Studies towards the catalytic anti-Markovnikov functionalisation of alkenes†
Christian J.
Richard
and
Adrian W.
Parkins
*
Department of Chemistry, King’s College London, Strand, London, UK WC2R 2LS. E-mail: awparkins@btinternet.com
First published on 14th September 2007
Abstract
In 1986 Jensen and Trogler reported that a biphasic system containing the mononuclear complex trans-[(Me3P)2PtHCl] and NaOH catalysed the anti-Markovnikov hydration of 1-hexene to 1-hexanol. The unusual method of preparation could also give rise to dinuclear complexes with bridging hydride ligands. We have tested some dinuclear complexes for catalytic activity and discovered that on heating these yield trinuclear complexes. Using the independently prepared trinuclear complex [(dppe)3Pt3H3]+, we found that it acted as a catalyst for the reaction between 1-octene and methanol to give 1-methoxyoctane, although the results were somewhat erratic. A cause of the erratic behaviour was found to be the presence of varying amounts of hydroperoxide impurities, which underwent a Hock rearrangement in acidic conditions.
Introduction
The anti-Markovnikov hydration of terminal alkenes is a very desirable objective of catalysis.1 In 1986 Jensen and Trogler claimed2 to have discovered a system based on a coordination compound of platinum, trans-[(Me3P)2PtHCl], which catalysed the anti-Markovnikov hydration of 1-hexene to 1-hexanol, eqn (1). | (1) |
Ramprasad and co-workers5 were unable to confirm Trogler’s claim, which was subsequently withdrawn.6 Grushin and co-workers7 suggested that the activity of Trogler’s system might be due to a catalytic impurity. We have retained an open mind on the genuineness of Trogler’s original claim, but regard his mechanism which involves a cationic platinum complex in the presence of [Et3NCH2Ph]Cl as extremely improbable. The purpose of the [Et3NCH2Ph]Cl was to act as a phase transfer catalyst to overcome the problem of immiscibility. Some workers have used water soluble sulfonated triphenylphosphine complexes as catalysts, but the outcomes were not very satisfactory.8,9
In common with Grushin,7 we also believe that the activity of Trogler’s system might be due to a catalytic impurity. Although Grushin did not speculate on the nature of the impurity, the presence of an impurity would explain why Ramprasad’s analytically pure samples of trans-[(Me3P)2PtHCl] showed no catalytic activity. In any case, Ramprasad synthesised trans-[(Me3P)2PtHCl] by sodium borohydride reduction of cis-[(Me3P)2PtCl2] in acidified methanol, and thus the product would not contain the same impurities present in Trogler’s synthesis. Trogler used a somewhat unusual method for the preparation of his catalyst precursor trans-[(Me3P)2PtHCl], which involved reducing cis-[(Me3P)2PtCl2] with sodium naphthalide in THF in the presence of H2.10 One possibility which we considered was that the sodium naphthalide was causing a methyl group on the trimethylphosphine to be cleaved off, giving rise to a secondary phosphine. While the cleavage of alkyl groups from phosphorus is generally thought to be more difficult than aryl group cleavage,11 Wilkinson and co-workers have reported the cleavage of a methyl group from a trimethylphosphine tungsten complex by sodium amalgam.12 Pursuing this idea of methyl cleavage led to our very active nitrile hydration catalyst,13 but complexes with secondary phosphines only gave erratic results for alkene hydration, which were not a significant improvement on Trogler’s results. The sodium naphthalide preparation of trans-[(Me3P)2PtHCl] is inconvenient, and Trogler later published a preparation of trans-[(Me3P)2PtHCl] based on the sodium borohydride reduction of cis-[(Me3P)2PtCl2] in methanol in the presence of diethylamine.14
In an attempt to understand the nature of the postulated catalytic impurity in Trogler’s system, we became interested in a reaction first reported by Minghetti and co-workers15 who found that styrene reacted with the dinuclear H-bridged cationic complex 1 to give the bridging alkylidene complex 2, as shown in eqn (2). This involved the transfer of a hydride in 1 to the benzylic carbon of styrene.
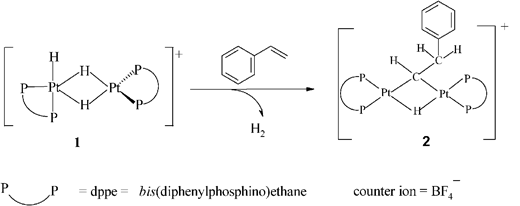 | (2) |
The reaction is not unique, as ethene also reacts with an analogue of 1 to form the corresponding bridged alkylidene complex.16 The complex 1 and its analogues are stereochemically non-rigid in solution, due to the rapid exchange of bridging and terminal hydrogens.17–19 This fluxionality is attributed to the presence of the chelating phosphines, which hold the phosphorus atoms in cis positions. The equivalent complexes with non-chelating phosphines do not display fluxionality in solution.20 Related work has been reported by Spencer and co-workers.21,22
The fact that the dinuclear complexes with bridging hydrides add hydrogen to the β-position of alkenes suggested to us that such complexes might be playing a role in Trogler’s system, i.e. might be the “impurity”. Zudin and co-workers23 suggested that a palladium complex with a bridging hydride, [(Ph3P)2Pd(µ-H)(µ-CO)Pd(PPh3)2]+, is possibly the catalytic species in the synthesis of diethyl ketone from ethene and carbon monoxide.
Results and discussion
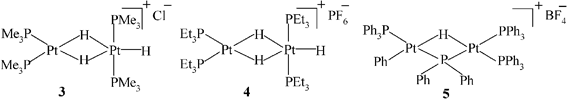
We modified the rather unusual method used by Trogler10 for the preparation of trans-[(Me3P)2PtHCl] by using two equivalents of NaC10H8 rather than one as used by Trogler in order to enhance the formation of (Me3P)2PtH2in situ, according to eqns (3) and (4). We hoped that the hydride bridged complex [(Me3P)2Pt(µ-H)2PtH(PMe3)2][Cl], 3, would then form according to eqn (5).| [(Me3P)2PtCl2] + NaC10H8 + ½H2 = [(Me3P)2PtHCl] + C10H8 + NaCl | (3) |
| [(Me3P)2PtHCl] + NaC10H8 + ½H2 = [(Me3P)2PtH2] + C10H8 + NaCl | (4) |
| [(Me3P)2PtH2] + [(Me3P)2PtHCl] = [(Me3P)2Pt(µ-H)2PtH(PMe3)2]Cl (3) | (5) |
We carried out tests for catalytic activity using the solution containing [(Me3P)2Pt(µ-H)2PtH(PMe3)2]Cl, 3, which might arise in Trogler’s system, and also [(dppe)Pt(µ-H)2PtH(dppe)]BF4, 1,15 and [(Et3P)2Pt(µ-H)2PtH(PEt3)2]PF6, 4.20,25 We used 1-octene rather than 1-hexene, because of the slight risk of losing volatile C6 products in the work up. The molar ratio of 1-octene : catalyst used for 1 and 4 was ∼200. The ratio for 3 was not known accurately as the complex was not isolated. (It may be worth mentioning that both isomeric cations [(Et3P)2HPt(µ-H)PtH(PEt3)2]+ and [(Et3P)2Pt(µ-H)2PtH(PEt3)2]+ can be synthesised, although we did not work with the former.25)
The first tests were carried out with 1, 3 and 4 with sodium hydroxide, water and 1-octene in either THF or methanol. These conditions corresponded most closely to those used by Trogler. Sodium dodecanesulfonate (SDS) was added to give a homogeneous solution. The reaction mixtures were analysed by GC-MS, in conjunction with the NIST database26 and the Eight Peak Index of Mass Spectra.27 No 1-octanol was detected, although the 1-octene did undergo double bond migration. This is evidence that the complexes interact with 1-octene. The catalytic reactions were carried out both with and without the addition of fluoroboric acid and sodium or potassium hydroxide. SDS was also added as a phase transfer catalyst when sodium or potassium hydroxide was added. When no acid or base was added the reaction mixture became acidic during the reaction. This may be due to decomposition of the catalyst.
An organic product we obtained from 1-octene using 528 as catalyst was the aldehydetrans-2-octenal and some trans-oct-2-en-1-ol, 2- and 3-octanone were also detected. Some trans-2-octenal was formed when small amounts of added acid were used, but with excess acid the formation of trans-2-octenal was inhibited. The addition of base also inhibited the formation of trans-2-octenal.
We encountered a problem in the catalytic testing concerned with the identification of an organic product obtained from 1-octene with 1 in acidified methanol. The product showed a base peak in its mass spectrum at m/z = 87, and the GC retention time was very similar to that of 4-methoxyoctane. The mass spectra of the 1-, 2- and 3-methoxyoctanes have been reported by Katritzky and co-workers,29 and we synthesised 4-methoxyoctane and measured its spectrum. None of the isomers shows a molecular ion and the main fragment is derived from α-cleavage.30 The fragmentation of the isomers is summarised in Scheme 1.
 | ||
| Scheme 1 Major fragmentation pathways in the mass spectra of isomeric methoxyoctanes. The base peak is shown in bold figures. Note that 4-methoxyoctane has a base peak at 87 and an additional peak at 101, but none at 115. | ||
It therefore appeared that the reaction had given 4-methoxyoctane, and we considered the possibility that there had been a C–H activation reaction of the type reported by Basickes and Sen.31 A more careful examination of the mass spectrum showed that the unknown compound also had a prominent peak at m/z = 115, not present in 4-methoxyoctane. Further work showed that the platinum catalyst was not necessary to form the unknown product. After some deliberation, it occurred to us that impurities of oct-1-ene-3-hydroperoxide in the acidified methanolic solution of 1-octene used might have undergone a Hock rearrangement32 which is also known as a hydroperoxide rearrangement.33 Using the known reaction mechanism34 on oct-1-ene-3-hydroperoxide, but with the carbocation capturing methanol rather than the usual water, 1-methoxy-1-vinyloxyhexane is formed as shown in Scheme 2. We were unable to find spectroscopic data for these compounds in the literature.
 | ||
| Scheme 2 Reaction mechanism showing the Hock rearrangement of oct-1-ene-3-hydroperoxide in acidic methanol. | ||
The mass spectrum of the product shows a base peak at m/z = 87 and also a peak at m/z = 115, both of which are readily explained by the 1-methoxy-1-vinyloxyhexane; see case (a) in Scheme 3. This interpretation was verified by repeating the reaction with (b) 1-octene and ethanol and (c) 1-hexene and methanol. The expected products are shown in Scheme 3, and the mass spectra obtained agree with the fragmentation schemes shown. See also ESI.†
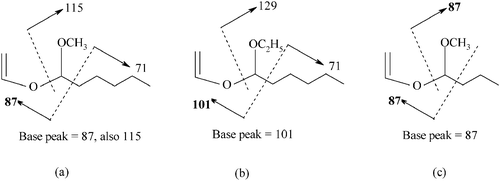 | ||
| Scheme 3 Products of the Hock rearrangement of hydroperoxides from (a) 1-octene and methanol, (b) 1-octene and ethanol and (c) 1-hexene and methanol, and their fragmentation pathways. | ||
The 1-octene used in the early stages of this work had been distilled under argon, but distillation was later found to be insufficient to remove hydroperoxide impurities. To overcome this problem, we removed the hydroperoxide impurities from our samples by passing the 1-octene though a column of activated Al2O3. When further reactions were carried out with treated 1-octene, the GCs showed that the peak due to 1-methoxy-1-vinyloxyhexane was very much reduced in intensity. Several purifications were found to be necessary to eliminate the hydroperoxide impurities.
There are few reports in the catalytic literature concerning problems arising from the presence of hydroperoxides in alkenes. One such example was recently reported by Süss-Fink and co-workers,35,36 who found that the catalytic activity of an arene hydrogenation catalyst, originally considered to be due to the trinuclear ruthenium compound, [(η6-C6H6)(η6-C6Me6)2Ru3(µ2-H)3(µ3-H)]BF4, was in fact due to RuO2. Oxidation by a hydroperoxide impurity in the ethyl benzene substrate produced the RuO2. We do not discount the possibility that the 1-hexene used by Trogler2 may have been contaminated with hydroperoxides. If this was the case, the presence or absence of hydroperoxide impurities may have been detrimental to the reproducibility of his results, since they may have reacted with any platinum containing species present. Pritzkow and co-workers37 have studied the autoxidation of all four n-octenes, but they did not report an acidic work up procedure analogous to that used here.
In many catalytic experiments, it was observed that 5, which is almost colourless, decomposed to products which gave red-orange solutions. This decomposition was rapid in the relatively high-boiling solvents 2-propanol (b.p. 82 °C), toluene (b.p. 111 °C) and [1,4]-dioxane (b.p. 101 °C), but occurred to a much lesser degree in the catalytic reactions run in methanol (b.p. 67.4 °C) or THF (b.p. 66 °C). The products from the reactions run in methanol rarely contained trans-2-octenal or trans-oct-2-en-1-ol. This suggests that the presence of these products might be linked to the abundance of platinum species derived from the decomposition of 5 in higher boiling solvents.
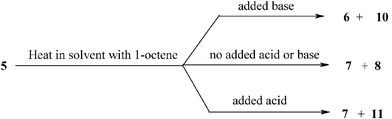
The red-orange solutions obtained using 5 were investigated by 31P{1H} NMR. The spectra were initially rather difficult to interpret because they contained mixtures, but some components were found to correspond to the known compounds 6, 7, 8, and possibly 9. Two other compounds 10 and 11 were detected and analysed by multinuclear NMR and found to contain unidentified NMR silent coordinating groupsX and Z. The structures proposed for 10 and 11 are very tentative. The nature of the products depended on the pH of the reaction mixture. Under basic conditions the products were 6 and 10. When no acid or base was added the products were 7 and 8, and under acid conditions the products were 7 and the dinuclear compound 11. The reactions are summarised in eqn (6) and the structures given in Scheme 4. See the ESI for details of the NMR spectra.†
 | (6) |
 | ||
| Scheme 4 Products arising from the decomposition of 5 in catalytic reactions (X and Z are unidentified NMR silent coordinating groups). | ||
When 5 was heated in acidified methanolin the absence of 1-octene a mixture of the trinuclear complex 7 and other unidentified products was obtained.
To the best of our knowledge, the formation of the orange-red complexes 6––9 from 5 is unprecedented. Complexes 6––9 are generally prepared (in varying yields) from the UV irradiation, pyrolysis or heating solutions of the mononuclear zero-valent compounds Pt(PPh3)4 and (Ph3P)2Pt(ethene).38–42 Although Braunstein and co-workers43 have commented that the opening of a phosphido bridge is rare, the formation of complexes 6––9 implies that the phosphido bridge in 5 was cleaved to give the mononuclear fragments (Ph3P)2Pt and (Ph3P)2PtH which in turn lead to the di- and trinuclear complexes 6––9. The formation of the trinuclear species is an important observation in that we must now consider the possibility that Trogler’s catalytic hydration system2 may have contained trinuclear species. To test this hypothesis we tested the trinuclear platinum cluster [(dppe)3Pt3H3]+, 12, which was reported by Minghetti and coworkers in 1981.44

Minghetti showed that depending on the ratio of the reducing agent KBH4 to the platinum dimethylpyrazole complex [(dppe)Pt(HPz-N)2](BF4)2 di- or trinuclear platinum hydride clusters could be prepared. There was some uncertainty concerning the number of hydride ions in the [(dppe)3Pt3(H)x]+ cation, but 195Pt NMR studies by Nixon and co-workers45 established the composition as [(dppe)3Pt3H3]+, (12). The X-ray diffraction study of 12 showed that it is an “open” cluster according to the definition proposed by Chini.46 The Pt2 and Pt3 inter-atomic distance is 4.01 Å, which implies that there is no metal–metal bond between Pt2 and Pt3.45
Tests for catalytic activity were carried out with 12 as described above for 1, 3, 4 and 5. The products were identified by GC-MS. Calibration experiments were carried out to quantify the peak height with the weight of each known component, and this allowed turnover numbers to be calculated. There were also some unidentified peaks in the GC traces.
1-Octanol was not detected in the products of any of the catalytic experiments using 12 as catalyst with the addition of acid or base or neither. However, the anti-Markovnikov product 1-methoxyoctane was detected by GC-MS, from experiments (3 and 4 and 9–11) in which the 1-octene used had been previously treated to remove hydroperoxide impurities. This observation suggests that a reaction between the hydroperoxide impurities and 12 deactivated it with respect to the catalytic formation of 1-methoxyoctane. Under our experimental conditions, the formation of 1-methoxyoctane was sometimes accompanied by 2-methoxyoctane, and the turnover number to the 1-methoxy isomer was typically 50 TON. We suspected that the rapid decomposition of 12 (or a catalytic species derived from it) was partly responsible for the low turnovers for 1-methoxyoctane.
To our knowledge, the catalytic formation of 1-methoxyoctane from methanol, 1-octene and a platinum hydride cluster is unprecedented and supports the suggestion that an unidentified platinum hydride cluster was the catalytic species responsible for the formation of 1-hexanol in Trogler’s reported biphasic system. Trogler’s catalytic system required the presence of base, but the catalytic reaction of 1-octene with 12 appeared to be inhibited by base. However, Trogler’s system was biphasic and a direct comparison may not be helpful. Excess acid also inhibited the formation of 1-methoxyoctane using 12 as catalyst. A speculative catalytic cycle showing the production of 1-methoxyoctane from 1-octene is shown in Scheme 5. Jensen and Trogler mention a deuterium labelling experiment in their report.2 Although it is not clear what their experiment involved, they report that the deuterium from the platinum catalyst finished up in the β-position in 1-hexanol, which is also consistent with our suggested mechanism involving 12.
 | ||
| Scheme 5 Suggested mechanism for the formation of 1-methoxyoctane using 12 as catalyst. | ||
While the detection of 1-methoxyoctane from the reaction between 1-octene and methanol using 12 as catalyst is the desired result, several other organic products were also observed. The experimental conditions are given in Table 1 and the organic products are summarised in Table 2.
| Run no., catalyst |
Molar ratio:
ca. [1-octene]/[12] |
Acid (HBF4) | Base (KOH) | Solvent |
Molar ratio:
[SDS]/[12] |
|---|---|---|---|---|---|
| 1, 12[OH] | 1000 | — | — | Water (7 cm3) | — |
| 2, 12[OH] | 1000 | — | — | Water (7 cm3) | ca. 12 |
| 3, 12[OH] | 275 | — | — | MeOH (8 cm3) | — |
| 4, 12[OH] | 275 | — | — | MeOH (8 cm3) | ca. 12 |
| 5, 12[OH] | 1000 | Added | — | Water (7 cm3) | — |
| 6, 12[BF4] | 1000 | Added | — | Water (7 cm3) | — |
| 7, 12[OH] | 1000 | Added | — | Water (7 cm3) | ca. 12 |
| 8, 12[BF4] | 1000 | Added | — | Water (7 cm3) | ca. 12 |
| 9, 12[OH] | 275 | Added | — | MeOH (8 cm3) | — |
| 10, 12[BF4] | 275 | Added | — | MeOH (8 cm3) | — |
| 11, 12[OH] | 275 | Added | — | MeOH (8 cm3) | ca. 12 |
| 12, 12[BF4] | 275 | Added | — | MeOH (8 cm3) | ca. 12 |
| 13, 12[OH] | 1000 | — | Added | Water (7 cm3) | — |
| 14, 12[BF4] | 1000 | — | Added | Water (7 cm3) | — |
| 15, 12[OH] | 1000 | — | Added | Water (7 cm3) | ca. 12 |
| 16, 12[BF4] | 1000 | — | Added | Water (7 cm3) | ca. 12 |
| 17, 12[OH] | 275 | — | Added | MeOH (8 cm3) | ca. 12 |
| 18, 12[BF4] | 275 | — | Added | MeOH (8 cm3) | ca. 12 |
| Run no., catalyst | 2-Octanone | 3-Octanone | 1-Methoxyoctane | 2-Methoxyoctane | trans-2-Octenal | Double bond isomers of 1-octene | Hock product |
|---|---|---|---|---|---|---|---|
| T = present when 1-octene used had been treated to remove hydroperoxides. U = present when 1-octene used was untreated. * = 1-octene virtually unreacted. | |||||||
| 1, 12[OH] | *T,U | ||||||
| 2, 12[OH] | T | T,U | U | ||||
| 3, 12[OH] | T,U | T | T | T,U | U | ||
| 4, 12[OH] | T,U | T | T | T,U | U | ||
| 5, 12[OH] | *T,U | ||||||
| 6, 12[BF4] | *T,U | ||||||
| 7, 12[OH] | T,U | U | U | T,U | U | ||
| 8, 12[BF4] | T,U | U | U | T,U | U | ||
| 9, 12[OH] | U | T | T | U | T,U | U | |
| 10, 12[BF4] | U | T | T | U | T,U | U | |
| 11, 12[OH] | U | T | T | U | T,U | U | |
| 12, 12[BF4] | T,U | T,U | U | ||||
| 13, 12[OH] | *T,U | ||||||
| 14, 12[BF4] | *T,U | ||||||
| 15, 12[OH] | T,U | T,U | |||||
| 16, 12[BF4] | T,U | T,U | |||||
| 17, 12[OH] | T,U | T,U | |||||
| 18, 12[BF4] | T,U | T,U | |||||
All the experiments in Table 1 were run twice: once using 1-octene which had been treated with Al2O3 to remove impurities of oct-1-ene-3-hydroperoxide, and a second time with untreated 1-octene. As the results in Table 2 show, there are considerable differences in the products obtained with treated and untreated 1-octene, and 1-methoxyoctene was only obtained with treated 1-octene. The formation of 1-methoxyoctane was erratic, and occasionally traces of the 1-methoxy-1-vinyloxyhexane were detected in the products from treated 1-octene, showing that the removal of the hydroperoxide was not perfect. Experiments 2, 7 and 8, which had a high 1-octene : catalyst ratio, showed an increase in the turnovers for 3-octanone (480–1000 TON) and trans-2-octenal (250–800 TON), although the latter was only present when untreated 1-octene was used. The increase in these turnovers might be linked to the increased occurrence of phosphido bridged clusters. In addition to this, the increased amount of 1-octene would have contained a greater quantity of oct-1-ene-3-hydroperoxide, which might account for the increased turnovers of trans-2-octenal. We did not measure the amount of hydroperoxide present in the untreated samples of 1-octene.
The anti-Markovnikov product trans-2-octenal was only detected in products derived from experiments in which 1-octene had not been previously treated to remove hydroperoxide impurities. At high concentrations of acid, trans-2-octenal was not detected. The turnover number of trans-2-octenal was in the range ca. 20–25 TON. When experiments 2, 7 and 8 were repeated with 1-octene which had been treated to remove hydroperoxide impurities, no trans-2-octenal was detected by GC-MS. However, 3-octanone was still detected with similar turnovers. A blank experiment without 12 was carried out and no trans-2-octenal was detected. The occurrence of trans-2-octenal and 2-octanone was favoured at low concentrations of acid, either added or formed adventitiously. 3-Octanone was probably formed in a Wacker-type reaction from 1-octene after double bond isomerisation. In the presence of base, trans-2-octenal and 3-octanone were not detected. Wacker-type reactions were presumably responsible for the varied yields of 2-octanone which was usually detected in our catalytic experiments.
We have included in the ESI† some comments on the fate of 12 during the catalytic reactions, and background information relating the newly discovered catalytic activity of 12 to Jensen and Trogler’s earlier work.
Conclusions
To summarise, we hope that we have shed some light on the problem of the anti-Markovnikov functionalisation of terminal alkenes. The crucial experiment was carried out by Minghetti and co-workers15 in 1985, and we have tried to bring this observation to the plenitude of its significance. The initial report by Jensen and Trogler2 was also an essential reference point in our work. Using these two studies as a starting point, we have devised a (somewhat erratic) catalytic system which uses a trinuclear platinum complex as the catalyst precursor. It is of course possible that compounds of even higher nuclearity are involved. The further association of trimetallic units has been reviewed by Imhof and Venanzi.47 In 1998 Adams and Cotton48 edited a monograph on catalysis by clusters, but the work reported here establishes a new direction for this field which could not be foreseen 10 years ago. We have not solved the problem of the addition of water to alkenes, one aspect of which involves another level of difficulty due to the immiscibility of water and alkenes.Experimental
General
All operations were carried out under an inert atmosphere of nitrogen or argon. NMR spectra were recorded using Brucker Avance 360 or 400 spectrometers. 31P{1H} chemical shifts are reported relative to external H3PO4, positive chemical shifts being downfield of the reference. GC-MS were recorded with a Varian CP-3800 Gas Chromatograph and a Saturn 2200 GC-MS, using a Varian Chromopack capillary column: CP-Sil 8 CB Lowbleed/MS. Toluene, diethyl ether and THF were distilled under nitrogen from blue solutions containing sodium benzophenone ketyl.Syntheses
cis-[(Me3P)2PtCl2],49trans-[(Et3P)2PtHCl],50(dppe)PtCl2,51 [(Et3P)2Pt(µ-H)2PtH(PEt3)2]PF6 (4),25 [(dppe)Pt(HPz-N)2](BF4)2,52 [(Ph3P)2Pt(µ-H)(µ-PPh2)Pt(Ph)(PPh3)]BF4 (5),26 were prepared by literature methods and characterised by comparison of their NMR spectra with the literature. In the preparation of 4, NH4PF6 was substituted for NaBPh4 to give a more soluble product. 2-Octene, 3-octene, 2-octanone, 3-octanone, trans-2-octenal and trans-oct-2-en-1-ol were commercial samples. The methoxyoctanes were prepared from the sodium53 or potassium54 salt of the alcohol and methyl iodide, as described here for 4-methoxyoctane which was previously reported by Olah55et al. but without preparative details. The preparation of 4-methoxyoctane was carried out using a procedure similar to that used by Letsinger and co-workers for 2-methoxyoctane.54If the presence of 3 was indicated by 31P{1H} NMR, the preparations of 3 in THF were not worked up, but used directly as catalytic solutions, in which an attempt was made to hydrate 1-octene to 1-octanol. It was not feasible to estimate yields and so the ratio of 1-octene to 3 was not known.

General procedure used to screen platinum complexes for catalytic activity
Combinations of the di- and trinuclear platinum complexes (ca. 20 mg), 1-octene (0.7 cm3), methanol (8.0 cm3) or water (7.0 cm3) were added to a 25 cm3 round-bottomed flask equipped with a magnetic stir bar, and further additions made to bring about the required experimental conditions. In the case of 3 a solution of the complex rather than the solid was used. If acid conditions were required HBF4 (48% w/w in water) was added and if basic conditions were required NaOH or KOH was added as an aqueous solution. In the case of basic conditions, sodium dodecanesulfonate (SDS) was added to homogenise the reaction mixture. Air was purged from the reaction system with nitrogen and the mixture heated under reflux and stirred for ca. 20 h, under nitrogen. On cooling to room temperature, the reaction mixture was diluted with water (40 cm3), extracted with dichloromethane (2 × 20 cm3) and the extracts dried over anhydrous MgSO4. The dichloromethane solution was then concentrated in vacuo, made up to the mark 5.0 cm3 in a volumetric flask with fresh dichloromethane and aliquots (0.1 µL) withdrawn for GC-MS analysis. In combination with calibration graphs made up from authentic samples of the organic products this enabled a quantitative determination of the turnover numbers of each product to be made. The gas chromatograms of authentic compounds were taken immediately after the GC run of the unknown reaction products, and their retention times compared. This ensured that no spurious peaks arose through cross-contamination. Subsequently, the remaining solution not used for GC-MS was dried in vacuo and all non-volatile residues submitted for infra-red, 31P{1H} and 1H NMR analysis. During the washing of all glassware, an ultrasonic bath was used to remove any traces of platinum residues that may have been deposited in a preceding reaction. To avoid the loss of products, a large reflux condenser (80 cm) was used and all the joints in the glassware were sealed with Teflon tape.Acknowledgements
We thank the Engineering and Physical Sciences Research Council for a studentship to C.J.R., and Johnson Matthey plc for a loan of platinum salts. We are grateful to Mr A. Cakebread and Mr R. Tye for assistance with the GC-MS measurements.References
- S. Borman, Chem. Eng. News, 2004, 82, 42–43.
- C. M. Jensen and W. C. Trogler, Science, 1986, 233, 1069–1071 CAS.
- Catalytic Heterofunctionalisation, ed. A. Togni and H. Grützmacher, Wiley-VCH, Weinheim, 2001 Search PubMed.
- K. C. Hultzsch, Adv. Synth. Catal., 2005, 347, 367–391 CrossRef CAS.
- D. Ramprasad, H. J. Yue and J. A. Marsella, Inorg. Chem., 1988, 27, 3151–3155 CrossRef CAS.
- W. C. Trogler, J. Chem. Educ., 1988, 65, 294–297 CrossRef CAS.
- V. V. Grushin, I. S. Akhrem and M. E. Volpin, J. Organomet. Chem., 1989, 371, 403–419 CrossRef CAS.
- D. S. Helfer, D. S. Phaho and J. Atwood, Abstracts of Papers, 226th ACS Annual Meeting, INOR-703, American Chemical Society, Washington, DC, 2003; D. S. Phaho, Towards the catalytic hydration of terminal alkenes by water-soluble platinum complexes, Thesis, State University of New York, Buffalo, 2000 Search PubMed.
- H. F. Koch, L. A. Girard and D. M. Roundhill, Polyhedron, 1999, 18, 2275–2279 CrossRef CAS.
- D. L. Packett, C. M. Jensen, R. L. Cowan, C. E. Strouse and W. C. Trogler, Inorg. Chem., 1985, 24, 3578–3583 CrossRef CAS ; US Patent 4,684,751.
- A. W. Parkins, Coord. Chem. Rev., 2006, 250, 449–467 CrossRef CAS.
- K. W. Chiu, R. A. Jones, G. Wilkinson, A. M. R. Galas and M. B. Hursthouse, J. Chem. Soc., Dalton Trans., 1981, 1892–1897 RSC.
- T. Ghaffar and A. W. Parkins, J. Mol. Catal. A: Chem., 2000, 160, 249–261 CrossRef CAS.
- J. R. Phillips and W. C. Trogler, Inorg. Synth., 1992, 29, 189–192 CAS.
- G. Minghetti, A. Albinati, A. L. Bandini and G. Banditelli, Angew. Chem., Int. Ed. Engl., 1985, 24, 120–121 CrossRef.
- A. L. Bandini, G. Banditelli and G. Minghetti, J. Organomet. Chem., 2000, 595, 224–231 CrossRef CAS.
- A. L. Bandini, G. Banditelli, M. Grassi and A. Ponti, Dalton Trans., 2004, 2027–2035 RSC.
- C. B. Knobler, H. D. Kaesz, G. Minghetti, A. L. Bandini, G. Banditelli and F. Bonati, Inorg. Chem., 1983, 22, 2324–2331 CrossRef CAS.
- T. H. Tulip, T. Yamagata, T. Yoshida, R. D. Wilson, J. A. Ibers and S. Otsuka, Inorg. Chem., 1979, 18, 2239–2250 CrossRef CAS.
- R. S. Paonessa and W. C. Trogler, Inorg. Chem., 1983, 22, 1038–1048 CrossRef CAS.
- N. Carr, B. J. Dunne, L. Mole, A. G. Orpen and J. L. Spencer, J. Chem. Soc., Dalton Trans., 1991, 863–871 RSC.
- N. N. Carr, L. Mole, A. G. Orpen and J. L. Spencer, J. Chem. Soc., Dalton Trans., 1992, 2653–2662 RSC.
- V. N. Zudin, V. D. Chinakov, V. M. Nekipelov, V. A. Likholobov and Y. I. Yermakov, J. Organomet. Chem., 1985, 289, 425–430 CrossRef CAS.
- D. L. Packett, A. Syed and W. C. Trogler, Organometallics, 1988, 7, 159–166 CrossRef CAS.
- S. Chaloupka and L. M. Venanzi, Inorg. Synth., 1990, 27, 30–38 CAS.
- Standard Reference Data Program, National Institute of Standards and Technology, Gaithersburg.
- Eight Peak Index of Mass Spectra, Royal Society of Chemistry, UK, 3rd edn, 1983.
- A. W. Parkins, C. J. Richard and J. W. Steed, Inorg. Chim. Acta, 2005, 358, 2827–2832 CrossRef CAS.
- A. R. Katritzky, Z. Dega-Szafran, M. L. Lopez-Rodriguez and R. W. King, J. Am. Chem. Soc., 1984, 106, 5577–5585 CrossRef CAS.
- F. W. McLafferty, Interpretation of Mass Spectra, University Science Books, Mill Valley, CA, 3rd edn, 1980, p. 209 Search PubMed.
- N. Basickes and A. Sen, Polyhedron, 1995, 14, 197–202 CrossRef CAS.
- H. Hock and H. Kropf, Angew. Chem., 1957, 69, 313–340 CAS.
- L. F. Fieser and M. Fieser, Advanced Organic Chemistry, Rheinhold, New York, 1961, p. 288 Search PubMed.
- H. Kropf, in Houben Weyl: Methoden der Organischen Chemie, G. Thieme, Stuttgart, 1988, vol. E13, p. 1085 Search PubMed.
- G. Süss-Fink, B. Therrien, L. Vieille-Petit, M. Tschan, V. B. Romakh, T. R. Ward, M. Dadras and G. Laurenczy, J. Organomet. Chem., 2004, 689, 1362–1369 CrossRef CAS.
- M. Faure, M. Jahncke, A. Neels, H. Stoeckli-Evans and G. Süss-Fink, Polyhedron, 1999, 18, 2679–2685 CrossRef CAS.
- W. Pritzkow, R. Radeglia and W. Schmidt-Renner, J. Prakt. Chem., 1979, 321, 813–826 CrossRef CAS.
- N. J. Taylor, P. C. Chieh and A. J. Carty, J. Chem. Soc., Chem. Commun., 1975, 448–449 RSC.
- F. Glockling, T. McBride and R. J. I. Pollock, J. Chem. Soc., Chem. Commun., 1973, 650–650 Search PubMed.
- R. Bender, S. E. Bouaoud, P. Braunstein, Y. Dusausoy, N. Merabet, J. Raya and D. Rouag, J. Chem. Soc., Dalton Trans., 1999, 735–741 RSC.
- C. Archambault, R. Bender, P. Braunstein and Y. Dusausoy, J. Chem. Soc., Dalton Trans., 2002, 4084–4090 RSC.
- M. A. Bennett, D. E. Berry, T. Dirnberger, D. C. R. Hockless and E. Wenger, J. Chem. Soc., Dalton Trans., 1998, 2367–2371 RSC.
- C. Archambault, R. Bender, P. Braunstein, A. DeCian and J. Fischer, Chem. Commun., 1996, 2729–2730 RSC.
- G. Minghetti, A. L. Bandini, G. Banditelli and F. Bonati, J. Organomet. Chem., 1981, 214, C50–C52 CrossRef CAS.
- D. Carmichael, P. B. Hitchcock, J. F. Nixon and A. Pidcock, J. Chem. Soc., Chem. Commun., 1988, 1554–1556 RSC.
- P. Chini, Inorg. Chim. Acta Rev., 1968, 2, 31–51 Search PubMed.
- D. Imhof and L. M. Venanzi, Chem. Soc. Rev., 1994, 23, 185–193 RSC.
- R. D. Adams and F. A. Cotton, Catalysis by di- and polynuclear metal cluster complexes, Wiley-VCH, Weinheim, 1998 Search PubMed.
- E. Matern, J. Pikies and G. Fritz, Z. Anorg. Allg. Chem., 2000, 626, 2136–2142 CrossRef CAS.
- G. W. Parshall, Inorg. Synth., 1970, 12, 26–33 CAS.
- K. Yasufuku, H. Noda and H. Yamazaki, Inorg. Synth., 1989, 26, 369–372 CAS.
- G. Minghetti, G. Banditelli and F. Bonati, J. Chem. Soc., Dalton Trans., 1979, 1851–1856 RSC.
- L. W. Devaney and G. W. Panian, J. Am. Chem. Soc., 1953, 75, 4836–4837 CrossRef CAS.
- R. L. Letsinger, A. W. Schnizer and E. Bobko, J. Am. Chem. Soc., 1951, 73, 5708–5710 CrossRef CAS.
- G. A. Olah, A. Husain, B. G. B. Gupta and S. C. Narang, Angew. Chem., Int. Ed. Engl., 1981, 20, 690–691 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Spectra and background information. See DOI: 10.1039/b708470f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2008 |