pharm. Bull., 2005, 53, 868 CrossRef CAS.
- M. Katoh, R. Matsune, H. Nagase and T. Honda, Tetrahedron Lett., 2004, 45, 6221 CrossRef CAS.
- M. Katoh, R. Matsune and T. Honda, Heterocycles, 2006, 67, 189 CrossRef CAS.
- Y. Takeuchi, K. Azuma, K. Takakura, H. Abe, H.-S. Kim, Y. Wataya and T. Harayama, Tetrahedron, 2001, 57, 1213 CrossRef CAS.
- J. P. Michael, C. B. de Koning and D. P. Pienaar, Synlett, 2006, 383 CrossRef CAS.
- A. Kamal, N. Shankaraiah, K. L. Reddy and V. Devaiah, Tetrahedron Lett., 2006, 47, 4253 CrossRef CAS.
- J.-F. Liu, P. Ye, K. Sprague, K. Sargent, D. Yohannes, C. M. Baldino, C. J. Wilson and S.-C. Ng, Org. Lett., 2005, 7, 3363 CrossRef CAS.
- J.-F. Liu, C. J. Wilson, P. Ye, K. Sprague, K. Sargent, Y. Si, G. Beletsky, D. Yohannes and S.-C. Ng, Bioorg. Med. Chem. Lett., 2006, 16, 686 CrossRef CAS.
- H. Hua, M. Cheng, X. Li and Y. Pei, Chem. Pharm. Bull., 2002, 50, 1393 CrossRef CAS.
- M. Cheng, Q. Li, B. Lin, Y. Sha, J. Ren, Y. He, Q. Wang, H. Hua and K. Ruud, Tetrahedron: Asymmetry, 2006, 17, 179 CrossRef CAS.
- S. H. Wiedemann, J. A. Ellman and R. G. Bergman, J. Org. Chem., 2006, 71, 1969 CrossRef CAS.
- A. Hamid, A. Elomri and A. Daïch, Tetrahedron Lett., 2006, 47, 1777 CrossRef CAS.
- E. S. Lee, S. I. Kim, S. H. Lee, T. C. Jeong, T. C. Moon, H. W. Chang and Y. Jahng, Bull. Korean Chem. Soc., 2005, 26, 1975 CAS.
- Z. Z. Ma, Y. Hano, T. Nomura and Y.-J. Chen, Heterocycles, 1997, 46, 541 CrossRef CAS.
- Z. Ma, Y. Hano and T. Nomura, Heterocycles, 2005, 65, 2203 CrossRef CAS.
- R. Tangirala, S. Antony, K. Agama, Y. Pommier and D. P. Curran, Synlett, 2005, 2843 CAS.
- E. S. Lee, J. G. Park, S. I. Kim and Y. Jahng, Heterocycles, 2006, 68, 151 CrossRef CAS.
- N. H. Al-Said and Z. N. Ishtaiwi, Acta Chim. Slov., 2005, 52, 328 CAS.
- N. H. Al-Said and L. S. Al-Qaisi, Tetrahedron Lett., 2006, 47, 693 CrossRef CAS.
- J.-F. Liu, M. Kaselj, Y. Isome, J. Chapnick, B. Zhang, G. Bi, D. Yohannes, L. Yu and C. M. Baldino, J. Org. Chem., 2005, 70, 10488 CrossRef CAS.
- M. G. Bock, R. M. DiPardo, S. M. Pitzenberger, C. P. Homnick, S. M. Springer and R. M. Freidinger, J. Org. Chem., 1987, 52, 1644 CrossRef CAS.
- J.-F. Liu, P. Ye, B. Zhang, G. Bi, K. Sargent, L. Yu, D. Yohannes and C. M. Baldino, J. Org. Chem., 2005, 70, 6339 CrossRef CAS.
- M. Penhoat, P. Bohn, G. Dupas, C. Papamicaël, F. Marsais and V. Levacher, Tetrahedron: Asymmetry, 2006, 17, 281 CrossRef CAS.
- W.-Y. Teng, Y.-L. Huang, C.-C. Shen, R.-L. Huang, R.-S. Chung and C-.C. Chen, J. Chin. Chem. Soc., 2005, 52, 1253 CAS.
- R. Roseghini, P. Moreira, V. Vale, A. M. Pinheiro, J. F. O. Costa, T. Bittencourt, I. Nascimento, R. Schaer, E. Velozo, R. El-Bachá, R. Meyer and S. Freire, Immonopharmacol. Immunotoxicol., 2006, 28, 361 ( Chem. Abstr. , 2007 , 146 , 243605 ) Search PubMed.
- J. D. Wansi, J. Wandji, L. M. Meva’a, A. F. K. Waffo, R. Ranjit, S. N. Khan, A. Asma, C. M. Iqbal, M.-C. Lallemand, F. Tillequin and Z. F. Tanee, Chem. Pharm. Bull., 2006, 54, 292 CrossRef CAS.
- J. D. Wansi, J. Wandji, A. F. K. Waffo, H. E. Ngeufa, J. C. Ndom, S. Fotso, R. P. Maskey, D. Njamen, T. Z. Fomum and H. Laatsch, Phytochemistry, 2006, 67, 475 CrossRef CAS.
- D. Naidoo, P. H. Coombes, D. A. Mulholland, N. R. Crouch and A. J. J. van den Bergh, Phytochemistry, 2005, 66, 1724 CrossRef CAS.
- M. H. Bahar, J. D. Shringarpure, G. H. Kulkarni and B. K. Sabata, Phytochemistry, 1982, 21, 2729 CrossRef CAS.
- K. M. Meepagala, K. K. Schrader, D. E. Wedge and S. O. Duke, Phytochemistry, 2005, 66, 2689 CrossRef CAS.
- S. L. MacNeil, B. J. Wilson and V. Snieckus, Org. Lett., 2006, 8, 1133 CrossRef CAS.
- B. Akagah, F. Estour, P. Vérité, E. Seguin, F. Tillequin and O. Lafont, J. Heterocycl. Chem., 2005, 42, 1267 CrossRef CAS.
- H. Doan Thi Mai, T. Gaslonde, S. Michel, M. Koch, F. Tillequin, C. Bailly, M.-H. David-Cordonnier, B. Pfeiffer, S. Léonce and A. Pierré, Chem. Pharm. Bull., 2005, 53, 919 CrossRef.
- J.-B. Bongui, A. Elomri, D. Cahard, F. Tillequin, B. Pfeiffer, A. Pierré and E. Seguin, Chem. Pharm. Bull., 2005, 53, 1540 CrossRef CAS.
- T. M. Nguyen, C. Sittisombut, S. Boutefnouchet, M.-C. Lallemand, S. Michel, M. Koch, F. Tillequin, R. Mazinghien, A. Lansiaux, M.-H. David-Cordonnier, B. Pfeiffer, L. Kraus-Berthier, S. Léonce and A. Pierré, J. Med. Chem., 2006, 49, 3383 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2008 |