Extending the porphyrin core: synthesis and photophysical characterization of porphyrins with π-conjugated β-substituents
Barbara
Ventura
*a,
Lucia
Flamigni
a,
Giancarlo
Marconi
a,
Fabio
Lodato
b and
David L.
Officer
*bc
aIstituto per la Sintesi Organica e la Fotoreattività (ISOF), CNR, Via P. Gobetti 101, 40129, Bologna, Italy
bNanomaterials Research Centre, Massey University, Palmerston North, Private Bag, 11222, New Zealand
cMacDiarmid Institute for Advanced Materials and Nanotechnology, Massey University, Palmerston North, Private Bag, 11222, New Zealand
First published on 17th September 2007
Abstract
New free-base and Zn-porphyrins with either vinyl groups or p-phenylene vinylene groups substituted on the β carbon have been synthesized and studied from a photophysical viewpoint in toluene. A bathochromic shift of all absorption bands together with a pronounced decrease in the molar absorption coefficient at the wavelength of maximum absorption (εmax) and a broadening of the Soret band was observed with increasing conjugation of the substituent. A similar bathochromic shift trend was also observed in the corresponding emission spectra. The results are discussed in terms of delocalization of the frontier molecular orbitals of the porphyrin ring onto the substituent π-orbitals, and semi-empirical calculations confirming this interpretation are presented. High reactivity with oxygen was displayed by the triplet excited states of both free-base and Zn-porphyrins, and sensitized singlet oxygen (1Δg), detected via its luminescence, was formed with high yields. Moreover, the transient absorption spectra of the lowest triplet displayed interesting features in the near-infrared region: a clear enhancement in the molar absorption coefficient in the range 600–900 nm was observed with increasing conjugation of the substituent.
Introduction
The search for porphyrin systems linked by π-conjugated connectors is receiving increasing attention in view of their potential application in the design of molecular wires for electronic and photonic applications.1–3 In this context, the analysis of the effect of π-conjugated substituents on the photophysical properties of porphyrins is of great relevance.The extent of conjugation introduced by a substituent in the aromatic ring of porphyrins depends on several factors, such as the type of substituent introduced, the position of the substituent on the ring and, above all, the degree of coplanarity that can be achieved between the porphyrin plane and the plane/s of the unsaturated system/s connected to it. In fact, several studies on multi-porphyrinic arrays have demonstrated that when the spacer contains or consists of a phenyl group directly connected to the ring, the orbital interaction between the porphyrins and the bridge is strongly reduced by the close-to-perpendicular position preferentially assumed by the groups in order to minimize steric hindrance.4–6 In these systems, the through-bond electronic communication between the porphyrin units is negligible and their spectral properties are usually characterized by exciton coupling originating from the Coulombic interactions between the dipole moments of the adjacent units, that results only in a split of the Soret band. On the contrary, when the spacer is connected to the porphyrin rings via a vinyl or an ethynyl group, the π systems can generally be assumed to be coplanar. This favors the extension of the π-conjugation over the bridge and the formation of highly delocalized excited states. The spectral characteristics of these arrays are a bathochromic shift of all absorption bands compared to those of the parent porphyrin monomer, a pronounced broadening and/or splitting of the Soret band, and an enhancement of the Q-bands.4,7–9 For example, in fused porphyrin arrays, wherein the individual porphyrins are triply linked to form long tapes, the extensive π-conjugation results in extremely red-shifted (up to 3000 cm–1) and intensified Q-bands.10,11
Since the electronic properties of the frontier orbitals of porphyrins depend both on the nature and the position of the substituents,12–14 it is important, in designing substituted porphyrins characterized by highly delocalized molecular orbitals, to understand the effect of varying the point of substituent attachment. While the electronic properties of meso-substituted conjugated porphyrins have been well studied,15–19 the effect of conjugated β-substituents on the photochemical and electrochemical properties of porphyrins has not been explored in detail, and only a small number of papers reports on this topic.20–26 We are interested in the use of vinylic substituents to affect electronic communication between the units of multi-porphyrin arrays and, therefore, we have undertaken a detailed photophysical study of a series of β-vinyl substituted porphyrins commensurate with our investigations into the photoinduced energy and electron transfer processes in porphyrinic assemblies of different types and topologies.27–33
We present here the synthesis (Scheme 1) and full photophysical characterization in toluene of a series of four free-base (1, 3, 5, 7) and a series of four Zn-tetraphenylporphyrins (2, 4, 6, 8) substituted at the β-carbon with either vinyl groups or p-phenylene vinylene groups (Chart 1). The properties of the two series of β-substituted porphyrins have been compared with those of the parent porphyrins, TPP (tetraphenylporphyrin) and ZnTPP (Zn-tetraphenylporphyrin), respectively. It is shown that increasing delocalization of the porphyrin π-system occurs as the length of the conjugated substituent increases, and this has been supported by semiempirical calculations. Moreover, the triplet states of all the examined porphyrins show high reactivity with molecular oxygen and formation of singlet oxygen occurs in high yields.
 | ||
| Scheme 1 Synthesis of porphyrins 1––8 from precursors 10–15 and vinylstilbene 9 from 16. | ||
 | ||
| Chart 1 Porphyrins 1––8, vinylstilbene 9, aldehyde 16 and precursors 10–15. | ||
Results and discussion
Synthesis
All the β-substituted porphyrins were designed to contain a terminal methylene group so that the extension of the conjugation was maximized. Okuma et al.34 have shown that terminal vinyl aromatic compounds can be prepared in very good yields using the Wittig reaction of methyl phosphonium salt with activated aromatic aldehydes using DBU as base. Less activated aldehydes required longer reaction times and higher temperatures (higher boiling solvents). This seemed ideally suited to the preparation of the required vinylic porphyrins because it has been shown that porphyrin aldehydes readily undergo Wittig reactions.35 All compounds used in this study (1––9) were prepared according to this method from the respective aldehydes (Scheme 1). Because most of the precursor aldehydes are not particularly reactive, all the Wittig reactions were performed in refluxing dry toluene under argon. Yields were optimized by dissolving/suspending the porphyrin aldehyde and three equivalents of the phosphonium salt in toluene, and adding DBU in excess (10 equivalents) at reflux.In order to make both free-base and Zn metallated porphyrins, Wittig reactions can either be performed on free-base or on Zn porphyrin aldehydes and the homologue obtained by metallation/demetallation. Both of these latter reactions proceed under very mild conditions and with almost quantitative yield.
The preparation of 2-vinyl-TPP 1 has already been described,36 but in this work it was made from 2-formyl-TPP 10 using the simple Wittig conditions shown in Scheme 1. The syntheses of the other members of the series 2––8 have not been previously reported and they were obtained in moderate to good yields using the Wittig approach. The precursor porphyrin aldehydes 10, 11, 12, 13 and 14 have been reported.21 Extended porphyrins 7 and 8, containing two phenylene groups, were synthesized in four and three steps, respectively, from 2-formyl-ZnTPP 11via the extended Zn porphyrin aldehyde 15 (Scheme 2). Zn insertions were performed according to the acetate method 37 and free-base porphyrins were obtained by treating a dichloromethane solution of the Zn porphyrin with 3 M HCl.
 | ||
| Scheme 2 Synthesis of extended porphyrin aldehyde 15. | ||
The characterisation of the porphyrins 1––8 was straightforward. The 1H NMR spectra of all the compounds displayed proton magnetic resonances typical of β-substituted tetraphenylporphyrins with the characteristic broad NH singlets around –2.5 ppm for the free-base porphyrins. As illustrated for 8 in Fig. 1, the spectra of the vinylporphyrins 1, 2, 5––8 showed the normal AMX coupling pattern for the protons of a terminal vinyl group (three doublets of doublets between 5 and 7 ppm, Fig. 1), with geminal coupling of the terminal protons typically from 1–2 Hz. The vinylic protons closest to the porphyrin core typically resonate around 7.0 and 7.3 ppm as can be seen in the COSY spectrum in Fig. 1. The seven β-pyrollic proton resonances all occur within 0.4 ppm of each other furthest downfield. LR-MALDI and HR-FAB mass spectra were easily obtained for all the compounds presented; either molecular ions (M+) or protonated ions (M + H+) were measured.
 | ||
| Fig. 1 1H NMR spectrum (500 MHz, CDCl3) of styryl porphyrin 8, with its COSY spectrum shown in the inset. | ||
Absorption spectroscopy
The absorption spectra of reference TPP and free-base porphyrins 1, 3, 5 and 7 are displayed in Fig. 2. From inspection of the spectra it is clear that passing from TPP to 7, i.e. by increasing the degree of conjugation of the β-substituent, a dramatic decrease in εmax and a red-shift and a broadening of the Soret band occur (Fig. 2 and Table 1). In particular, there is a strong decrease in εmax compared to TPP, for 1, 3 and 5 where, respectively, one vinyl group, two vinyl groups and a divinylbenzene group are attached to the tetrapyrrolic ring. In porphyrin 7, that has a further phenylene vinylene group in the β-substituent, the Soret εmax remains unchanged compared to 5 but a further broadening of the band is observed. The red-shift of the Soret band is more pronounced in passing from TPP to 1 (from 419 to 424 nm) than to the other porphyrins in the series and in the Q-bands a general red-shift of all bands along the series occurs with increasing conjugation. | ||
| Fig. 2 Molar absorption coefficients of TPP and free-base porphyrins in toluene, with an expansion of the 470–700 nm region shown in the inset. | ||
| λ max/nm | 10–5ε/M–1 cm–1 | λ max/nm | 10–5ε/M–1 cm–1 | ||
|---|---|---|---|---|---|
| TPP | 419 | 4.43 | ZnTPP | 423 | 5.37 |
| 514 | 0.19 | 550 | 0.24 | ||
| 548 | 0.08 | 589 | 0.05 | ||
| 592 | 0.05 | ||||
| 648 | 0.04 | ||||
| 1 | 424 | 3.69 | 2 | 425 | 3.93 |
| 519 | 0.22 | 552 | 0.22 | ||
| 554 | 0.08 | 590 | 0.04 | ||
| 596 | 0.06 | ||||
| 653 | 0.03 | ||||
| 3 | 426 | 2.57 | 4 | 430 | 2.16 |
| 522 | 0.21 | 556 | 0.18 | ||
| 560 | 0.09 | 591 | 0.05 | ||
| 600 | 0.07 | ||||
| 659 | 0.02 | ||||
| 5 | 427 | 1.67 | 6 | 433 | 2.18 |
| 524 | 0.17 | 559 | 0.23 | ||
| 564 | 0.10 | 594 | 0.11 | ||
| 602 | 0.06 | ||||
| 659 | 0.01 | ||||
| 7 | 344 (sh) | 0.32 | 8 | 350 (sh) | 0.40 |
| 373 (sh) | 0.34 | 375 (sh) | 0.35 | ||
| 428 | 1.59 | 433 | 1.96 | ||
| 524 | 0.18 | 560 | 0.24 | ||
| 568 | 0.13 | 597 | 0.14 | ||
| 602 | 0.07 | ||||
| 659 | 0.02 | ||||
In the spectra of the Zn-porphyrins, Table 1, the Soret bands present a shift toward lower energies similarly to what is observed in the free-base series; the decrease of εmax is evident for 2 and 4 compared to the model ZnTPP whereas 6 and 8, with respect to 4, mainly show a broadening of the band around 470 nm. The Q bands, on the other hand, in addition to exhibiting the trend in the bathochromic shifts, also show a variation in the relative intensities: the Q(0,0) band increases its intensity while the Q(1,0) band is almost unaffected. No split of the Soret and of the Q bands can be observed in the absorption spectra of all examined porphyrins, a split that usually occurs when a significant distorsion of the D4h symmetry is induced by the presence of a substituent.38
It is quite evident that the absorption spectrum of the free-base porphyrin 7, that carries the longest substituent, is different from the plain superposition of the spectra of the components TPP and vinylstilbene unit 9, that can be considered a model for the β-substituent present in 7: while the TPP Soret band has a strongly decreased molar absorption coefficient and a broadening which extends to 500 nm (Fig. 2), the bands of 9 around 300–350 nm (ε of the order of 30000, data not shown) shift around 350–400 nm in 7. The same occurs for the Zn homologue 8 compared with the models ZnTPP and 9.
The observed bathochromic shifts can be explained by considering that conjugation generally reduces the HOMO–LUMO gap. In particular, the a1u and a2u HOMO orbitals of porphyrins have different electron-density on the β and the meso carbons and substituents with different electron donating or withdrawing capacity can modulate in different ways the energetic distribution of the porphyrin frontier orbitals (i.e. the a2u/a1u energies) according to their position.12–14,39 Thus, in the examined β-substituted porphyrins, the decrease of the HOMO–LUMO gap along the series could be due both to the stabilization of the LUMO orbitals as a consequence of the increased delocalization and to the destabilization of the filled HOMO orbitals (a1u in particular) as a result of mixing with the substituent π-orbitals that lie at lower energies.
In order to obtain support for the above rationalisations, a series of simple MO calculations based on the ZINDO/S Hamiltonian was performed, having first optimized the geometries with the MM+ method; 40 the results are reported in Table 2. Out of the components of the first band Q(0,0) (i.e.Qx and Qy), we observe a red shift in going from the unsubstituted models to the substituted porphyrins of both series, while the energy of the emitting state of compounds 1––8, (Qx), is calculated to be almost constant. In the case of the Zn-porphyrins, the oscillator strength of the whole band is predicted to increase with the extension of conjugation of the substituent especially for the Qx component, the Qy component being almost unaffected. The largest effect brought about by the substituent is predicted to be on the B (Soret) bands (here again with the Bx component much more likely affected than the By): in agreement with the experimental features, one observes a large red shift with the increase of the substituent size and a noticeable intensity drop in going from TPP (f = 6.50) and ZnTPP (f = 4.98) to the terms with larger substituent groups. These effects can be understood by examining the composition of the states in terms of electronic delocalization.
| λ/nm, f (S0 → S1) | λ/nm, f (S0 → S2) | ||||
|---|---|---|---|---|---|
| Qx | Qy | Bx | By | Δµ(S1 – S0)/D | |
| TPP | 646, 0.16 | 599, 0.34 | 355, 3.36 | 352, 3.14 | –1.37 |
| 1 | 654, 0.14 | 613, 0.36 | 359, 2.17 | 357, 2.27 | 0.22 |
| 3 | 657, 0.14 | 616, 0.35 | 389, 3.09 | 371, 0.79 | 0.68 |
| 5 | 658, 0.13 | 616, 0.38 | 414, 3.00 | 357, 1.68 | 0.65 |
| 7 | 655, 0.15 | 618, 0.44 | 449, 3.53 | 360, 1.78 | 0.72 |
| ZnTPP | 634, 0.26 | 634, 0.26 | 362, 2.66 | 358, 2.32 | –0.65 |
| 2 | 646, 0.32 | 640, 0.22 | 375, 2.46 | 365, 1.94 | –1.79 |
| 4 | 648, 0.34 | 641, 0.21 | 414, 1.79 | 374, 1.57 | 2.83 |
| 6 | 650, 0.35 | 642, 0.17 | 444, 1.80 | 381, 1.29 | 2.67 |
| 8 | 648, 0.39 | 640, 0.16 | 468, 2.67 | 388, 1.49 | 2.79 |
As it is well known from Gouterman’s four orbital theory,38 the Q and B bands derive from destructive and constructive promotions from the two lowest occupied MOs (a1u and a2u) to the degenerate LUMO e2g, in a D4h symmetry. In distorted symmetries, such as that of TPP, where the central hydrogen interaction causes a splitting of the e2g orbitals, one observes two distinct components of the Q band, as it is evident comparing the calculated energy of TPP and ZnTPP in Table 2. Moreover, the intensity ratio Q(1,0)/Q(0,0), where Q(1,0) refers to a vibronic quantum added to Q(0,0), has been shown to be proportional to the square of the HOMO–LUMO gap.41 While the substituents in the meso position influence mainly the a2u (HOMO) orbital, leaving almost unaffected the a1u (HOMO–1), which presents a node in the correspondence of the meso carbons of the pyrrole rings, the reversal occurs for β substitution. This is the reason why the x components of the Q and Soret bands are much more affected than the y counterparts; these components in fact derive, for example in the series of the Zn-porphyrins, from HOMO–1 (a1u) → LUMO+1 (e2g″) promotions by 35 and 55%, respectively. The destabilization of the a1u orbital is calculated as –0.39 and –0.24 eV going from ZnTPP to 8 and from TPP to 7, respectively, whereas the difference of energy of a2u is –0.02 eV and –0.03 eV for the same compounds. The increase in size of the substituent, beyond extending the delocalization of π electrons with consequent red shift of the fluorescence bands (see below), also causes a large delocalization of charges reflected in increased dipole moment differences between S1 and S0 (last column of Table 2). This feature could be exploited, in principle, to tune the emitting properties of this class of compounds using the appropriate substituents for particular functional applications.
Our results are in agreement with the observed progressive broadening and red-shifting of the Q bands with increase in conjugation chain length reported recently by Walsh et al.25 in a DFT study on the optical properties of Zn-tetraphenylporphyrins β-substituted with conjugated carboxylic acid side chains.
The shift to lower energies, of the order of 0.23 eV, of the bands of vinylstilbene 9 when it is attached to the porphyrin as in 7 and 8, can be taken as a further indication of the effective electronic delocalisation that occurs between the conjugated substituent and the porphyrin ring. It is well known, in fact, that the lowest energy absorption peak of p-phenylene vinylene oligomers shifts toward lower energies when the length of the oligomer, i.e. the π-conjugation, increases.42–44
Emission spectroscopy
The emission spectra of optically matched solutions of TPP and free-base porphyrins 1, 3, 5 and 7 in toluene at room temperature upon excitation at 520 nm are displayed in Fig. 3(a). The emission of the free-base porphyrins shifts to lower energy with respect to the emission of TPP when the conjugation of the β-substituent increases, with a trend that follows the behavior of the absorption spectra. A shift of 5–7 nm, in fact, occurs in the emission peaks for the first members of the series (1 and 3, see Table 3) while 5 and 7 are at the same wavelengths as 3. From inspection of the spectra obtained in a glassy matrix (Fig. 3(b)) where the emission bands are better resolved, the trend of the emission maximum moving to higher wavelengths with the increased conjugation of the substituent is confirmed and more evident also for the last two members of the series (see Table 3). This indicates a decrease of the energies of the lowest singlet excited state in the free-base porphyrin series, in agreement with the postulated extension of the π-conjugation of the porphyrin ring over the substituent. Such an effect was also recently reported for a series of β-substituted Zn-tetraphenylporphyrins with different linkers: beyond the effect of π-conjugation, a dependence on the electron-withdrawing or donating property of the terminal group was observed.22 | ||
| Fig. 3 Uncorrected emission spectra of optically matched solutions (A = 0.073) of TPP and free-base porphyrins in toluene excited at 520 nm at room temperature (a) and normalized spectra at 77 K (b). | ||
| 295 K | 77 K | ||||||
|---|---|---|---|---|---|---|---|
| λ max a/nm | Φ fl a | τ b | 10–7kr/s–1 | λ max a/nm | τ b/ns | E c/eV | |
| a Emission maxima derived from non-corrected emission spectra. Absolute quantum yields were determined by comparing corrected emission spectra, using TPP in aerated toluene as a standard (Φfl = 0.1112). Excitation at 520 nm for the free-base porphyrin series, at 560 nm for the Zn-porphyrin series and at 329 nm for model 9. Values in parentheses relate to the triplet state. b Excitation at 560 nm for the porphyrin series and at 331 nm for model 9. c Derived from the emission maxima at 77 K. Values in parentheses relate to the triplet state. | |||||||
| TPP | 650, 715 | 0.110 | 9.3 | 1.2 | 643, 712 | 13.4 | 1.92 |
| 1 | 655, 721 | 0.091 | 8.5 | 1.1 | 648, 718 | 12.3 | 1.91 |
| 3 | 662, 726 | 0.091 | 8.5 | 1.1 | 652, 721 | 12.4 | 1.90 |
| 5 | 663, 729 | 0.090 | 8.1 | 1.1 | 655, 724 | 12.2 | 1.89 |
| 7 | 662, 730 | 0.091 | 9.0 | 1.0 | 657, 726 | 13.7 | 1.89 |
| ZnTPP | 594, 642 | 0.047 | 1.9 | 2.5 | 598, 654, (780) | 2.7 | 2.07 (1.59) |
| 2 | 599, 645 | 0.044 | 1.7 | 2.6 | 602, 656, (777) | 2.5 | 2.06 (1.59) |
| 4 | 605, 654 | 0.050 | 1.8 | 2.8 | 607, 663, (785) | 2.6 | 2.04 (1.58) |
| 6 | 603, 659 | 0.063 | 1.9 | 3.3 | 611, 667, (789) | 2.6 | 2.03 (1.57) |
| 8 | 603, 660 | 0.073 | 1.8 | 4.1 | 613, 669, (792) | 2.4 | 2.02 (1.57) |
| 9 | 355, 375, 391 (sh) | 0.350 | 0.72 | 49.0 | 357, 376, 398 | 2.07 | 3.47 |
The fluorescence quantum yields Φfl (Table 3), calculated by comparing emission spectra corrected for the instrumental response, appear uniform within the series (Φfl = 0.090–0.091) and lower than that of reference TPP (Φfl = 0.11). The fluorescence lifetimes remain almost uniform along the series and similar to that of TPP, with a value that ranges from 8.1 to 9.0 ns at room temperature and from 12.2 ns to 13.7 ns at 77 K. As a consequence, the radiative rate constant (kr) of all the free-base porphyrins substantially equals that of unsubstituted TPP (1.2 × 107 s–1, see Table 3). The calculated oscillator strengths, reported in the first column of Table 2, are also consistent with the constant values extracted for the radiative constants along this series.
The emission spectra of optically matched solutions of ZnTPP and Zn porphyrins 2, 4, 6 and 8 in toluene at room temperature upon excitation at 560 nm are displayed in Fig. 4(a). All the Zn porphyrin emissions are red-shifted compared to ZnTPP with 6 and 8 significantly more so (Fig. 4(a) and Table 3), in a trend that follows the absorption behavior. Quite interestingly, a clear trend in the inversion of the relative intensities of the vibronic bands is also observed. It can be noted that the calculated wavelengths reported in Table 2 are in satisfactory agreement with the experimental ones (Table 3), showing a larger stabilization of the Qx band in both series going from the unsubstituted compounds (TPP and ZnTPP) to the substituted ones (8–12 nm) than among the last terms of the series (4 nm at most). The bathochromic shift of the emission bands of the Zn-porphyrins along the series is confirmed by the spectra obtained in glassy toluene solutions at 77 K, reported in Fig. 4(b). The calculated energies of the lowest singlet excited states are reported in Table 3 and their values decrease from 2.07 eV for the model ZnTPP to 2.02 eV for 8. Moreover, measurements at 77 K allowed the detection of the phosphorescence of all the examined Zn porphyrins that occurs at 780–790 nm, as can be observed in Fig. 4(b). We were unable to detect the phosphorescence in the porphyrins of the free-base series, but triplet states were identified in both free-base and Zn-porphyrins by their transient absorption spectra (see below). The measured energies of the Zn-porphyrin triplet states, reported in parentheses in Table 3, indicate a modest but continuous decrease in energy, indicating that, in addition to stabilizing the singlet states of porphyrins, an enhanced delocalization of the molecular orbitals also stabilizes the triplet states.
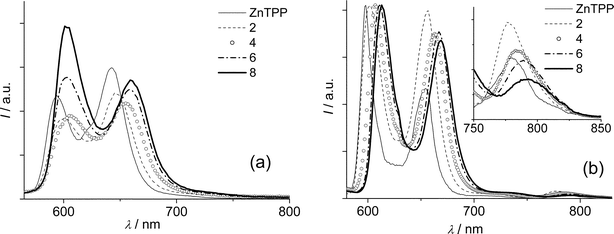 | ||
| Fig. 4 Uncorrected emission spectra of optically matched solutions (A = 0.077) of ZnTPP and Zn porphyrins in toluene excited at 560 nm at room temperature (a) and normalized spectra at 77 K (b). Expansion of the 750–850 nm region is shown in the inset. | ||
In contrast to the free-base series, the fluorescence quantum yields of the Zn-porphyrins 2 and 4 remain almost unchanged compared to ZnTPP (Φfl = 0.047) and only 6 (Φfl = 0.063) and 8 (Φfl = 0.073, see Table 3) show a significant increase. The measured lifetimes remain almost unchanged for all the Zn-porphyrins, varying from 1.7 to 1.9 ns at room temperature and from 2.4 ns to 2.7 ns at 77 K (Table 3). As a consequence an increment of the calculated radiative rate constant can be observed along the series, going from 2.5 × 107 s–1 for ZnTPP to 4.1 × 107 s–1 for 8. The presence of phenylene vinylene units, therefore, has a drastic effect on the emitting properties of the Zn-porphyrins, and the calculated oscillator strengths reported for this series (Table 2) show good agreement with this behaviour, with an increase of ca. 70% going from the unsubstituted ZnTPP to compound 8.
As discussed in the previous section, the spectral changes observed both in the absorption and in the emission properties along the two series indicate a progressively increased delocalization of the frontier molecular orbitals of the porphyrin unit. The extension of the π-conjugation over the substituent is possible if the molecule can assume a conformation where the porphyrin ring and the plane/s of the aromatic unit/s of the substituent are co-planar. For a β-substituted free-base tetraphenylporphyrin similar to porphyrin 5 (with an aldehyde terminal group attached to the phenyl instead of a vinyl one), the X-ray characterization demonstrated the co-planarity of the porphine ring and the phenyl substituent, with no participation in the porphine conjugation by the twisted meso-phenyl substituents.45 We observed the same feature in the calculated geometries of compounds 1––8, where an almost planar geometry of the porphine-substituent system is obtained even for the porphyrins with the longest conjugated substituents such as 7 and 8, supporting our interpretation of the experimental data. The meso-phenyls are twisted by ca. 42° with respect to the plane defined by the pyrroles and this deformation remains constant along the series.
As a model of the appended substituent in the most conjugated porphyrins, compound 9 has been characterized and the luminescence data are summarized in Table 3. Vinylstilbene 9 is a good emitter (Φfl = 0.35) with a lifetime of 0.72 ns at room temperature and 2.07 ns at 77 K, and the emitting excited state is localized at 3.47 eV, in agreement with previously reported oligo(p-phenylenevinylene)s with two phenyl rings.42–44 Steady-state experiments with excitation on the vinylstilbene band evidenced the complete quenching of the luminescence of unit 9, indicative of a very efficient energy transfer to the porphyrin, as previously reported for oligo(p-phenylenevinylene)s meso-substituted porphyrins.46,47
Transient absorption spectroscopy
The triplet states of the two porphyrin series have been probed by nanosecond flash photolysis. The transient absorption spectra obtained upon 532 nm excitation at ambient temperature in air-free toluene solutions at the end of the laser pulse are shown in Fig. 5 for the free-base porphyrins TPP, 3, 5 and 7 and in Fig. 6 for Zn-porphyrins ZnTPP, 4, 6 and 8. The spectral features of 1 and 2, with one vinyl group at the β-position of the porphyrin ring, are very similar to those of their analogs with two vinyl groups, 3 and 4, respectively (data not shown). The main absorption maxima wavelengths are summarized in Table 4 together with the triplet lifetimes obtained both in air-free and air-equilibrated toluene. The spectrum of the model TPP (Fig. 5) is characterized by a strong absorption band around 440 nm, a weaker band at 780 and a modest bleaching of the ground state features in the region 500–700 nm, in agreement with previously reported data.48,49 The spectra of the other porphyrins of the series show interesting differences. The first absorption band shifts increasingly along the series from 440 nm for TPP to 500 nm for 7, and its maximum absorption value reduces within the series. It must be borne in mind that the absorption properties in this region are determined by the ground state absorbance (Fig. 5 and Fig. 6 reports ΔA), so the apparent shift to higher wavelengths and the decrease in absorbance of the band is a consequence of the increasing absorbance of the ground state in the region 450–500 nm along the series (see Fig. 2). The bands in the near IR region, on the contrary, where no ground state absorbance takes place, are genuine: we can see that the band centered at 780 nm in TPP is present almost unchanged in the other porphyrins, except for 7 where it drastically increases and red-shifts (see Table 4 and Fig. 5). The existence of this intense band at 800 nm seems to be related to the presence of two phenylenevinylene groups attached to the TPP ring, since it is peculiar to porphyrin 7; this may provide application in the field of non linear optics in the near IR region.2,50 | ||
| Fig. 5 Transient absorption difference spectra in air-free toluene at the end of the pulse after excitation at 532 nm (18 ns pulse, 0.6 mJ pulse–1) for free-base porphyrins TPP, 3, 5, 7 (A532 = 0.53). | ||
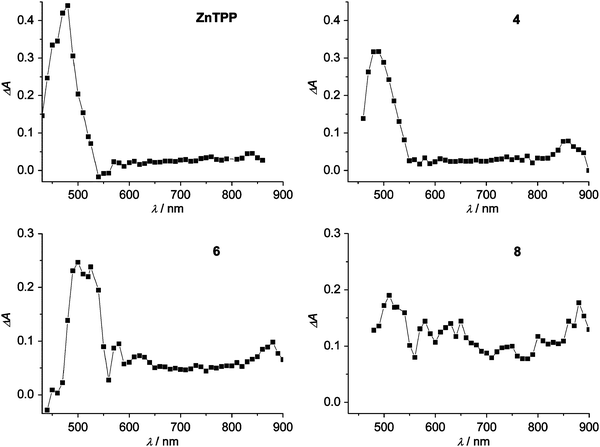 | ||
| Fig. 6 Transient absorption difference spectra in air-free toluene at the end of the pulse after excitation at 532 nm (18 ns pulse, 0.6 mJ pulse–1) for Zn-porphyrins ZnTPP, 4, 6, 8 (A532 = 0.55). | ||
| λ max/nm | τ a/µs | τ b/ns | 10–9kq/M–1 s–1 | Φ Δ c | |
|---|---|---|---|---|---|
| a Air-free solutions. b Air-equilibrated solutions. c The standard used is TPP in aerated toluene at room temperature (ΦΔ = 0.7058). Excitation at 520 nm for the free-base porphyrin series and at 560 nm for the Zn-porphyrin series. | |||||
| TPP | 440, 780 | 115 | 279 | 2.0 | 0.70 |
| 1 | 450, 780 | 110 | 272 | 2.0 | 0.79 |
| 3 | 460, 780 | 122 | 259 | 2.1 | 0.80 |
| 5 | 490, 780 | 110 | 260 | 2.1 | 0.79 |
| 7 | 500, 800 | 130 | 203 | 2.7 | 0.77 |
| ZnTPP | 480, 840 | 136 | 497 | 1.1 | 0.64 |
| 2 | 480, 850 | 126 | 554 | 1.0 | 0.80 |
| 4 | 490, 860 | 85 | 503 | 1.1 | 0.68 |
| 6 | 510, 870 | 90 | 433 | 1.3 | 0.79 |
| 8 | 510, — | 73 | 275 | 2.0 | 0.76 |
The ZnTPP spectrum (Fig. 6 and Table 4) has a strong band at 480 nm and a less pronounced band at 840 nm, in agreement with previous reports.49,51 Inspection of the spectra of the Zn series reveals a trend similar to the free-base series in the apparent decrease and bathochromic shift of the first absorption band. The small band at 840 nm present in ZnTPP shifts to longer wavelengths in the other porphyrins, although in porphyrin 8 there is no clear indication of a band in the range explored (up to 900 nm). A clear increase in the molar absorption coefficient in the range 600–900 nm is evident with increasing conjugation along the series. It should be noted that the observed spectra result from transitions from the lowest triplet to the upper levels of the same multiplicity and they cannot be a source of information on the energy of the triplet states, which are derived from the 77 K phosphorescence spectra, as reported above. They are, however, interesting since the substituted porphyrins exhibit clear spectral features that are quite different from the parent porphyrins (Table 4), indicating the involvement in the transitions of excited triplet states at energies different from TPP and ZnTPP. It is interesting to note that, in agreement with our data, in a study where the π-conjugation of tetraphenylmetalloporphyrins is increased through aromatic ring condensation at the pyrroles, together with an overall red-shift in the ground state absorption spectra, a red-shift of the T1–Tnabsorption spectra and an increase of the triplet molar extinction coefficient through the near-infrared region with annulation was found.52
The triplet lifetimes of the free-base porphyrins are similar and range from 110 to 130 µs in air-free solutions whereas in Zn-porphyrins the effect of the substituents on the triplet lifetimes is evident; the lifetimes decrease from 136 µs for ZnTPP to 73 µs for 8 in air-free solutions. The oxygen quenching reaction rate constant, kq, can be derived from the lifetimes in air-equilibrated solutions (see Experimental Section and Table 4). In agreement with previously reported data, 48–49,51,53 most of the free base porphyrins have kq values of about 2 × 109 M–1 s–1and most of the Zn-porphyrins have rate constants of the order of 1 × 109 M–1 s–1, but within the series an increased reactivity of the last member, either 7 or 8, is observed.
Singlet oxygen photosensitization
Since both series showed a high reactivity with ground state molecular oxygen, in order to get information on the capability of conjugated porphyrins to generate singlet oxygen, we explored their photosensitizing ability by determining the 1Δg quantum yield (ΦΔ) via the detection of its luminescence at 1270 nm. There are, in fact, reports on the use of vinyl-substituted porphyrins in photodynamic therapy.54,55 The quantum yields of 1Δg production of the free-base and Zn-porphyrin series are collected in Table 4 and representative cases of 1Δg luminescence are reported in Fig. 7. It can be seen that for the free-base porphyrins the yields are uniform within the series, around a value of 0.80, and higher than that of the reference TPP. This result can be related to the previously described homogeneity in the emission quantum yields of the free-base series (Table 3) where Φfl are all moderately reduced compared to that of TPP and an increased intersystem crossing, with a consequent higher yield of triplet formation, probably occurs. For the Zn series a more inhomogeneous behavior is observed but the values are similar to that of ZnTPP (Table 4). These data indicate that the yield of singlet oxygen production is independent on the type of substitution in the examined Zn-porphyrins. However, the high values obtained in both series, confirm the strong reactivity of these β-substituted porphyrins with molecular oxygen and their possible use as photosensitizers. | ||
| Fig. 7 Singlet oxygen luminescence detected in air-equilibrated toluene for optically matched solutions of 3, 7 and TPP after excitation at 520 nm. | ||
Conclusions
A series of four free-base porphyrins and a series of four Zn-porphyrins β-substituted with either vinyl groups or p-phenylene vinylene groups have been synthesized and photophysically characterized in toluene solutions. The bathochromic shift of both absorption and emission bands of the examined porphyrins is consistent with an increasing delocalization of the frontier molecular orbitals of the porphyrin unit when the π-conjugation of the substituent increases, delocalization that was confirmed by semiempirical calculations. The singlet excited state lifetimes and fluorescence quantum yields were similar to those of the unsubstituted porphyrin references for both series and no clear trend was observed along the series, except for an increase of fluorescence quantum yield in the Zn-porphyrins in case of phenylene–vinylenic substitution. The triplet states, with lifetimes of the order of one hundred microseconds, displayed an interesting increase in the molar absorption coefficient of their transient absorption spectra above 700 nm along both series, with possible applications in the field of non linear optics in the near IR. These results suggest that conjugated β-substituents can be considered as useful interporphyrin linkers in the design of multiporphyrinic arrays with enhanced electronic communication. The reaction of the triplet states with oxygen, moreover, is fast and the yield of singlet oxygen sensitization is high for all the examined porphyrins; this could open promising applications in the use of conjugated porphyrins as therapeutic agents in photodynamic therapy.Experimental
Synthesis
High-resolution FAB mass spectra were recorded on a VG-70SE instrument at University of Auckland, New Zealand and on a VG ZAB 2 SEQ VG-Micromass at the Australian National University, Australia. 1H NMR spectrometry experiments were performed using 400 and 500 MHz Bruker Avance instruments running TOPSPIN 1.3 software. Proton chemical shifts in CDCl3 are relative to TMS. Chemical shifts in other solvents are relative to residual protons (tetrahydrofuran-d8, 3.58 ppm; dioxane-d8, 3.53 ppm). Data are expressed as position (in ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad, app = apparent), relative integral, coupling constant (JP indicating 1H–31P coupling) and assignment. Coupling constants were not reported when smaller than 1 Hz.Column chromatography employed silica gel (0.032–0.063 mm, Merck Kieselgel 60) or equivalent. Thin layer chromatography (TLC) was performed using pre-coated silica gel plates (Merck Kieselgel 60F254). Flash chromatography was used for compound purification. AR grade solvents and reagents were used for synthesis, and distilled laboratory grade solvents were used for chromatography. DBU (diazabicycloundecene), tBuOK and methyltriphenylphosphonium bromide (Me–ps) were purchased from Acros. All reagents were used as purchased. Porphyrin aldehydes 10–14 were prepared as previously reported.21 The preparation of toluene(diethylphosphonate)56 and 1,4-xylenebis(diethylphosphonate)57 were carried out according to literature procedures from the respective commercial bromides.
Spectroscopy and photophysics
Spectroscopic grade toluene (C. Erba) was used without further purification. Absorption spectra were recorded with a Perkin-Elmer Lambda 9 spectrophotometer and emission spectra were detected by a Spex Fluorolog II spectrofluorimeter equipped with a Hamamatsu R928 photomultiplier. Standard 10 mm fluorescence cells were used at 295 K whereas experiments at 77 K made use of capillary tubes in a homemade quartz dewar filled with liquid nitrogen. Emission quantum yields were determined after correction for the photomultiplier response, with reference to TPP in aerated toluene (Φfl = 0.1112). An FLS920 spectrofluorimeter (Edinburgh) equipped with an Hamamatsu R5509-72 supercooled photomultiplier tube at 193 K and a TM300 emission monochromator with a NIR grating blazed at 1000 nm was used to measure the singlet oxygen luminescence quantum yield ΦΔ using that for TPP in toluene as reference (ΦΔ = 0.7058). The excitation was at 520 nm for the free-base porphyrins and at 560 nm for the Zn-porphyrins. Luminescence lifetimes in the nanosecond range were obtained with an IBH single photon counting equipment with excitation at 560 nm and 331 nm from a pulsed diode source (resolution 0.5 ns).Transient absorbance in the nanosecond range made use of a laser flash photolysis apparatus based on a Nd:YAG laser (JK Lasers) delivering pulses of 18 ns.59 The second harmonic (532 nm) was used for excitation. The absorbance of the solutions at the exciting wavelength was about 0.5 and the pulse energy used was 0.6 mJ pulse–1 for the determination of the spectra and of the order of 0.1 mJ pulse–1 for the triplet lifetime determination. Experiments were conducted in homemade, 10 mm optical path cuvettes, the solution bubbled with argon for 10 minutes to remove dissolved oxygen and sealed. The oxygen quenching reaction rate constant, kq, was determined from the equation: k = k0 + kq[O2], by measuring the rate constant, k, for the air-free and the air-saturated cases. The oxygen solubility in air-equilibrated toluene was taken to be 1.8 × 10–3 M.60
Estimated uncertainties are 10% for transient absorbance lifetimes, 8% for luminescence lifetimes, 10% for molar absorption coefficients and 20% for quantum yields, and the working temperature, if not otherwise specified, was 295 ± 2 K.
The excited state properties of the molecules were calculated using the ZINDO/S (singly excited configurations in a space of 14 × 14 promotions), having optimized the ground state geometries with the Molecular Mechanics (MM + force field) program included in the Hyperchem package.40
Acknowledgements
We would like to acknowledge financial support from Italian CNR, (PM.P04.010 MACOL) and Ministero dell’Istruzione, dell’Università e della Ricerca, the New Zealand New Economy Research Fund (MAUX0202) and the New Zealand Marsden Fund (MAUX0407).References
- H. L. Anderson, Chem. Commun., 1999, 2323–2330 RSC.
- M. J. Frampton, H. Akdas, A. R. Cowley, J. E. Rogers, J. E. Slagle, P. A. Fleitz, M. Drobizhev, A. Rebane and H. L. Anderson, Org. Lett., 2005, 7, 5365–5368 CrossRef CAS.
- K. Susumu, T. V. Duncan and M. J. Therien, J. Am. Chem. Soc., 2005, 127, 5186–5195 CrossRef CAS.
- J. J. Piet, P. N. Taylor, B. R. Wegewijs, H. L. Anderson, A. Osuka and J. M. Warman, J. Phys. Chem. B, 2001, 105, 97–104 CrossRef CAS.
- A. Osuka, N. Tanabe, S. Kawabata, I. Yamazaki and Y. Nishimura, J. Org. Chem., 1995, 60, 7177–7185 CrossRef CAS.
- K. Pettersson, A. Kyrychenko, E. Ronnow, T. Ljungdahl, J. Martensson and B. Albinsson, J. Phys. Chem. A, 2006, 110, 310–318 CrossRef CAS.
- D. P. Arnold and D. A. James, J. Org. Chem., 1997, 62, 3460–3469 CrossRef CAS.
- F. Odobel, S. Suresh, E. Blart, Y. Nicolas, J. P. Quintard, P. Janvier, J. Y. Le Questel, B. Illien, D. Rondeau, P. Richomme, T. Haupl, S. Wallin and L. Hammarström, Chem.–Eur. J., 2002, 8, 3027–3046 CrossRef CAS.
- T. H. Huang, Y. J. Chen, S. S. Lo, W. N. Yen, C. L. Mai, M. C. Kuo and C. Y. Yeh, Dalton Trans., 2006, 2207–2213 RSC.
- A. Tsuda and A. Osuka, Science, 2001, 293, 79–82 CrossRef CAS.
- D. F. Perepichka and M. R. Bryce, Angew. Chem., Int. Ed., 2005, 44, 5370–5373 CrossRef CAS.
- P. G. Seybold and M. Gouterman, J. Mol. Spectrosc., 1969, 31, 1–13 CrossRef CAS.
- R. A. Binstead, M. J. Crossley and N. S. Hush, Inorg. Chem., 1991, 30, 1259–1264 CrossRef CAS.
- J. A. Shelnutt and V. Ortiz, J. Phys. Chem., 1985, 89, 4733–4739 CrossRef CAS.
- D. P. Arnold, Synlett, 2000, 12, 296–305.
- S. M. LeCours, S. G. DiMagno and M. J. Therien, J. Am. Chem. Soc., 1996, 118, 11854–11864 CrossRef CAS.
- T. E. O. Screen, I. M. Blake, L. H. Rees, W. Clegg, S. J. Borwick and H. L. Anderson, J. Chem. Soc., Perkin Trans. 1, 2002, 320–329 RSC.
- M. Drobizhev, F. Q. Meng, A. Rebane, Y. Stepanenko, E. Nickel and C. W. Spangler, J. Phys. Chem. B, 2006, 110, 9802–9814 CrossRef CAS.
- W. N. Yen, S. S. Lo, M. C. Kuo, C. L. Mai, G. H. Lee, S. M. Peng and C. Y. Yeh, Org. Lett., 2006, 8, 4239–4242 CrossRef CAS.
- C.-T. Chen, H.-C. Yeh, X. Zhang and J. Yu, Org. Lett., 1999, 1, 1767–1770 CrossRef CAS.
- Q. Wang, W. M. Campbell, E. E. Bonfantani, K. W. Jolley, D. L. Officer, P. J. Walsh, K. Gordon, R. Humphry-Baker, M. K. Nazeeruddin and M. Gratzel, J. Phys. Chem. B, 2005, 109, 15397–15409 CrossRef CAS.
- E. Annoni, M. Pizzotti, R. Ugo, S. Quici, T. Morotti, M. Bruschi and P. Mussini, Eur. J. Inorg. Chem., 2005, 3857–3874 CrossRef CAS.
- A. Lembo, P. Tagliatesta and D. M. Guldi, J. Phys. Chem. A, 2006, 110, 11424–11434 CrossRef CAS.
- M. Morone, L. Beverina, A. Abbotto, F. Silvestri, E. Collini, C. Ferrante, R. Bozio and G. A. Pagani, Org. Lett., 2006, 8, 2719–2722 CrossRef CAS.
- P. J. Walsh, K. C. Gordon, D. L. Officer and W. M. Campbell, J. Mol. Struct. (THEOCHEM), 2006, 759, 17–24 CrossRef CAS.
- P. J. Walsh, K. C. Gordon, P. Wagner and D. L. Officer, ChemPhysChem, 2006, 7, 2358–2365 CrossRef CAS.
- L. Flamigni, A. M. Talarico, J. C. Chambron, V. Heitz, M. Linke, N. Fujita and J. P. Sauvage, Chem.–Eur. J., 2004, 10, 2689–2699 CrossRef CAS.
- I. M. Dixon, J. P. Collin, J. P. Sauvage and L. Flamigni, Inorg. Chem., 2001, 40, 5507–5517 CrossRef CAS.
- L. Flamigni, M. R. Johnston and L. Giribabu, Chem.–Eur. J., 2002, 8, 3938–3947 CrossRef CAS.
- L. Flamigni, A. M. Talarico, S. Serroni, F. Puntoriero, M. J. Gunter, M. R. Johnston and T. P. Jeynes, Chem.–Eur. J., 2003, 9, 2649–2659 CrossRef CAS.
- L. Flamigni, A. M. Talarico, B. Ventura, G. Marconi, C. Sooambar and N. Solladie, Eur. J. Inorg. Chem., 2004, 2557–2569 CrossRef CAS.
- L. Flamigni, A. M. Talarico, B. Ventura, R. Rein and N. Solladie, Chem.–Eur. J., 2006, 12, 701–712 CrossRef CAS.
- A. K. Burrell, D. L. Officer, P. G. Plieger and D. C. W. Reid, Chem. Rev., 2001, 101, 2751–2796 CrossRef CAS.
- K. Okuma, O. Sakai and K. Shioji, Bull. Chem. Soc. Jpn., 2003, 76, 1675–1676 CrossRef CAS.
- A. K. Burrell and D. L. Officer, Synlett, 1998, 1297–1307 CrossRef CAS.
- H. J. Callot, Tetrahedron, 1973, 29, 899–901 CrossRef CAS.
- J. W. Buchler, in Porphyrins and Metalloporphyrins, ed. K. M. Smith, Elsevier, Amsterdam, 1975, ch. 5 Search PubMed.
- M. Gouterman, J. Mol. Spectrosc., 1961, 6, 138–163 CrossRef CAS.
- C. Djerassi, Y. Lu, A. Waleh, A. Y. L. Shu, R. A. Goldbeck, L. A. Kehres, C. W. Crandell, A. G. H. Wee, A. Knierzinger, R. Gaeteholmes, G. H. Loew, P. S. Clezy and E. Bunnenberg, J. Am. Chem. Soc., 1984, 106, 4241–4258 CrossRef.
- Hyperchem 6.02, Hypercube Inc., Gainesville, FL, 2000 Search PubMed.
- P. J. Spellane, M. Gouterman, A. Antipas, S. Kim and Y. C. Liu, Inorg. Chem., 1980, 19, 386–391 CrossRef CAS.
- O. Narwark, A. Gerhard, S. C. J. Meskers, S. Brocke, E. Thorn-Csanyi and H. Bassler, Chem. Phys., 2003, 294, 17–30 CrossRef CAS.
- W. B. Davis, W. A. Svec, M. A. Ratner and M. R. Wasielewski, Nature, 1998, 396, 60–63 CrossRef CAS.
- H. C. Lin, C. M. Tsai, G. H. Huang and J. M. Lin, J. Polym. Sci. Polym. Chem., 2006, 44, 783–800 Search PubMed.
- A. K. Burrell, D. L. Officer and D. C. W. Reid, Angew. Chem., Int. Ed. Engl., 1995, 34, 900–902 CAS.
- M. Wolffs, F. J. M. Hoeben, E. H. A. Beckers, A. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2005, 127, 13484–13485 CrossRef CAS.
- T. G. Jiu, Y. J. Li, H. Y. Gan, Y. L. Li, H. B. Liu, S. Wang, W. D. Zhou, C. R. Wang, X. F. Li, X. F. Liu and D. B. Zhu, Tetrahedron, 2007, 63, 232–240 CrossRef CAS.
- L. Flamigni, N. Armaroli, F. Barigelletti, V. Balzani, J. P. Collin, J. O. Dalbavie, V. Heitz and J. P. Sauvage, J. Phys. Chem. B, 1997, 101, 5936–5943 CrossRef CAS.
- L. Flamigni, A. M. Talarico, B. Ventura, C. Sooambar and N. Solladié, Eur. J. Inorg. Chem., 2006, 11, 2155–2165 CrossRef.
- D. Beljonne, G. E. Okeefe, P. J. Hamer, R. H. Friend, H. L. Anderson and J. L. Bredas, J. Chem. Phys., 1997, 106, 9439–9460 CrossRef CAS.
- L. Flamigni, A. M. Talarico, M. J. Gunter, M. R. Johnston and T. P. Jeynes, New J. Chem., 2003, 27, 551–559 RSC.
- J. E. Rogers, K. A. Nguyen, D. C. Hufnagle, D. G. McLean, W. J. Su, K. M. Gossett, A. R. Burke, S. A. Vinogradov, R. Pachter and P. A. Fleitz, J. Phys. Chem. A, 2003, 107, 11331–11339 CrossRef CAS.
- S. L. Murov, I. Carmichael and G. L. Hug, in Handbook of Photochemistry, ed. Marcel Dekker Inc., New York, 2nd edn, 1993, p. 250 Search PubMed.
- B. Aveline, O. Delgado and D. Brault, J. Chem. Soc., Faraday Trans., 1992, 88, 1971–1976 RSC.
- E. M. P. Silva, F. Giuntini, M. A. F. Faustino, J. P. C. Tome, M. Neves, A. C. Tome, A. M. S. Silva, M. G. Santana-Marques, A. J. Ferrer-Correia, J. A. S. Cavaleiro, M. F. Caeiro, R. R. Duarte, S. A. P. Tavares, I. N. Pegado, B. d'Almeida, A. P. A. De Matos and M. L. Valdeira, Bioorg. Med. Chem. Lett., 2005, 15, 3333–3337 CrossRef CAS.
- A. Michaelis and R. Haehne, Chem. Dtsch. Ber. Ges., 1898, 31, 1048 Search PubMed.
- J. K. Lee, R. R. Schrock, D. R. Baigent and R. H. Friend, Macromolecules, 1995, 28, 1966–1971 CrossRef CAS.
- F. Wilkinson, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1993, 22, 113–262 CAS.
- L. Flamigni, J. Phys. Chem., 1992, 96, 3331–3337 CrossRef CAS.
- S. L. Murov, I. Carmichael and G. L. Hug, in Handbook of Photochemistry, ed. Marcel Dekker Inc., New York, 2nd edn, 1993, p. 293 Search PubMed.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2008 |