DOI:
10.1039/B712081H
(Paper)
New J. Chem., 2008,
32, 159-165
Large photoactive supramolecular ensembles prepared from C60–pyridine substrates and multi-Zn(II)–porphyrin receptors†
Received
(in Montpellier, France)
1st August 2007
, Accepted 29th August 2007
First published on 17th September 2007
Abstract
Fullerene derivatives bearing a pyridine sub-unit have been prepared. Their ability to form self-assembled supramolecular structures with mono- and polytopic Zn(II)–porphyrin receptors has been first evidenced by UV-vis studies. These supramolecular complexes are multi-component photoactive devices, in which the emission of the porphyrinic receptor is dramatically quenched by the fullerene units. This new property, resulting from the association of the different molecular sub-units, also allowed us to investigate in detail the self-assembly process using fluorescence titrations. The binding studies revealed positive cooperative effects for the assembly of the C60–pyridine derivatives with polytopic receptors as a result of intramolecular C60–C60 interactions between the different guests assembled onto the multi-Zn(II)–porphyrin hosts.
Introduction
Self-assembly is an incredibly powerful concept in modern chemistry.1 The ability of simple molecules to spontaneously assemble into discrete nanostructures offers unlimited possibilities for fundamental discoveries and practical applications. Furthermore, the self-assembly of carefully designed building blocks can generate new properties. In particular, this principle has been used to produce sophisticated photoactive supramolecular devices.2,3 As part of this research, non-covalent fullerene –porphyrin conjugates have generated significant research efforts in the past few years.4 In most cases, these supramolecular arrays have been obtained from a C60 derivative bearing a pyridyl moiety and a metalloporphyrin through coordination to the metal center.5 However, the binding constants of such systems, initially developed by Diederich et al.6 and D’Souza et al.,7 are usually rather low. More recently, related supramolecular ensembles of improved stability have been obtained by designing systems with additional recognition elements,8 or by applying the supramolecular click chemistry principle.9 A few examples of structures resulting from the apical coordination of C60–pyridine substrates to receptors appended with multiple Zn(II)–porphyrin sub-units have also been described.10,11 Interestingly, photophysical investigations of such systems have revealed a more efficient electron transfer compared to simple porphyrin –fullerene supramolecular dyads.10 Indeed, the larger number of C60 units enhances the probability of electron transfer from the Zn(II)–porphyrin units. In addition, efficient energy migration along the densely packed Zn(II)porphyrin array may also enhance the opportunity for this electron transfer. These considerations prompted us to study multi-component supramolecular edifices resulting from the self-assembly of fullerene –pyridine substrates onto multi-Zn(II)–porphyrin receptors. Interestingly, detailed spectrophotometric investigations revealed higher stability when the number of Zn(II)–porphyrin sub-units was increased. This can be explained by positive cooperative effects resulting from intramolecular C60–C60 interactions between the different fullerene –pyridine guests assembled onto the multi-Zn(II)–porphyrin receptors. We thus show that an increase in the number of components within supramolecular fullerene –porphyrin conjugates is not only interesting from the photophysical point of view, but improves also the overall thermodynamic stability of the assembly.
Results and discussion
Synthesis
The porphyrinic building blocks used in this study are shown in Fig. 1. Compounds LZn,12LZn212 and LZn613 were prepared according to previously reported procedures. Their NMR spectra were identical to those described in the literature. In addition, mass spectrometry analysis confirmed their structures.
 |
| | Fig. 1 Chemical structures of the porphyrin derivatives used in this study. | |
The synthesis of fullerene substrates S1 and S2 is depicted in Scheme 1. The synthetic approach to prepare compound S1 relies upon the 1,3-dipolar cycloaddition of an azomethine ylide generated in situ from 4-pyridinecarboxaldehyde (3) and a N-alkylglycine derivative. This methodology has proven to be a powerful procedure for the functionalization of C60 due to its versatility and the ready availability of the starting materials.14 In the present study, we decided to use N-(3,5-didodecyloxybenzyl) glycine (4)15 rather than the more commonly used N-methylglycine. Indeed, the 3,5-didodecyloxybenzyl group has proven to be a good solubilizing group for fullerene derivatives16 and should prevent solubility problems in the target pyrrolidinofullerene. Thus, the reaction of aldehyde 3 with 4 and C60 in refluxing ortho-dichlorobenzene (ODCB) gave S1 in 47% isolated yield after column chromatography on silica gel. Fullerene substrate S2 was obtained by taking advantage of the versatile cyclopropanation reaction developed by Bingel.17 To this end, malonate 7 was prepared by the reaction of alcohol 5 with acid 618 under esterification conditions using N,N′-dicyclohexylcarbodiimide (DCC), 1-hydroxybenzotriazole (HOBt) and 4-dimethylaminopyridine (DMAP). The reaction of C60 with compound 7, iodine and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) under Bingel conditions17,19 then gave methanofullerene S2 in 47% yield.
 |
| | Scheme 1
Reagents and conditions: (i) C60, ODCB, Δ (47%); (ii) DCC, DMAP, HOBt, CH2Cl2, 0 °C to r.t. (91%); (iii) C60, I2, DBU, PhMe, r.t. (47%). | |
Owing to the presence of the 3,5-didodecyloxybenzyl unit, compounds S1 and S2 are very soluble in common organic solvents such as toluene, CH2Cl2, CHCl3 and THF, and complete spectroscopic characterization was easily achieved. The structures of both S1 and S2 were further confirmed by mass spectrometry. The expected molecular ion peaks were observed at m/z 1299.5 for S1 ([M]+, calc. for C98H62N2O2 1299.58) and m/z 1401.4 for S2 ([M + H]+, calc. for C102H66NO6 1401.65).
Absorption and emission binding studies
The association of LZn with fullerene substrates S1 and S2 was studied in CH2Cl2 at 25 °C by UV-vis binding studies. The addition of S1 or S2 to LZn resulted in a bathochromic shift of the Zn(II)–porphyrin absorption bands, in agreement with the axial ligation of the pyridyl moieties (Fig. 2).5Luminescence titrations were also carried out. Indeed, a strong quenching of the porphyrin emission was observed upon addition of the C60–pyridine derivatives to CH2Cl2 solutions of LZn (Fig. 2). At this point, it must be emphasized that both intramolecular and intermolecular (collisions and re-absorption events) quenching processes can occur. In order to obtain a suitable reference, all the investigations on mixtures of LZn and S1 or S2 were carried out in parallel with mixtures of the porphyrin receptor and model fullerene derivative 8 (Fig. 3), which is unable to form a supramolecular complex with Zn(II)–porphyrins (i.e. a C60 derivative with no pyridine unit).6,9 Since a comparison with the reference solution is always made, the intermolecular quenching processes can be ignored, and the difference in emission intensity between the two solutions only accounts for intramolecular quenching. The titrations were performed at constant concentration of porphyrin LZn. The spectral changes observed in the emission spectra upon addition of S1 or S2 were recorded. Excitation occurred at an isosbestic point, at a wavelength where both complexed and uncomplexed species exhibit the same molar absorption coefficient. In any case, the porphyrin emission bands were quenched and red-shifted with successive addition of S1 or S2 to LZn. Finally, for the sake of comparison and to emphasize the role of the fullerene units in the final conjugates, the binding properties of pyridine (Py) were also examined. In all of these cases, the titrations allowed the characterization of a single supramolecular complex: [LZn·py] (log K1 = 3.55(4)), [LZn·S1] (log K1 = 3.50(8)) and [LZn·S2] (log K1 = 3.66(8)). The binding constants were not strongly dependent on the nature of the substrate, thus pointing out the absence of additional interactions between the porphyrinic π-system and the C60 unit within the supermolecules obtained from S1 and S2.
![Top: UV-vis absorption spectrophotometric titration of LZn with S1: l = 0.2 cm (l: optical cell pathlength); (a) [LZn]tot = 1.85 × 10–4 M, (b) [S1]tot/[LZn]tot = 6.8. Bottom: Luminescence spectrophotometric titration of LZn with S1; λexc = 559 nm, emission and excitation slit widths 15 and 20 nm, respectively; (a) [LZn]tot = 1.79 × 10–6 M, (b) [S1]tot/[LZn]tot = 63; solvent CH2Cl2, T = 25.0(2) °C.](/image/article/2008/NJ/b712081h/b712081h-f2.gif) |
| | Fig. 2 Top: UV-vis absorption spectrophotometric titration of LZn with S1: l = 0.2 cm (l: optical cell pathlength); (a) [LZn]tot = 1.85 × 10–4 M, (b) [S1]tot/[LZn]tot = 6.8. Bottom: Luminescence spectrophotometric titration of LZn with S1; λexc = 559 nm, emission and excitation slit widths 15 and 20 nm, respectively; (a) [LZn]tot = 1.79 × 10–6 M, (b) [S1]tot/[LZn]tot = 63; solvent CH2Cl2, T = 25.0(2) °C. | |
 |
| | Fig. 3 Model compound 8. | |
The binding behavior of Py, S1 and S2 to bis-metalloporphyrin LZn2 was also investigated by UV-vis absorption and luminescence in CH2Cl2. The results of the thermodynamic studies of LZn2 with the different substrates are summarized in Table 1. As a typical example, the UV-vis and luminescence spectrophotometric titrations of LZn2 with substrate S2 are shown in Fig. 4.
![Top: UV-vis absorption spectrophotometric titration of LZn2 with S2; l = 0.2 cm; (a) [LZn2]tot = 5.65 × 10–5 M; (b) [S2]tot/[LZn2]tot = 5.5. Bottom: Luminescence spectrophotometric titration of LZn2 with S2. λexc = 557 nm; emission and excitation slit widths 15 nm and 20 nm, respectively; (a) [LZn2]tot = 1.16 × 10–6 M; (b) [S2]tot/[LZn2]tot = 46.8. Solvent: CH2Cl2; T = 25.0(2) °C.](/image/article/2008/NJ/b712081h/b712081h-f4.gif) |
| | Fig. 4 Top: UV-vis absorption spectrophotometric titration of LZn2 with S2; l = 0.2 cm; (a) [LZn2]tot = 5.65 × 10–5 M; (b) [S2]tot/[LZn2]tot = 5.5. Bottom: Luminescence spectrophotometric titration of LZn2 with S2. λexc = 557 nm; emission and excitation slit widths 15 nm and 20 nm, respectively; (a) [LZn2]tot = 1.16 × 10–6 M; (b) [S2]tot/[LZn2]tot = 46.8. Solvent: CH2Cl2; T = 25.0(2) °C. | |
Table 1 Stability constants determined for LZn2 and substrates S1, S2 and Py by UV-vis and luminescence binding studiesa
| Substrate |
log K1 |
log K2 |
log β2 |
K
2/K1 |
|
Solvent CH2Cl2, T = 25.0(2) °C, error = 3σ.
Determined from the UV-vis absorption titration .
Determined from the luminescence titration (σ: standard deviation; β2: global stability constant).
|
|
Py
|
4.1(1)b |
3.4(3)b |
7.4(3)b |
0.3(1) |
| 4.1(3)c |
3.6(9)c |
7.7 (9)c |
|
S1
|
— |
— |
8.4(5)b |
3.2(7) |
| 4.02(2)c |
4.5(2)c |
8.5(2)c |
|
S2
|
— |
— |
8.6(1)b |
10.0(3.3) |
| 3.7(3)c |
4.7(3)c |
8.4(3)c |
Both the UV-vis and luminescence studies of LZn2 with substrate Py showed the presence of two complexes: [LZn2·Py] and [LZn2·Py2]. In contrast, only the global stability constants of complexes [LZn2·S12] and [LZn2·S22] were deduced from the UV-vis titrations, suggesting that the 1 : 1 complexes are minor species under our experimental conditions. This observation is a first indication of positively-cooperative interactions in the 1 : 2 associates. Processing of the luminescence data allowed us to precisely determine the successive stability constants (K1 and K2), defined by equilibria (1) and (2).
| | 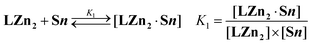 | (1) |
| | 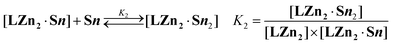 | (2) |
A thorough examination of these thermodynamic parameters thus confirmed the strong positive cooperativity in the self-assembly of the 1 : 2 ensembles. Indeed, the
K2/
K1 ratio provides a criterion to quantify the interactions between the two identical and independent binding sites.
20 For the binding of
S1 and
S2 to
LZn2, the
K2/
K1 values summarized in
Table 1 are significantly larger than 0.25, which is the value expected for a statistical model, clearly indicating positive intramolecular interactions in the 1 : 2 associates. The observed cooperativity may be ascribed to strong intramolecular
fullerene –
fullerene interactions between the two guests within [
LZn2·
S12] and [
LZn2·
S22] (
Fig. 5). This hypothesis is in line with previous observations made on supramolecular
C60–oligophenylenevinylene conjugates,
21 and is further supported by the absence of any positive interactions for the 2 : 1 complex obtained from
LZn2 and
Py, for which the
K2/
K1 ratio ≈ 0.3(1). It is also important to highlight that the
K2/
K1 ratio is significantly greater for substrate
S2 than
S1. Indeed, the chain connecting the
C60 moiety to the pyridine binding unit in
S2 gives a larger degree of flexibility, thus allowing optimization of the contacts between the two
C60 spheres in the 1 : 2 complex.
![Calculated structure of the supramolecular complex [LZn2·S12] showing the intramolecular fullerene–fullerene interactions (the dodecyl chains have been replaced by methyl groups in the calculations.](/image/article/2008/NJ/b712081h/b712081h-f5.gif) |
| | Fig. 5 Calculated structure of the supramolecular complex [LZn2·S12] showing the intramolecular fullerene –fullerene interactions (the dodecyl chains have been replaced by methyl groups in the calculations. | |
Upon addition of Py, the UV-vis spectrum of LZn6 changed substantially, and the observed bathochromic shifts of the B and Q bands were in full agreement with the apical coordination of the Py substrate to the Zn(II)–porphyrin moieties of LZn6. As shown in Fig. 6, Job’s plot22 revealed a 1 : 6 stoichiometry for the complex of Py with LZn6. The UV-vis titration results are also presented in Fig. 6. Interestingly, clear isosbestic points are observed. This result may reflect the fact that the Zn(II)–porphyrin units in LZn6 behave as independent binding sites. Indeed, the absorption and emission spectra of LZn6 were similar to those of LZn and LZn2, thus providing further evidence that the porphyrin sub-units behave independently in LZn6.
![UV-vis spectrophotometric titration of LZn6 with substrate Py; l = 0.5 cm; (a) [LZn6]tot = 1.17 × 10–6 M; (b) [Py]tot/[LZn6]tot = 735; solvent CH2Cl2, T = 25.0(2) °C. Inset: Job’s plots (ΔA/ΔAmax at 564 nm) upon mixing LZn6 and Py; ([LZn6]tot + [Py]tot) = 3.0 × 10–5 M.](/image/article/2008/NJ/b712081h/b712081h-f6.gif) |
| | Fig. 6 UV-vis spectrophotometric titration of LZn6 with substrate Py; l = 0.5 cm; (a) [LZn6]tot = 1.17 × 10–6 M; (b) [Py]tot/[LZn6]tot = 735; solvent CH2Cl2, T = 25.0(2) °C. Inset: Job’s plots (ΔA/ΔAmax at 564 nm) upon mixing LZn6 and Py; ([LZn6]tot + [Py]tot) = 3.0 × 10–5 M. | |
The same studies were carried out with compounds S1 and S2. In both cases, a Job’s plot provided evidence for 1 : 6 complex formation, and the UV-vis titrations were closely similar to that discussed for Py. The apparent association constants,11 deduced from the spectrophotometric titrations of LZn6 with Py, S1 and S2, are summarized in Table 2. The complexation of LZn6 with ligands Py, S1 and S2 was further investigated by luminescence experiments. As a typical example, the luminescence spectrophotometric titration of LZn6 with substrate S2 is shown in Fig. 7. The apparent association constants derived from the emission data (Table 2) are higher by about one order of magnitude for the fullerene -substituted ligands S1 and S2 compared to those obtained from the UV-vis titrations. These differences may be ascribed to the partial quenching of the emission of free Zn(II)–porphyrin units in LZn6 upon binding of the first fullerene ligand. However, the binding studies clearly revealed a significant increase in the binding constants for the fullerene -functionalized substrates relative to Py. As discussed for LZn2 (vide supra), the latter observation can be rationalized by the existence of intramolecular π–π interactions between the different fullerene sub-units within the complexes, resulting from the association of LZn6 with S1 or S2. This positive cooperative effect is more pronounced for substrate S2. As seen for binding to the ditopic receptor LZn2, the spacer connecting the C60 unit to the pyridine moiety in S2 brings sufficient flexibility to optimize the fullerene –fullerene interactions.
![Luminescence
titration of LZn6 with S2. Solvent CH2Cl2, T = 25.0(2) °C, λex = 428 nm, emission and excitation slit widths 15 and 20 nm, respectively; (a) [LZn6]tot = 4.91 × 10–8 M; (b) [S2]/[LZn6]tot = 277.](/image/article/2008/NJ/b712081h/b712081h-f7.gif) |
| | Fig. 7
Luminescence
titration of LZn6 with S2. Solvent CH2Cl2, T = 25.0(2) °C, λex = 428 nm, emission and excitation slit widths 15 and 20 nm, respectively; (a) [LZn6]tot = 4.91 × 10–8 M; (b) [S2]/[LZn6]tot = 277. | |
Table 2 Apparent successive stability constants (log K*) determined for LZn6 and substrates Py, S1 and S2 by UV-vis and luminescence binding studiesa
Conclusion
Fullerene derivatives bearing a pyridine sub-unit have been prepared. Their ability to form self-assembled supramolecular structures with mono- and polytopic Zn(II)–porphyrin receptors has been first evidenced by UV-vis studies. Further binding studies were easily carried out in solution thanks to efficient intramolecular quenching of the emission of the Zn(II)–porphyrin receptors by the C60 acceptor within the non-covalent arrays. The presence of the fullerene sub-unit in the guests is not only important for their ability to act as energy and/or electron acceptors in photoactive assemblies, but is also of significant importance in the increased stability of the 2 : 1 and 6 : 1 complexes containing C60–pyridine derivatives, and the polytopic receptors LZn2 and LZn6, respectively, due to positive cooperative interactions. The results reported in this paper demonstrate that increasing the number of building blocks does not constitute a severe limitation in the self-assembly of large supramolecular architectures. However, it shows that the largest assemblies are strengthened due to increasing numbers of secondary interactions (π–π stacking, hydrophobic interactions) within the self-assembled photoactive devices.
Experimental
General methods
Reagents and solvents were purchased as reagent grade and used without further purification. Compounds LZn,12LZn2,12LZn6,13415 and 618 were prepared according to previously reported procedures. All reactions were performed in standard glassware under an inert Ar atmosphere. Evaporation and concentration were performed at water aspirator pressure with drying in vacuo at 10–2 Torr. Column chromatography : Silica gel 60 (230–400 mesh, 0.040–0.063 mm) was purchased from E. Merck. Thin Layer Chromatography (TLC) was performed on glass sheets coated with silica gel 60 F254 purchased from E. Merck, visualized by UV light. NMR spectra were recorded on a Bruker AM 300 (300 MHz) instrument with solvent peaks as a reference. FAB mass spectra (MS) were obtained on a ZA HF instrument with 4-nitrobenzyl alcohol as the matrix. Elemental analyses were performed by the analytical service at the Laboratoire de Chimie de Coordination, Toulouse.
Synthesis
Compound S1.
A solution of 3 (45 mg, 0.42 mmol), C60 (300 mg, 0.42 mmol) and 4 (333 mg, 0.63 mmol) in ODCB (60 mL) was refluxed for 12 h. The mixture was cooled to r.t., filtered and evaporated to dryness under reduced pressure. Column chromatography (SiO2, CH2Cl2/MeOH 95 : 5) yielded brown glassy product S1 (256 mg, 47%). 1H NMR (CDCl3, 300 MHz): δ 0.88 (t, J = 7 Hz, 6 H), 1.30–1.43 (m, 32 H), 1.58 (m, 4 H), 1.79 (m, 4 H), 3.62 (d, J = 13.5 Hz, 1 H), 4.01 (m, 4 H), 4.19 (d, J = 9.5 Hz, 1 H), 4.43 (d, J = 13.5 Hz, 1 H), 4.92 (d, J = 9.5 Hz, 1 H), 5.20 (s, 1 H), 6.49 (t, J = 2 Hz, 1 H), 6.80 (d, J = 2 Hz, 2 H), 7.89 (br s, 2 H) and 8.72 (d, J = 6 Hz, 2 H). FAB-MS: m/z 1299.5 ([M]+, calc. for C98H62N2O2 1299.58). Elemental analysis calc. for C98H62N2O2·MeOH: C, 89.30; H, 5.00; N, 2.10; found C, 88.90; H, 5.04; N, 2.24%.
Compound 7.
DCC (2.90 mg, 14.19 mmol) and DMAP (173 mg, 1.42 mmol) were added to a solution of alcohol 5 (536 mg, 3.91 mmol) and acid 6 (2.0 g, 3.55 mmol) in CH2Cl2 stabilized by amylene (100 mL) at 0 °C under argon. After 1 h, the cooling bath was removed. The mixture was allowed to slowly warm to r.t. After 24 h, the mixture was filtered and the solvent removed under reduced pressure. Column chromatography (SiO2, CH2Cl2/MeOH 95 : 5) afforded 7 (2.20 mg, 91%) as a colorless, glassy product. IR (KBr, cm–1): 1749 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). 1H NMR (CDCl3, 300 MHz): δ 0.87 (t, J = 7 Hz, 6 H), 1.27 (m, 36 H), 1.67 (m, 4 H), 2.02 (m, 2 H), 2.71 (t, J = 6 Hz, 2 H), 3.69 (s, 2 H), 3.88 (t, J = 6.5 Hz, 4 H), 4.26 (t, J = 6 Hz, 2 H), 5.07 (s, 2 H), 6.42 (m, 3 H), 7.23 (m, 2 H) and 8.56 (m, 2 H).
O). 1H NMR (CDCl3, 300 MHz): δ 0.87 (t, J = 7 Hz, 6 H), 1.27 (m, 36 H), 1.67 (m, 4 H), 2.02 (m, 2 H), 2.71 (t, J = 6 Hz, 2 H), 3.69 (s, 2 H), 3.88 (t, J = 6.5 Hz, 4 H), 4.26 (t, J = 6 Hz, 2 H), 5.07 (s, 2 H), 6.42 (m, 3 H), 7.23 (m, 2 H) and 8.56 (m, 2 H).
Compound S2.
DBU (0.2 mL, 1.39 mmol) was added to a solution of C60 (400 mg, 0.56 mmol), compound 7 (378 mg, 0.56 mmol) and iodine (155 mg, 0.61 mmol) in toluene (600 mL) under argon. After 12 h, the mixture was filtered over SiO2 (CH2Cl2 then CH2Cl2/MeOH 95 : 5) and the solvent removed under reduced pressure. Purification by column chromatography (SiO2, CH2Cl2/MeOH 95 : 5) afforded S2 (545 mg, 47%) as a dark red, glassy product. IR (KBr, cm–1): 1752 (CO). 1H NMR (CDCl3, 300 MHz): δ 0.87 (t, J = 7 Hz, 6 H), 1.27 (m, 36 H), 1.67 (m, 4 H), 2.05 (m, 2 H), 2.75 (t, J = 6 Hz, 2 H), 3.88 (t, J = 6.5 Hz, 4 H), 4.35 (t, J = 6 Hz, 2 H), 5.44 (s, 2 H), 6.35 (t, J = 1.5 Hz, 1 H), 6.52 (d, J = 1.5 Hz, 2 H), 7.15 (m, 2 H) and 8.47 (m, 2 H). FAB-MS: m/z 1401.4 ([M + H]+, calc. for C102H66NO6 1401.65). Elemental analysis calc. for C102H65NO6·H2O: C, 86.36; H, 4.76; N, 2.10; found C, 86.14; H, 4.77; N, 0.99%.
Compound 8.
As described for S1, with benzaldehyde (59 mg, 0.56 mmol), C60 (400 mg, 0.56 mmol) and 4 (333 mg, 0.63 mmol) in ODCB (100 mL). Column chromatography (SiO2, hexane/CH2Cl2 8 : 2) yielded 8 (327 mg, 45%) as a brown, glassy product. 1H NMR (CDCl3, 300 MHz): δ 0.88 (t, J = 7 Hz, 6 H), 1.30–1.43 (m, 32 H), 1.58 (m, 4 H), 1,79 (m, 4 H), 3.61 (d, J = 13.5 Hz, 1 H), 3.96 (m, 4 H), 4.15 (d, J = 9.5 Hz, 1 H), 4.43 (d, J = 13.5 Hz, 1 H), 4.92 (d, J = 9.5 Hz, 1 H), 5.21 (s, 1 H), 6.45 (t, J = 2 Hz, 1 H), 6.76 (d, J = 2 Hz, 2 H), 7.40 (m, 1 H), 7.48 (m, 2 H) and 7.85 (m, 2 H). FAB-MS: m/z 1299.5 ([M + H]+, calc. for C99H64NO2 1299.60). Elemental analysis calc. for C99H63NO2·H2O: C, 90.25; H, 5.05; N, 1.06; found C, 90.38; H, 5.01; N, 1.21%.
Binding studies
All the binding studies were carried out with spectroscopic grade CH2Cl2 (E. Merck, 99.9% for spectroscopy). To prevent any photochemical degradation, all the solutions were protected from daylight exposure. All stock solutions were prepared using an AG 245 Mettler Toledo analytical balance (precision 0.01 mg), and complete dissolution in CH2Cl2 was achieved using an ultrasonic bath. The concentrations of stock solutions of the receptors and substrates (≈10–4 M) were calculated by the quantitative dissolution of solid samples in CH2Cl2.
UV-vis titrations
The spectrophotometric titrations of LZn, LZn2 and LZn6 with S1 ([LZn]tot = 1.84 × 10–4 M, [LZn2]tot = 5.65 × 10–5 M, [LZn6]tot = 2.93 × 10–6 M) and S2 ([LZn]tot = 5.82 × 10–5 M, [LZn2]tot = 5.65 × 10–5 M, [LZn6]tot = 3.04 × 10–6 M) were carried out in a Hellma quartz optical cell (l = 0.2 cm). To evaluate the influence of the fullerene units on the binding constants, spectrophotometric titrations of the same receptors with Py were conducted under similar experimental conditions ([LZn]tot = 1.79 × 10–6 M, l = 1 cm; [LZn2]tot = 1.13 × 10–6 M, l = 1 cm; [LZn6]tot = 1.17 × 10–6 M, l = 0.5 cm). Microvolumes of a concentrated solution of S1, S2 or Py were added to 2, 1 or 0.4 mL of LZn, LZn2 or LZn6 with µL Hamilton syringes (#710 and #750). The [substrate]tot/[receptor]tot ratios were varied within ranges from 0 to 863 for Py, from 0 to 108 for S1 and from 0 to 24.5 for S2 (see the ESI for detailed conditions† ). Special care was taken to ensure that complete equilibration was attained. The corresponding UV-vis spectra were recorded from 290 to 700 nm on a Cary 300 (Varian) spectrophotometer maintained at 25.0(2) °C by the flow of a Haake NB 22 thermostat. The spectrophotometric data were processed using the Specfit program,23 which adjusts the stability constants and the corresponding extinction coefficients of the species formed at equilibrium. Specfit uses factor analyses to reduce the absorbance matrix and extract the eigenvalues prior to the multi-wavelength fit of the reduced data set according to the Marquardt algorithm.24
Luminescence titrations were carried out on solutions of LZn, LZn2 and LZn6 with absorbances smaller than 0.1 at wavelengths ≥ λexc in order to avoid any errors due to the inner filter effect. The titrations of 2 mL of LZn, LZn2 and LZn6 with Py ([LZn]tot = 1.79 × 10–6 M; [LZn2]tot = 1.13 × 10–6 M; [LZn6]tot = 1.17 × 10–6 M), S1 ([LZn]tot = 1.79 × 10–6 M; [LZn2]tot = 1.13 × 10–6 M; [LZn6]tot = 4.91 × 10–8 M) and S2 ([LZn]tot = 1.90 × 10–6 M; [LZn2]tot = 1.16 × 10–6 M; [LZn6]tot = 4.91 × 10–8 M) were carried out in a 1 cm Hellma quartz optical cell by the addition of known microvolumes of solutions of S1, S2 and Py with µL Hamilton syringes (#710 and #750). The [substrate]tot/[receptor]tot ratios were varied within ranges from 0 to 531 for Py, from 0 to 220 for S1 and from 0 to 277 for S2 (see ESI for detailed conditions† ). Special care was taken to ensure that complete equilibration was attained. The excitation wavelengths were set at 557(1) or 559(1) nm for LZn, at 557(1) or 559(1) nm for LZn2 and 558(1) or 428(1) nm for LZn6, respectively, and correspond, in most of the cases, to the isosbestic points between the electronic spectra of the free Zn(II)porphyrinic receptors and the corresponding pentacoordinated complexes. The Zn(II)porphyrin -centered luminescence spectra were recorded from 500 to 800 nm on a Perkin-Elmer LS-50B instrument maintained at 25.0(2) °C by the flow of a Haake FJ thermostat. The slit widths were set at 15 and 20 nm for the emission and excitation, respectively. Luminescence titrations of LZn, LZn2 and LZn6 receptors were conducted under precise and identical experimental conditions using a model fullerene derivative that was unable to form complexes (C60 derivative with a phenyl group as a substitute for the pyridine unit) in order to separate the variation of the luminescence intensity that results from dynamic and re-absorption phenomena. For the Py substrate, the spectrofluorimetric data were processed with the Specfit program.23 For guests S1 and S2, the luminescence data sets were analyzed25,26 with the Microcal Origin program.27
Acknowledgements
This research was supported by the CNRS (UMR 7177 and UPR 8241), the French Ministry of Research (ACI Jeunes Chercheurs to N. S., doctoral fellowships to A. T. and M. U.) and the EU (HPRN-CT-2002-00171, FAMOUS).
References
-
(a)
J.-M. Lehn, Supramolecular Chemistry: Concepts and Perspectives, VCH, Weinheim, 1995 Search PubMed;
(b)
F. Vögtle, Supramolecular Chemistry, Wiley, Chichester, 1991 Search PubMed;
(c)
Supramolecular Chemistry of Fullerenes, ed. N. Martin and J.-F. Nierengarten, Tetrahedron Symposium-in-Print, 2006, vol. 62, pp. 1905–2132 Search PubMed;
(d)
Supramolecular Chemistry Anniversary, ed. P. A. Gale, Chem. Soc. Rev., 2007, 36, 125–440 Search PubMed.
- For recent reviews, see:
(a) L. Sanchez, N. Martin and D. M. Guldi, Angew. Chem., Int. Ed., 2005, 44, 5374 CrossRef CAS;
(b) V. Balzani, A. Credi, S. Silvi and M. Venturi, Chem. Soc. Rev., 2006, 35, 1135 RSC;
(c) J.-F. Nierengarten, U. Hahn, T. M. Figueira-Duarte, F. Cardinali, N. Solladié, M. E. Walther, A. Van Dorsselaer, H. Herschbach, E. Leize, A.-M. Albrecht-Gary, A. Trabolsi and M. Elhabiri, C. R. Chim., 2006, 9, 1022 CrossRef CAS;
(d) T. M. Figueira-Duarte, A. Gégout and J.-F. Nierengarten, Chem. Commun., 2007, 109 RSC;
(e) Y. Nakamura, N. Aratani and A. Osuka, Chem. Soc. Rev., 2007, 36, 831 RSC.
- For selected examples, see:
(a) M. Linke, J.-C. Chambron, V. Heitz, J.-P. Sauvage, S. Encinas, F. Barigelletti and L. Flamigni, J. Am. Chem. Soc., 2000, 122, 11834 CrossRef CAS;
(b) J. Brettar, J.-P. Gisselbrecht, M. Gross and N. Solladié, Chem. Commun., 2001, 733 RSC;
(c) E. H. A. Beckers, A. P. H. J. Schenning, P. A. van Hal, A. El-ghayoury, L. Sánchez, J. C. Hummelen, E. W. Meijer and R. A. Janssen, Chem. Commun., 2002, 1, 2888 RSC;
(d) E. H. A. Beckers, P. A. van Hal, A. P. H. J. Schenning, A. El-ghayoury, E. Peeters, M. T. Rispens, J. C. Hummelen, E. W. Meijer and R. A. J. Janssen, J. Mater. Chem., 2002, 12, 2054 RSC;
(e) F. Hauke, A. Swartz, D. M. Guldi and A. Hirsch, J. Mater. Chem., 2002, 12, 2088 RSC;
(f) S. R. Wilson, S. MacMahon, F. T. Tat, P. D. Jarowski and D. I. Schuster, Chem. Commun., 2003, 226 RSC;
(g) U. Hahn, M. Elhabiri, A. Trabolsi, H. Herschbach, E. Leize, A. Van Dorsselaer, A.-M. Albrecht-Gary and J.-F. Nierengarten, Angew. Chem., Int. Ed., 2005, 44, 5338 CrossRef CAS;
(h) M. Koepf, A. Trabolsi, M. Elhabiri, J. A. Wytko, D. Paul, A.-M. Albrecht-Gary and J. Weiss, Org. Lett., 2005, 7, 1279 CrossRef CAS;
(i) L. Flamigni, A. M. Talarico, B. Ventura, R. Rein and N. Solladié, Chem.–Eur. J., 2006, 12, 701 CrossRef CAS;
(j) A. Hosseini, S. Taylor, G. Accorsi, N. Armaroli, C. A. Reed and P. D. W. Boyd, J. Am. Chem. Soc., 2006, 128, 15904.
-
(a) D. M. Guldi, Chem. Soc. Rev., 2002, 31, 22 RSC;
(b) H. Imahori and Y. Sakata, Eur. J. Org. Chem., 1999, 2445 CrossRef CAS;
(c) D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2001, 34, 40 CrossRef CAS;
(d) H. Imahori, J. Phys. Chem. B, 2004, 108, 6130 CrossRef CAS;
(e) H. Imahori, Org. Biomol. Chem., 2004, 2, 1425 RSC.
-
(a) F. D’Souza and O. Ito, Coord. Chem. Rev., 2005, 249, 1410 CrossRef CAS;
(b) A. Mateo-Alonso, C. Sooambar and M. Pratro, C. R. Chim., 2006, 9, 944 CrossRef CAS.
- N. Armaroli, F. Diederich, L. Echegoyen, L. Flamigni, G. Marconi and J.-F. Nierengarten, New J. Chem., 1999, 23, 77 RSC.
- F. D’Souza, G. R. Deviprasad, M. S. Rahman and J. P. Choi, Inorg. Chem., 1999, 38, 2157 CrossRef CAS.
-
(a) F. D’Souza, S. Gadde, M. E. Zandler, M. Ito, Y. Araki and O. Ito, Chem. Commun., 2004, 2276 RSC;
(b) F. D’Souza, R. Chitta, S. Gadde, M. E. Zandler, A. S. D. Sandanayaka, Y. Araki and O. Ito, Chem. Commun., 2005, 1279 RSC;
(c) F. D’Souza, R. Chitta, S. Gadde, M. E. Zandler, A. L. McCarty, A. S. D. Sandanayaka, Y. Araki and O. Ito, Chem.–Eur. J., 2005, 11, 4416 CrossRef.
- A. Trabolsi, M. Elhabiri, M. Urbani, J. L. Delgado de la Cruz, F. Ajamaa, N. Solladié, A.-M. Albrecht-Gary and J.-F. Nierengarten, Chem. Commun., 2005, 5736 RSC.
- W. S. Li, K. S. Kim, D. L. Jiang, H. Tanaka, T. Kawai, J. H. Kwon, D. Kim and T. Aida, J. Am. Chem. Soc., 2006, 128, 10527 CrossRef CAS.
- J. L. Hou, H. P. Yi, X. B. Shao, C. Li, Z. Q. Wu, X. K. Jiang, L. Z. Wu, C. H. Tung and Z. T. Li, Angew. Chem., Int. Ed., 2006, 45, 796 CrossRef CAS.
- F. Ajamaa, A.-M. Albrecht-Gary, J. L. Delgado de la Cruz, M. Elhabiri, J.-F. Nierengarten, N. Solladié, A. Trabolsi and M. Urbani, J. Porphyrins Phthalocyanines, 2006, 10, 1337 CrossRef CAS.
- M. Takase, R. Ismael, R. Murakami, M. Ikeda, D. Kim, H. Shinmori, H. Furuta and A. Osuka, Tetrahedron Lett., 2002, 43, 5157 CrossRef CAS.
- M. Prato and M. Maggini, Acc. Chem. Res., 1998, 31, 519 CrossRef CAS.
- F. Ajamaa, T. M. Figueira-Duarte, C. Bourgogne, M. Holler, P. W. Fowler and J.-F. Nierengarten, Eur. J. Org. Chem., 2005, 3766 CrossRef CAS.
-
(a) J.-F. Nierengarten, Top. Curr. Chem., 2003, 228, 87 CAS;
(b) U. Hahn, F. Cardinali and J.-F. Nierengarten, New J. Chem., 2007, 31, 1128 RSC.
- C. Bingel, Chem. Ber., 1993, 126, 1957 CrossRef CAS.
- J.-F. Nierengarten and J.-F. Nicoud, Tetrahedron Lett., 1997, 38, 7737 CrossRef CAS.
- Modified Bingel conditions, see:
(a) J.-F. Nierengarten, V. Gramlich, F. Cardullo and F. Diederich, Angew. Chem., Int. Ed. Engl., 1996, 35, 2101 CrossRef CAS;
(b) X. Camps and A. Hirsch, J. Chem. Soc., Perkin Trans. 1, 1997, 1595 RSC.
-
(a) B. Perlmutter-Hayman, Acc. Chem. Res., 1986, 19, 90 CrossRef;
(b) G. Ercolani, J. Am. Chem. Soc., 2003, 125, 16097 CrossRef CAS.
-
(a) M. Elhabiri, A. Trabolsi, F. Cardinali, U. Hahn, A.-M. Albrecht-Gary and J.-F. Nierengarten, Chem.–Eur. J., 2005, 11, 4793 CrossRef CAS;
(b) J.-F. Nierengarten, U. Hahn, A. Trabolsi, H. Herschbach, F. Cardinali, M. Elhabiri, E. Leize, A. Van Dorsselaer and A.-M. Albrecht-Gary, Chem.–Eur. J., 2006, 12, 3365 CrossRef CAS.
-
K. A. Connors, Binding Constants, J. Wiley and Sons, New York, 1987, pp. 78 Search PubMed.
-
(a) H. Gampp, M. Maeder, C. J. Meyer and A. D. Zuberbühler, Talanta, 1985, 32, 95 CrossRef CAS;
(b) F. J. C. Rossoti, H. S. Rossoti and R. J. Whewell, J. Inorg. Nucl. Chem., 1971, 33, 2051 CrossRef CAS;
(c) H. Gampp, M. Maeder, C. J. Meyer and A. D. Zuberbühler, Talanta, 1985, 32, 257 CAS;
(d) H. Gampp, M. Maeder, C. J. Meyer and A. D. Zuberbühler, Talanta, 1986, 33, 943 CrossRef CAS.
-
(a) D. W. Marquardt, J. Soc. Ind. Appl. Math., 1963, 11, 431 Search PubMed;
(b) M. Maeder and A. D. Zuberbühler, Anal. Chem., 1990, 62, 2220 CrossRef CAS.
-
(a)
A. G. Marshall, Biophysical Chemistry, J. Wiley and Sons, New York, 1978, pp. 70 Search PubMed;
(b)
D. M. Freifelder, Physical Biochemistry, W. H. Freeman & Co., New York, 1982, pp. 659 Search PubMed;
(c) G. Scatchard, Ann. N. Y. Acad. Sci., 1949, 51, 660 CrossRef CAS.
-
(a) H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703 CrossRef CAS;
(b) M. Barra, C. Bohne and J. C. Scaiano, J. Am. Chem. Soc., 1990, 112, 8075 CrossRef CAS;
(c) H. Htun, J. Fluoresc., 2004, 14, 217 CrossRef.
-
Origin™ 7.5, Microcal Software, Inc., Northampton, USA, 2006 Search PubMed.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2008 |
Click here to see how this site uses Cookies. View our privacy policy here.