Synthesis and electronic properties of fullerene derivatives substituted with oligophenylenevinylen–ferrocene conjugates
Teresa M.
Figueira-Duarte
a,
Yannick
Rio
b,
Andrea
Listorti
b,
Béatrice
Delavaux-Nicot
a,
Michel
Holler
c,
Filippo
Marchioni
d,
Paola
Ceroni
*d,
Nicola
Armaroli
*b and
Jean-François
Nierengarten
*a
aGroupe de Chimie des Fullerènes et des Systèmes Conjugués, Laboratoire de Chimie de Coordination du CNRS, 205 route de Narbonne, 31077 Toulouse Cedex 4, France. E-mail: jfnierengarten@lcc-toulouse.fr
bMolecular Photoscience Group, Istituto per la Sintesi Organica e la Fotoreattività, Consiglio Nazionale delle Ricerche, via Gobetti 101, 40129 Bologna, Italy. E-mail: nicola.armaroli@isof.cnr.it
cLaboratoire de Physico-Chimie Bioinorganique, Université Louis Pasteur et CNRS (UMR 7509), Ecole Européenne de Chimie, Polymères et Matériaux (ECPM), 25 rue Becquerel, 67087 Strasbourg Cedex 2, France. E-mail: mholler@chimie.u-strasbg.fr
dDipartimento di Chimica “G. Ciamician”, Università di Bologna, via Selmi 2, I-40126 Bologna, Italy. E-mail: paola.ceroni@unibo.it
First published on 22nd August 2007
Abstract
C60-bridge-Fc arrays (C60-Fc, C60-PV-Fc, C60-2PV-Fc, C60-3PV-Fc), bearing a fulleropyrrolidine and a ferrocene (Fc) unit connected via an oligophenylenevinylene (OPV) bridge have been prepared. Suitable dyads (PV-Fc, 2PV-Fc, 3PV-Fc) to be used as references for the study of the electronic properties of the largest arrays have been also synthesized. The electrochemical properties of all the dyad and triad multicomponent arrays have been studied by cyclic voltammetry, evidencing a rich and complex electrochemical pattern due to the presence of several electroactive moieties. The first reduction is always assigned to the fullerene moiety and the first oxidation is centred on the Fc group, making the triad systems suitable candidates for photoinduced electron transfer via the interposed bridge. Photophysical studies evidence a complete quenching of the fluorescence of organic conjugated moieties in PV-Fc, 2PV-Fc, 3PV-Fc, possibly via energy transfer to the Fc unit. In the more complex C60/Fc arrays the quenching of the C60 moieties is ultrafast in CH2Cl2 solution and most likely attributable to electron transfer via the OPV wire. In toluene, the dynamic process of singlet and triplet fullerene quenching can be traced via time resolved fluorescence and transient absorption spectroscopy and the values of the rate constants are smaller with increasing donor–acceptor distance. Definitive assignment of the intercomponent quenching mechanism between the fullerene and the ferrocene moiety (energy or electron transfer) can hardly be obtained. Several repeated attempts to detect the radical anion fingerprint of fulleropyrrolidine with transient absorption failed, even in bimolecular quenching experiments. This supports the view that distance-dependent C60 → Fc singlet–triplet and triplet–triplet energy transfer may compete successfully with the desired charge separation step.
Introduction
In recent years, the use of C60 as an electron and/or energy acceptor in photochemical molecular devices has been intensively investigated.1 As part of this research, the combination of the carbon sphere with π-conjugated oligomers for the construction of donor–fullerene arrays is of particular interest.2 Such hybrid systems have effectively shown excited state interactions making them excellent candidates for fundamental photophysical studies.3 In addition, covalently linked fullerene –(π-conjugated oligomer) derivatives offer interesting perspectives for optical limiting or photodynamic therapy applications.4 Furthermore, they can also be used as the active layer in organic photovoltaic cells.5 More elaborated systems in which an additional donor subunit has been connected to the C60–(π-conjugated oligomer) system have also been described.6–8 For example, a series of structurally well-defined arrays that incorporate a π-extended tetrathiafulvalene (ex-TTF) as electron donor and C60 as electron acceptor, linked by oligophenylenevinylene (OPV) moieties of different lengths has been prepared.6 The electrochemical studies reveal a lack of significant electronic communication between the donor (ex-TTF) and the acceptor (C60) moieties through the π-conjugated oligomer. However, photoinduced electron transfer leading to the radical pair ex-TTF+-OPV-C60– has been evidenced in all these compounds and the charge-recombination dynamics indicates a very low influence of the oligomer length on the electron transfer rate. These findings clearly reveal the nanowire behaviour of the OPV units.9A variety of C60-bridge-Fc triads has been also prepared and characterized photochemically. Several bridge units have been exploited such as olefins ,10ethers ,11 oligothiophenes,12 OPV dendrons,13 and even mixed bridges.12,14 An extensively investigated class of photoactive arrays with C60 and Fc is also that involving a photoactive porphyrin moiety as central bridge. In these systems the central porphyrin chromophore acts as primary light absorbing unit and undergoes electron transfer to the C60 fragment.15 Eventually charge shift from the Fc to the porphyrin moiety leads to long-lived charge separated species, but typically with low yield due to a competitive energy transfer deactivation to the low lying Fc triplet.16
In this paper we report the preparation and the electronic properties of four novel C60-bridge-Fc arrays (C60-Fc, C60-PV-Fc, C60-2PV-Fc, C60-3PV-Fc), bearing a fulleropyrrolidine and a ferrocene (Fc) unit connected via a oligophenylenevinylene bridge, in an attempt to demonstrate the occurrence of a Fc → fullerene photoinduced electron transfer.8 Indeed the terminal Fc unit promotes ultrafast quenching of the fullerene moiety in CH2Cl2 for all of the multicomponent arrays, likely attributable to efficient photoinduced electron transfer via the OPV wire. By contrast, in less polar toluene, fullerene singlet and triplet quenching can be resolved and the rate constants show a substantial distance dependence. Combining the present experimental findings on C60-Fc, C60-PV-Fc, C60-2PV-Fc, C60-3PV-Fc and some suitably designed reference systems with recent literature data, electron and energy transfer quenching pathways between C60 and Fc are discussed.
Results and discussion
Synthesis
The preparation of compounds C60-Fc, C60-PV-Fc and C60-2PV-Fc is shown in Scheme 1. Compound 1 was obtained in five steps from p-bromobenzaldehyde as previously reported.17 Reaction of 1 with ferrocenecarboxaldehyde under Wadsworth–Emmons conditions gave PV-Fc in 96% yield. Subsequent treatment with CF3CO2H afforded 2. It is worth nothing that this deprotection step must be carried out under oxygen free conditions to prevent decomposition, resulting most probably from the easy oxidation of the Fc unit under acidic conditions. Reaction of 2 with phosphonate 1 in the presence of t-BuOK afforded compound 2PV-Fc with 99% yield. Treatment with CF3CO2H in CH2Cl2/H2O under oxygen free conditions then gave 3. The synthetic approach to prepare compounds C60-PV-Fc and C60-2PV-Fc relies upon the 1,3-dipolar cycloaddition of the azomethine ylides generated in situ from the corresponding aldehydes and N-benzylglycine derivative 4.18 Reaction of aldehydes 2 and 3 with 4 and C60 in o-dichlorobenzene (ODCB) at 100 °C gave the corresponding pyrrolidinofullerene derivatives C60-PV-Fc and C60-2PV-Fc in 40 to 33% isolated yield, respectively, after column chromatography on silica gel. Compound C60-Fc was prepared under similar conditions from C60, 4 and ferrocenecarboxaldehyde. Owing to the presence of the 3,5-didodecyloxybenzyl subunit, the pyrrolidinofullerenes C60-Fc, C60-PV-Fc and C60-2PV-Fc are well soluble in common organic solvents such as CH2Cl2, CHCl3, or THF and complete spectroscopic characterization was easily achieved. Their structure was further confirmed by mass spectrometry. In all the cases, the expected molecular ion peak was clearly observed.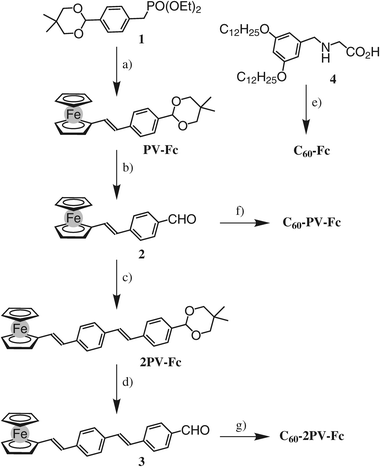 | ||
| Scheme 1 Reagents and conditions: (a) ferrocenecarboxaldehyde, t-BuOK, THF, 0 °C to rt, 2 h, 96%; (b) CF3CO2H, H2O, CH2Cl2, rt, 5 h, 80%; (c) 1, t-BuOK, THF, 0 °C to rt, 2 h, 99%; (d) CF3CO2H, H2O, CH2Cl2, rt, 5 h, 70%; (e) ferrocenecarboxaldehyde, C60, ODCB, Δ, 12 h, 32%; (f) 4, C60, ODCB, 100 °C, 20 h, 40%; (g) 4, C60, ODCB, 100 °C, 20 h, 33%. | ||
The length of the OPV-Fc system was further increased (Scheme 2). In this case, phosphonate 519 with two alkyl chain was used in order to prevent any solubility problems. Reaction of aldehyde 3 with phosphonate 5 in the presence of t-BuOK in THF afforded 3PV-Fc in 50% yield. Subsequent treatment with CF3CO2H in CH2Cl2/H2O gave aldehyde 6 in a moderate yield (37%) which after reaction with C60 and 4 in ODCB at 100 °C gave C60-3PV-Fc in 26% yield.
 | ||
| Scheme 2 Reagents and conditions: (a) 3, t-BuOK, THF, 0 °C to rt, 2 h, 50%; (b) CF3CO2H, H2O, CH2Cl2, rt, 5 h, 37%; (c) 4, C60, ODCB, 100 °C, 20 h, 26%. | ||
The characterization of compound C60-3PV-Fc was found more difficult when compared to that of the shorter analogues C60-PV-Fc and C60-2PV-Fc. Indeed, two diastereoisomers (A and B) can exist in principle for compound C60-3PV-Fc (Fig. 1 and 2). However, the 1H and 13C NMR spectra (CDCl3, room temperature) reveal the existence of a single C1 symmetric compound. In addition, 1H-NMR spectra recorded at temperatures from –50 to 120 °C are all similar and reveal no dynamic exchange between A and B. Therefore only one of the two possible conformers exists for C60-3PV-Fc. As already observed for related ortho-substituted-phenyl pyrrolidinofullerenes, 2D-NOESY experiments revealed that C60-3PV-Fc adopts a conformation in which the unsubstituted ortho position is located atop the C60 (conformer A).18 Therefore, the reaction of 6 with C60 is diastereoselective, affording only one isomer.
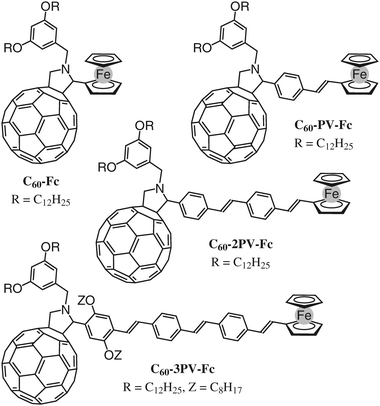 | ||
| Fig. 1 Compounds C60-Fc, C60-PV-Fc, C60-2PV-Fc and C60-3PV-Fc. | ||
 | ||
| Fig. 2 Schematic representation of the two possible atropisomers of compound C60-3PV-Fc. | ||
Electrochemical properties
The electrochemical properties of C60-Fc, C60-PV-Fc, C60-2PV-Fc, C60-3PV-Fc, PV-Fc, 2PV-Fc, 3PV-Fc, and fullerene model compound 7 were studied by cyclic voltammetry in THF/TBAPF6 solutions. The results are presented in Table 1.
| A | I | II | III | IV | V | VI | VII | |
|---|---|---|---|---|---|---|---|---|
| a Electrochemical process superimposed to solvent discharge. b Chemically irreversible process; Epa value at 0.2 V s–1. c Two-electron transfer process. | ||||||||
| Fc | +0.46 | |||||||
| FP | –0.47 | –1.04 | –1.68 | –2.15 | –2.96 | |||
| (–0.44) | (–0.99) | (–1.60) | (–2.10) | (–2.83) | ||||
| 7 | –0.44 | –1.00 | –1.61 | –2.07 | –2.76 | |||
| (–0.41) | (–0.96) | (–1.60) | (–2.07) | (–2.83) | ||||
| C60-Fc | +0.67 | –0.45 | –1.02 | –1.63 | –2.09 | –2.8a | ||
| (+0.60) | (–0.44) | (–1.00) | (–1.63) | (–2.14) | (–2.92) | |||
| C60-PV-Fc | +0.58 | –0.47 | –1.03 | –1.64 | –2.10 | –2.45 | –2.8a | |
| (+0.52) | (–0.42) | (–0.97) | (–1.60) | (–2.07) | (–2.44) | (–2.87) | ||
| PV-Fc | +0.58 | –2.34 | ||||||
| (+0.51) | (–2.36) | |||||||
| C60-2PV-Fc | +0.57 | –0.45 | –1.02 | –1.62 | –2.01 | –2.09 | –2.31 | — |
| (+0.51) | (–0.43) | (–0.98) | (–1.62) | (–2.01) | (–2.15) | (–2.41) | (–2.91) | |
| 2PV-Fc | +0.57 | –1.99 | — | |||||
| (+0.51) | (–2.03) | (–2.29) | ||||||
| C60-3PV-Fc | +0.56 | –0.48 | –1.06 | –1.67 | –1.86 | –2.04 | –2.16 | — |
| (+0.51) | (–0.42) | (–0.99) | (–1.65) | (–1.81) | (–1.98) | (–2.17) | (–2.98)bc | |
| 3PV-Fc | +0.57 | –1.75 | –2.39 | –2.79b | ||||
| (+0.51) | (–1.73) | (–2.39) | (–2.95)b | |||||
A very rich and complex electrochemical pattern has been evidenced for these compounds. Indeed, multiple redox sites are present, namely C60, OPV, Fc and dialkyloxybenzene. The comparison with proper model compounds allows the assignment of the electron transfer processes to the different redox sites, as depicted in Fig. 3. In all the fullerene derivatives, we can observe five reversible one-electron reduction processes typical of the pyrrolidinofullerene core (solid squares in Fig. 3). In the case of C60-3PV-Fc the last process is bielectronic and one of the two reduction is assigned to the fullerene moiety. The presence of the electron withdrawing dialkyloxybenzene group on the nitrogen atom of the pyrrolidinofullerene moiety causes a very small positive shift of the E1/2 values, as evidenced by the comparison of N-methylpyrrolidinofullerene (FP) with 7. In the case of all the compounds containing C60 and Fc, the first E1/2 value are very similar (Table 1 and Fig. 3).
 | ||
| Fig. 3 Diagram showing the half-wave potential values of the investigated compounds in THF/TBAPF6 at 218 K. | ||
In the cathodic region, besides the fullerene -based reductions, reversible one-electron OPV centred reductions are also observed for C60-PV-Fc, C60-2PV-Fc, and C60-3PV-Fc (open triangles in Fig. 3). In particular, peak V of C60-PV-Fc, peaks V and VI of C60-2PV-Fc have been assigned to the OPV moieties, on the basis of the close similarity to the reduction potentials of the corresponding model compounds. In particular, in both these fullerene derivatives a negative shift of the OPV reduction potentials has been observed, compared to the model OPV compounds (PV-Fc and 2PV-Fc, respectively). This behaviour is expected on the basis of the electrostatic repulsion with the negative charges on the fullerene cage. In the case of C60-3PV-Fc (a representative cyclic voltammetric curve is shown in Fig. 4), the assignment of the redox processes is less straightforward: we tentatively assign peaks IV, VI and VII (bielectronic process visible only at 218 K) to the OPV reductions on the basis of the good correlation of the other peaks with those of FP. It is worth noticing that the second OPV reduction of 3PV-Fc is not chemically reversible, contrary to the corresponding peak VI of C60-3PV-Fc. This result may explain the unexpected positive shift observed only for the second OPV reduction in the triad compared to the model 3PV-Fc. Upon increasing the length of the OPV chain (C60-PV-Fc < C60-2PV-Fc < C60-3PV-Fc), the first reduction process of the OPV moiety occurs at less negative potentials, as expected by the extension of the π orbital conjugated system.20 For compound C60-3PV-Fc, the observed positive shift of the first and second OPV reductions compared to C60-2PV-Fc is not only due to the extension of the π system, but also to the presence of two electron-withdrawing –OC8H17 substituents on one end of the OPV moiety.
 | ||
| Fig. 4 Cyclic voltammogram of compound C60-3PV-Fc in THF/TBAPF6 at scan rate of 0.2 V s–1 and T = 298 K. | ||
In the anodic region, only the one-electron oxidation of the Fc moiety can be observed, since the dialkyloxybenzene one is outside the available potential window. In a medium offering a wider anodic potential region such as CH2Cl2/TBAPF6, a chemically irreversible electron transfer process with Epa ∼ +1.5 V (vs. SCE) at 0.2 V s–1 has been attributed to the dialkyloxybenzene oxidation.21 The constancy of the E1/2 values corresponding to the ferrocene oxidation in compounds C60-PV-Fc, C60-2PV-Fc, and C60-3PV-Fc compared to the corresponding model compounds (PV-Fc, 2PV-Fc, and 3PV-Fc, respectively) and to pristine Fc demonstrates that the ferrocene oxidation process is not affected by the presence of the other redox sites. On the other hand, in the case of C60-Fc, the ferrocene moiety is directly connected to the electron-acceptor pyrrolidinofullerene core and a positive shift of the Fc oxidation is observed (ΔE = 100 mV), together with a decrease of the heterogeneous electron transfer rate. Simulation of the voltammetric curves in the range of scan rates between 0.1 and 2 V s–1 yielded the following values: ksh = 0.007 cm–1 s–1 and α = 0.7 much smaller than that reported for pristine ferrocene.22 On the contrary, no significant perturbation of the fullerene -centred reductions has been observed. This electrochemical behaviour would suggest that the HOMO orbital is localized not only on the Fc, but also partially on the fullerene core, conversely the LUMO is mainly localized on the fullerene .
In summary, for all the investigated C60-OPV-Fc compounds the first reduction is due to the fullerene moiety and the first oxidation is centred on the Fc group.
Photophysical properties
The absorption spectrum of the C60 model-compound 7 is virtually identical to similar pyrrolidinofullerene derivatives reported in the literature with the two characteristic peaks at 255 nm and 430 nm.23 Pristine Fc in CH2Cl2 solution exhibits a weak absorption band centred at 443 nm (ε = 100 M–1 cm–1), also in line with literature data.24The ferrocene reference compounds with the spacer attached (PV-Fc, 2PV-Fc, 3PV-Fc) show the characteristic absorption band of the phenylenevinylene fragment, progressively red-shifted with the increasing conjugation: PV-Fc (λmax = 312 nm, ε = 23 600 M–1 cm–1), 2PV-Fc (λmax = 356 nm, ε = 53 200 M–1 cm–1, Fig. 5), 3PV-Fc (λmax = 398 nm, ε = 71 000 M–1 cm–1). The potentially strong fluorescence of the organic conjugated moiety in 2PV-Fc and 3PV-Fc25 is completely quenched by the ferrocene unit.
 | ||
| Fig. 5 Absorption spectra (CH2Cl2) of C60-2PV-Fc (light gray), 7 (black), 2PV-Fc (gray) and the spectral trace obtained by summing the two model compounds 7 and 2PV-Fc (black dashed). | ||
In principle, the spectral overlap between the fluorescence profiles of OPV (peaked around 380–400 nm)25 and the weak lowest absorption band of the Fc acceptor attributed to d–d spin allowed transitions (ca. 2.7 eV, i.e. 460 nm)24 makes it possible a singlet energy transfer via Förster mechanism. Eventually, the lowest triplet electronic state of Fc, which was recently located as low as 1.16 eV26 (to be compared to ca. 1.75 eV of previous assignments),27 might act as final energy sink. Unfortunately excited electronic levels of ferrocenes are known to be rather elusive from the spectroscopic point of view and their energy content has been the object of debate for decades in the photochemical community.24,26,27 These ultrashort-lived levels do not display significant absorption or emission signatures and only indirect proof for their generation can be obtained. Quenching by electron transfer following excitation of the OPV moieties above 3.0 eV cannot be discarded for 2PV-Fc and 3PV-Fc which have, respectively, a thermodynamic driving force for charge separation of about 2.6 and 2.3 eV, as obtainable28 by the one-electron oxidation and reduction potentials in Table 1.
The UV-visible absorption spectra of the four fullerene multicomponent arrays C60-Fc, C60-PV-Fc, C60-2PV-Fc, and C60-3PV-Fc in CH2Cl2 are depicted in Fig. 6. In the UV region there is overlap between fullerene and OPV absorption features, whereas in the Vis spectral window (λ > 450 nm) only the fullerene moiety absorbs, with a minor contribution of Fc until 530 nm. The characteristic fulleropyrrolidine peak at 430 nm is recorded in all cases except C60-3PV-Fc, where it is masked by the band of the OPV fragment.
 | ||
| Fig. 6 Absorption spectra (CH2Cl2) of C60-Fc (black), C60-PV-Fc (gray), C60-2PV-Fc (light gray), and C60-3PV-Fc (black dashed). | ||
The absorption spectra of the fullerene hybrid systems were compared to the spectra obtained by summing the absorption spectra of the C60-model compound 7 with those of the corresponding Fc-OPV model systems (for C60-2PV-Fc, see Fig. 5). Excellent match between experimental and arithmetical traces is found for C60-3PV-Fc. With shorter spacers the matching is progressively lost and the largest difference is found for C60-Fc in the 400–500 nm region. This trend suggests the presence of interactions between the C60 and the Fc moiety, possibly mediated by the short OPV molecular wire,9,29 progressively weaker with longer distance. This explanation is supported by the electrochemical data (Table 1) which show a substantial interaction between the two moieties in C60-Fc.
Whatever the C60-OPV-Fc array, it is possible to excite selectively the C60 moiety at λ > 530 nm. In contrast, selective excitation of the weakly absorbing24ferrocene moiety is always precluded. The occurrence of photoinduced processes in the triads has been probed via excitation of the fulleropyrrolidine moiety at 610 nm in dichloromethane and toluene solution. The fullerene fluorescence spectra of the C60-OPV-Fc systems compared to the C60 model-compound 7 in CH2Cl2 are displayed in Fig. 7 (top panel).
 | ||
| Fig. 7 Fullerene fluorescence spectra of 7 (black), C60-Fc (gray dashed), C60-PV-Fc (black dashed), C60-2PV-Fc (light gray), and C60-3PV-Fc (gray) in CH2Cl2 (top) and toluene (bottom) with λexc = 610 nm. Inset (top and bottom left): sensitized singlet oxygen luminescence at λexc = 610 nm. Inset (bottom right): fluorescence decay at λexc = 635 nm. | ||
The fluorescence quantum yield and singlet lifetimes of 7 are typical for pyrrolidinofullerene derivatives in CH2Cl2 (φ = 3.0 × 10–4, τ = 1.3 ns). The fullerene emission is completely suppressed for C60-Fc, C60-PV-Fc and reduced by 90% for C60-2PV-Fc, and C60-3PV-Fc. The lifetime of the quenched singlet state (<300 ps) is below our experimental resolution in all cases. However, from the steady state luminescence (Fig. 7) and according to eqn (1), a rate constant for the quenching process can be estimated for the two longest arrays C60-2PV-Fc and C60-3PV-Fc, k = 7 × 109 s–1
 | (1) |
Photoexcitation of the four multicomponent systems in CH2Cl2 does not generate fullerene triplet, as inferred by the absence of the characteristic sensitized singlet oxygen luminescence peaked at 1268 nm,30 which is observed for the reference molecule 7. This indicates that the reactive excited state species is the fullerene singlet, in line with several examples of ferrocene–fullerene arrays reported to date.11,14
Spectral data in CH2Cl2 suggest that there is some (small) difference in the extent of the fulleropyrrolidine quenching between the two longest (90% yield) and the two shortest (100% yield) arrays. This prompted us to make the same experiment in toluene, in order to test if a less polar environment could better resolve the behaviour of the four hybrids. Fulleropyrrolidine fluorescence spectra in toluene are reported in Fig. 7, bottom panel. As observed in CH2Cl2, fullerene fluorescence is almost completely quenched for the shortest systems C60-Fc and C60-PV-Fc. On the contrary, a quenched, but still well detectable fullerene fluorescence is recorded for C60-2PV-Fc and C60-3PV-Fc and the corresponding lifetimes are progressively shorter, indicating a dynamic quenching of the fullerene singlet level in toluene: 7 (1.6 ns), C60-3PV-Fc (1.1 ns), C60-2PV-Fc (0.6 ns), C60-PV-Fc and C60-Fc (<0.3 ns).
The observed fullerene singlet quenching process in the two investigated solvents might be due to photoinduced electron transfer leading to Fc+-OPV-C60–, in analogy with previously reported fullerene –ferrocene systems with aliphatic,10ether ,11 or oligothiophene12 linkers. Such charge separated states, according to electrochemical data, are placed at 1.12 (C60-Fc), 1.05 (C60-PV-Fc), 1.02 (C60-2PV-Fc) and 1.04 eV (C60-3PV-Fc). Repeated attempts to evidence the formation of the charge separated state in both CH2Cl2 and toluene through the C60 radical anion fingerprint in the NIR spectral region (900–1600), down to a time resolution of 10 ns, turned out to be unsuccessful, suggesting an ultrafast charge recombination process favoured by the good electronic coupling offered by the OPV wire.9,29 On a shorter time scale (35 ps) the available spectral window of our apparatus is 400–900 nm and the C60 anion cannot be traced. No evidence of charge separated products was previously obtained with ferrocene–fullerene donor–acceptor partners connected via olefinic chains.10 On the contrary, relatively longer lived charge separated states were detected with ether (hundreds of ns)11 and oligothiophene (up to 50 ns)12 spacers.
We wish to emphasize that the lack of experimental evidence for electron transfer from the fullerene singlet might also be due to an energy transfer quenching process. Fullerene → ferrocene singlet (Förster) energy transfer is ruled out since endoergonic, whereas the singlet to triplet process via Dexter mechanism might occur (for the triplet energy of Fc, see above), as recently proposed in porphyrin–Fc systems where porphyrin fluorescence quenching is observed.16
In an attempt to evidence electron transfer quenching, we also investigated the fate of the residual fullerene triplet of C60-2PV-Fc and C60-3PV-Fc in toluene solution. In this solvent, for some C60-bridge-Fc systems, relatively long-lived CS states have been detected, because the charge recombination process is located in a Marcus-inverted region regime with longer-lived CS states compared to polar solvents.14 Indeed, for C60-2PV-Fc and C60-3PV-Fc, the residual fullerene triplet state evidenced in toluene air equilibrated solution via singlet-oxygen sensitization (Fig. 7, bottom panel, left inset), is substantially quenched in oxygen free solutions, where triplet lifetimes of 0.23 and 0.62 µs are found, to be compared to 15.7 µs for the reference system 7 (Fig. 8). Again, no evidence for electron transfer was found. The same result was obtained through bimolecular quenching studies between 7 and 2PV-Fc, providing again no evidence for electron transfer products.31
 | ||
| Fig. 8 Transient absorption spectra of 7 (black), C60-2PV-Fc (light gray), and C60-3PV-Fc (gray) in oxygen free toluene solution immediately after the laser pulse (full line) and after 1.5 ms (dashed line). Inset: absorbance decays at 710 nm of oxygen free (top) and air-equilibrated samples. λexc = 532 nm. | ||
These results strongly suggest that quenching of the fullerene triplet by the ferrocene moiety occurs via triplet energy transfer with a rate constant of 4.3 × 106 and 1.5 × 106 µs–1 for, respectively, C60-2PV-Fc and C60-3PV-Fc. Notably, the poor (if any) photoinduced electron transfer efficiency between ferrocene derivatives and the triplet of C60 has been highlighted recently by Araki et al. even in polar benzonitrile,26 and our results are in line with these findings.
It is worth noticing that, in toluene solution, there is a 3.5-fold and a 3-fold decrease in the rate of the quenching of the fullerene singlet and triplet states, respectively, between C60-2PV-Fc and C60-3PV-Fc (Table 2). It has been demonstrated that electron transfer through OPV bridges identical to those here reported, has no D–A distance dependence up to four phenylene bridges,9 thus a different mechanism (i.e. energy transfer) could be responsible for the photoinduced process in toluene. On the contrary, the much weaker distance effects in more polar CH2Cl2 might be an indication of an electron transfer process through the OPV wire.
Conclusion
Four novel multicomponent architectures C60-Fc, C60-PV-Fc, C60-2PV-Fc, C60-3PV-Fc along with suitable reference compounds for spectroscopic and electrochemical characterization have been prepared. The target systems are made of fulleropyrrolidine (C60) and ferrocene (Fc) units directly connected (C60-Fc) or separated through an oligophenylenevinylene (OPV) bridge of increasing length. Photophysical studies show a different behaviour in CH2Cl2 and toluene. In the former virtually complete quenching of the fullerene singlet excited states in all the multicomponent arrays is observed, tentatively attributed to ultrafast charge separation and recombination mediated by the efficient OPV molecular wire9 and in line with analogous triads with olefine short bridges.10 In toluene, instead, fullerene singlet and triplet quenching process are slower and can be traced via time resolved luminescence or transient absorption spectroscopy. Both singlet and triplet quenching show distance dependence along the series, differently from previous reports which demonstrate an excellent wire-like behaviour of OPV bridges with distance independent electron transfer rates up to 28 Å.9 This suggests that fullerene → ferrocene singlet–triplet and triplet–triplet energy transfer might be the operative quenching mechanism in apolar toluene solvent, although direct spectroscopic evidence for these processes is not obtainable. However, repeated attempts to detect transient electron transfer products (even via bimolecular quenching, which turned out to be successful with a variety of donors) failed, and no traces of the fulleropyrrolidine radical anion have been found. The present results along with the recent findings of Araki et al.,26 suggest some caution in choosing Fc units as electron donors to promote efficient charge separation in multicomponent arrays, also due to its optical elusiveness as a triplet or a radical cation.32 Notably, very recent results have also evidenced the tendency of Fc to undergo energy transfer, thus leading to a substantial abatement of the long-distance charge separation in a Fc-porphyrin-C60 array.16Experimental
General methods
Reagents and solvents were purchased as reagent grade and used without further purification. THF was distilled over sodium benzophenoneketyl . Compounds 1,174,18 and 519 were prepared according to previously reported procedures. All reactions were performed in standard glassware under an inert Ar atmosphere. Evaporation and concentration were done at water aspirator pressure and drying in vacuo at 10–2 Torr. Column chromatography : silica gel 60 (230–400 mesh, 0.040–0.063 mm) was purchased from E. Merck. Thin layer chromatography (TLC) was performed on glass sheets coated with silica gel 60 F254 purchased from E. Merck, visualization by UV light. NMR spectra were recorded on a Bruker AC 200 (200 MHz) or a Bruker AM 400 (400 MHz) with solvent peaks as reference. FAB-mass spectra (MS) were obtained on a ZA HF instrument with 4-nitrobenzyl alcohol as matrix. Elemental analyses were performed by the analytical service at the Institut Charles Sadron, Strasbourg.Compound PV-Fc
t-BuOK (0.33 g, 2.9 mmol) was added to a degassed solution of ferrocenecarboxaldehyde (0.57 g, 2.6 mmol) and 1 (1.0 g, 2.9 mmol) in dry THF (50 mL) at 0 °C under argon atmosphere. The mixture was stirred for 2 h and filtered over celite. The filtrate was evaporated to dryness and was taken up in CH2Cl2. The organic layer was washed with H2O, dried over MgSO4, filtered and evaporated to dryness. Column chromatography (SiO2, hexane/CH2Cl2 3 : 2) yielded PV-Fc (1.02 g, 96%) as an orange powder (mp 204 °C). 1H NMR (300 MHz, CDCl3): 7.45 (m, 4 H), 6.79 (AB, J = 17 Hz, 2 H), 5.39 (s, 1 H), 4.46 (t, J = 2 Hz, 2 H), 4.28 (t, J = 2 Hz, 2 H), 4.13 (s, 5 H), 3.72 (AB, J = 11 Hz, 4 H), 1.31 (s, 3 H), 0.81 (s, 3 H). 13C NMR (75 MHz, CDCl3): 138.32, 127.27, 126.35, 126.04, 125.75, 101.58, 77.65, 69.71, 69.49, 67.10, 30.23, 23.05, 21.90. Anal. calcd for C24H26O2Fe: C 71.65, H 6.51. Found: C 71.52, H 6.54%. UV/Vis (CH2Cl2): 266 (19 000), 311 (30 400), 456 (1700).Compound 2
A 1 : 1 mixture of CF3CO2H/H2O (30 mL) was added to a degassed solution of PV-Fc (1.02 g, 2.53 mmol) in CH2Cl2 (15 mL) under argon. The mixture was stirred at room temperature for 5 h under argon atmosphere. The reaction mixture was then washed with H2O (until pH was near neutrality), dried over MgSO4, filtered and evaporated to dryness. Column chromatography (SiO2, CH2Cl2/hexane 1 : 1) yielded 2 (0.64 g, 80%) as a dark red powder. IR (neat): 1691 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 2733, 2822 (C–H). 1H NMR (300 MHz, CDCl3): 9.98 (s, 1 H), 7.83 (d, J = 8 Hz, 2H), 7.56 (d, J = 8 Hz, 2H), 6.91 (AB, J = 17 Hz, 2 H), 4.51 (t, J = 2 Hz, 2 H), 4.36 (t, J = 2 Hz, 2 H), 4.16 (s, 5 H). 13C NMR (75 MHz, CDCl3): 191.57, 144.00, 134.56, 131.47, 130.30, 126.02, 124.51, 82.18, 69.72, 69.33, 67.31. Anal. calcd for C19H16OFe: C 72.18, H 5.10. Found: C 71.99, H 5.18%.
O), 2733, 2822 (C–H). 1H NMR (300 MHz, CDCl3): 9.98 (s, 1 H), 7.83 (d, J = 8 Hz, 2H), 7.56 (d, J = 8 Hz, 2H), 6.91 (AB, J = 17 Hz, 2 H), 4.51 (t, J = 2 Hz, 2 H), 4.36 (t, J = 2 Hz, 2 H), 4.16 (s, 5 H). 13C NMR (75 MHz, CDCl3): 191.57, 144.00, 134.56, 131.47, 130.30, 126.02, 124.51, 82.18, 69.72, 69.33, 67.31. Anal. calcd for C19H16OFe: C 72.18, H 5.10. Found: C 71.99, H 5.18%.
Compound 2PV-Fc
t-BuOK (0.25 g, 2.2 mmol) was added to a degassed solution of 2 (0.64 g, 2.0 mmol) and 1 (0.76 g, 2.2 mmol) in dry THF (50 mL) at 0 °C under argon. The mixture was stirred for 2 h and filtered under celite. The filtrate was evaporated to dryness and was taken up in CH2Cl2. The organic layer was washed with H2O, dried over MgSO4, filtered and evaporated to dryness. Column chromatography (SiO2, hexane/CH2Cl2 3 : 2) yielded 2PV-Fc (1.02 g, 99%) as an orange powder. 1H NMR (300 MHz, CDCl3): 7.55–7.41 (m, 8 H), 7.11 (s, 2 H), 6.80 (AB, J = 17 Hz, 2 H), 5.41 (s, 1 H), 4.48 (t, J = 2 Hz, 2 H), 4.30 (t, J = 2 Hz, 2 H), 4.14 (s, 5 H), 3.73 (AB, J = 11 Hz, 4 H), 1.32 (s, 3 H), 0.82 (s, 3 H). Anal. calcd for C32H32O2Fe: C 76.19, H 6.39. Found: C 76.36, H 6.47%. UV/Vis (CH2Cl2): 231 (25 700), 356 (55 200).Compound 3
A 1 : 1 mixture of CF3CO2H/H2O (30 mL) was added to a degassed solution of 2PV-Fc (1.0 g, 1.98 mmol) in CH2Cl2 (100 mL) under argon. The mixture was stirred at room temperature for 5 h under argon atmosphere. The reaction mixture was then washed with H2O (until pH was near neutrality), dried over MgSO4, filtered and evaporated to dryness. Column chromatography (SiO2, CH2Cl2/hexane 1 : 1) yielded 3 (0.58 g, 70%) as a dark red powder. IR (neat): 1690 (CO), 2719 (C–H). 1H NMR (300 MHz, CDCl3): 10.00 (s, 1 H), 7.77 (AB, J = 8 Hz, 4 H), 7.49 (AB, J = 8 Hz, 4 H), 7.21 (AB, J = 16 Hz, 2 H), 6.82 (AB, J = 16 Hz, 2 H), 4.50 (t, J = 2 Hz, 2 H), 4.33 (t, J = 2 Hz, 2 H), 4.16 (s, 5 H). Anal. calcd for C27H22OFe: C 77.52, H 5.30. Found: C 76.74, H 5.45%.
Compound 3PV-Fc
t-BuOK (0.1 g, 0.84 mmol) was added to a degassed solution of 3 (0.3 g, 0.7 mmol) and 5 (0.5 g, 0.84 mmol) in dry THF (150 mL) at 0 °C under argon. The mixture was stirred for 2 h and filtered under celite. The filtrate was evaporated to dryness and was taken up in CH2Cl2. The organic layer was washed with H2O, dried over MgSO4, filtered and evaporated to dryness. Column chromatography (SiO2, hexane/CH2Cl2 3 : 2) yielded 3PV-Fc (0.3 g, 50%) as a red glassy product. 1H NMR (300 MHz, CDCl3): 7.54–7.40 (m, 8 H), 7.19 (s, 2 H), 7.10 (m, 4 H), 6.78 (AB, J = 17 Hz, 2 H), 5.74 (s, 1 H), 4.50 (s, 2 H), 4.32 (s, 2 H), 4.16 (s, 5 H), 4.02 (m, 4 H), 3.71 (AB, J = 11 Hz, 4 H), 1.84 (m, 4 H), 1.33-1.26 (m, 23 H), 0.88 (m, 9 H), 0.80 (s, 3 H). 13C NMR (75 MHz, CDCl3): 151.12, 150.29, 137.21, 136.53, 135.89, 128.61, 128.08, 127.74, 127.32, 126.99, 126.83, 126.79, 126.69, 126.05, 125.66, 123.53, 111.49, 110.59, 97.04, 83.41, 77.85, 69.56, 69.34, 69.23, 69.11, 66.87, 31.80, 30.26, 29.66, 29.45, 29.41, 29.37, 29.33, 29.29, 26.25, 26.09, 23.20, 22.66, 21.85, 14.10. UV/Vis (CH2Cl2): 398 (71 000).Compound 6
A 1 : 1 mixture of CF3CO2H/H2O (10 mL) was added to a degassed solution of 3PV-Fc (0.24 g, 0.28 mmol) in CH2Cl2 (20 mL) under argon. The mixture was stirred at room temperature for 5 h under argon atmosphere. The reaction mixture was then washed with H2O (until pH was near neutrality), dried over MgSO4, filtered and evaporated to dryness. Column chromatography (SiO2, CH2Cl2/hexane 1 : 1) yielded 6 (0.08 g, 37%) as a dark red powder that was used in the next step as received. 1H NMR (300 MHz, CDCl3): 10.38 (s, 1 H), 7.32 (m, 8H), 7.05 (AB, J = 16 Hz, 2H), 7.03 (s, 4 H), 6.72 (AB, J = 17 Hz, 2 H), 4.40 (s, 2 H), 4.22 (s, 2 H), 4.07 (s, 5 H), 4.01 (t, J = 6 Hz, 2 H), 3.95 (t, J = 6 Hz, 2 H), 1.78 (m, 4 H), 1.43–1.19 (m, 20 H), 0.83 (m, 6 H).Compound 7
A mixture of formaldehyde (0.08 g, 0.1 mmol), C60 (0.07 g, 0.1 mmol) and 4 (0.11 g, 0.2 mmol) in ODCB (25 mL) was refluxed for 12 h under argon. After cooling, the resulting solution was evaporated to dryness and column chromatography (SiO2, hexane/CH2Cl2 4 : 1) gave 7 (39%) as a brown glassy product. 1H NMR (300 MHz, CDCl3): 6.88 (d, J = 2 Hz, 2 H), 6.48 (t, J = 2 Hz, 1 H), 4.45 (s, 4 H), 4.25 (s, 2 H), 4.03 (t, J = 6 Hz, 4 H), 1.82 (m, 4 H), 1.44 (m, 36 H), 0.88 (t, J = 7 Hz, 6 H). 13C NMR (75 MHz, CDCl3): 160.53, 155.03, 147.26, 146.20, 146.09, 146.02, 145.65, 145.37, 145.24, 144.52, 143.06, 142.58, 142.21, 142.03, 141.83, 140.17, 140.10, 136.22, 107.04, 100.54, 70.78, 68.16. 67.31, 58.61, 31.92, 29.71, 29.58, 29.55, 29.47, 29.36, 29.33, 26.13, 22.69, 14.14. Anal. calcd for C93H59O2N: C 91.37, H 4.86, N 1.15. Found: C 90.99, H 5.17, N 0.91%.Compound C60-Fc
A mixture of ferrocenecarboxaldehyde (0.119 g, 0.55 mmol), C60 (0.4 g, 0.55 mmol) and 4 (0.59 g, 1.11 mmol) in ODCB (100 mL) was refluxed for 12 h. After cooling to room temperature, the resulting solution was evaporated to dryness and column chromatography (SiO2, hexane/CH2Cl2 4 : 1) gave C60-Fc (0.25 g, 32%) as a brown glassy product. 1H NMR (300 MHz, CDCl3): 7.04 (d, J = 2 Hz, 2 H), 6.51 (t, J = 2 Hz, 1 H), 6.17 (d, J = 14 Hz, 1 H), 5.18 (s, 1 H), 4.86 (d, J = 9 Hz, 1 H), 4.65 (s, 1 H), 4.57 (s, 1 H), 4.31 (s, 5 H), 4.26 (t, J = 2 Hz, 2 H), 4.13 (d, J = 10 Hz, 1 H), 4.07 (m, 4 H), 3.79 (d, J = 14 Hz, 1 H), 1.84 (m, 4 H), 1.44 (m, 36 H), 0.89 (t, J = 7 Hz, 6 H). 13C NMR (75 MHz, CDCl3): 160.80, 156.46, 154.29, 153.97, 153.30, 147.61, 147.29, 147.25, 147.19, 146.53, 146.29, 146.25, 146.21, 146.12, 146.05, 145.92, 145.75, 145.56, 145.49, 145.25, 145.21, 145.15, 144.70, 144.65, 144.43, 144.41, 143.09, 142.99, 142.67, 142.61, 142.56, 142.24, 142.20, 142.17, 142.14, 142.07, 142.04, 141.88, 141.77, 141.59, 141.44, 140.16, 140.02, 139.47, 138.86, 136.32, 136.26, 136.05, 135.80, 106.39, 100.21, 86.58, 78.08, 75.47, 69.38, 68.45, 68.29, 68.23, 67.68, 67.46, 67.22, 57.65, 31.94, 29.72, 29.66, 29.50, 29.38, 26.17, 22.71, 14.15. Anal. calcd for C103H68O2NFe: C 87.89, H 4.87, N 1.00. Found: C 87.30, H 4.77, N 1.04. FAB-MS: calcd. for C103H68O2NFe 1406.46; found 1406.5 [M]+. UV/Vis (CH2Cl2): 255 (133 700), 306 (42 260), 430 (4300), 704 (360).Compound C60-PV-Fc
A mixture of 2 (0.105 g, 0.33 mmol), C60 (0.24 g, 0.33 mmol) and 4 (0.35 g, 0.66 mmol) in ODCB (60 mL) was heated at 100 °C for 20 h under argon. After cooling, the resulting solution was evaporated to dryness and column chromatography (SiO2, hexane/CH2Cl2 4 : 1) gave C60-PV-Fc (0.2 g, 40%) as a brown glassy product. 1H NMR (300 MHz, CDCl3): 7.84 (br s, 2 H), 7.50 (d, J = 8 Hz, 2 H), 6.84 (d, J = 2 Hz, 2 H), 6.80 (AB, J = 17 Hz, 2 H), 6.49 (t, J = 2 Hz, 1 H), 5.20 (s, 1 H), 4.92 (d, J = 9 Hz, 1 H), 4.51 (d, J = 13 Hz, 1 H), 4.45 (t, J = 2 Hz, 2 H), 4.28 (t, J = 2 Hz, 2 H), 4.17 (d, J = 10 Hz, 1 H), 4.14 (s, 5 H), 4.04 (t, J = 6 Hz, 4 H), 3.62 (d, J = 14 Hz, 1 H), 1.84 (m, 4 H), 1.44 (m, 36 H), 0.88 (t, J = 7 Hz, 6 H). 13C NMR (75 MHz, CDCl3): 160.11, 156.07, 153.68, 153.12, 153.03, 146.87, 146.42, 146.03, 145.90, 145.86, 145.77, 145.73, 145.69, 145.65, 145.51, 145.48, 145.30, 145.12, 145.08, 144.89, 144.84, 144.79, 144.70, 144.27, 144.18, 143.98, 143.94, 142.71, 142.54, 142.23, 142.14, 142.11, 141.89, 141.81, 141.70, 141.66, 141.57, 141.55, 141.51, 141.40, 141.21, 141.11, 139.72, 139.66, 139.59, 139.47, 139.11, 137.69, 136.41, 136.00, 135.53, 135.32, 134.94, 129.35, 127.16, 125.72, 125.21, 106.84, 99.83, 82.81, 80.65, 68.78, 68.65, 68.34, 67.77, 66.50, 66.46, 66.11, 56.17, 31.49, 29.27, 29.22, 29.06, 28.94, 25.72, 22.26, 13.71. Anal. calcd for C111H73O2NFe: C 88.37, H 4.88, N 0.93. Found: C 87.91, H 4.90, N 1.11%. FAB-MS: calcd. for C111H73O2NFe 1508.65; found 1508.3 [M]+. UV/Vis (CH2Cl2): 229 (154 600), 255 (168 000), 319 (85 900), 431 (7500), 703 (750).Compound C60-2PV-Fc
A mixture of 3 (0.23 g, 0.55 mmol), C60 (0.4 g, 0.55 mmol) and 4 (0.59 g, 1.11 mmol) in ODCB (100 mL) was heated at 100 °C for 20 h under argon. After cooling, the resulting solution was evaporated to dryness and column chromatography (SiO2, hexane/CH2Cl2 4 : 1) gave C60-2PV-Fc (0.3 g, 33%) as a brown glassy product. 1H NMR (300 MHz, CDCl3): 7.90 (br s, 2 H), 7.61 (d, J = 8 Hz, 2 H), 7.44 (AB, J = 8 Hz, 4 H), 7.12 (s, 2 H), 6.84 (d, J = 2 Hz, 2 H), 6.79 (AB, J = 17 Hz, 2 H), 6.49 (t, J = 2 Hz, 1 H), 5.22 (s, 1 H), 4.93 (d, J = 9 Hz, 1 H), 4.52 (d, J = 13 Hz, 1 H), 4.47 (t, J = 2 Hz, 2 H), 4.29 (t, J = 2 Hz, 2 H), 4.19 (d, J = 10 Hz, 1 H), 4.14 (s, 5 H), 4.04 (t, J = 6 Hz, 4 H), 3.64 (d, J = 13 Hz, 1 H), 1.84 (m, 4 H), 1.43 (m, 36 H), 0.88 (t, J = 7 Hz, 6 H). 13C NMR (75 MHz, CDCl3): 160.57, 156.48, 154.08, 153.47, 153.37, 147.31, 146.83, 146.46, 146.30, 146.27, 146.22, 146.18, 146.14, 146.10, 145.95, 145.74, 145.53, 145.32, 145.26, 145.15, 144.71, 144.62, 144.42, 144.38, 143.15, 142.98, 142.68, 142.55, 142.33, 142.25, 142.15, 142.10, 142.07, 142.02, 141.92, 141.85, 141.65, 141.55, 140.16, 140.11, 139.95, 139.90, 139.57, 137.70, 137.37, 136.84, 136.44, 136.28, 136.00, 135.77, 129.83, 129.78, 128.92, 127.62, 127.16, 126.88, 126.14, 125.80, 107.32, 100.30, 80.99, 69.44, 69.31, 68.77, 68.23, 66.98, 56.62, 31.94, 29.72, 29.67, 29.51, 29.38, 26.17, 22.71, 14.15. Anal. calcd for C119H79O2NFe.H2O: C 87.75, H 5.01, N 0.86. Found: C 87.75, H 5.03, N 1.03%. FAB-MS: calcd. for C119H79O2NFe 1610.79; found 1610.4 [M]+. UV/Vis (CH2Cl2): 229 (154 600), 255 (168 000), 319 (859 00), 431 (7500), 703 (750).Compound C60-3PV-Fc
A mixture of 6 (0.08 g, 0.1 mmol), C60 (0.07 g, 0.1 mmol) and 4 (0.11 g, 0.2 mmol) in ODCB (25 mL) was heated at 100 °C for 20 h under argon. After cooling, the resulting solution was evaporated to dryness and column chromatography (SiO2, hexane/CH2Cl2 4 : 1) gave C60-3PV-Fc (0.05 g, 26%) as a brown glassy product. 1H NMR (300 MHz, CDCl3): 7.75 (s, 1 H), 7.49 (s, 4 H), 7.45 (d, J = 3 Hz, 1 H), 7.38 (d, J = 6 Hz, 4 H), 7.15 (s, 1 H), 7.14 (d, J = 12 Hz, 1 H), 7.09 (d, J = 2 Hz, 2 H), 6.83 (d, J = 2 Hz, 2 H), 6.74 (AB, J = 17 Hz, 2 H), 6.48 (t, J = 2 Hz, 1 H), 5.82 (s, 1 H), 4.91 (d, J = 7 Hz, 1 H), 4.56 (s, 2 H), 4.52 (d, J = 11 Hz, 1 H), 4.37 (s, 2 H), 4.25 (d, J = 7 Hz, 1 H), 4.20 (s, 5 H), 4.09 (m, 3 H), 4.02 (t, J = 5 Hz, 4 H), 3.80 (m, 1 H), 3.70 (d, J = 10 Hz, 1 H), 1.82 (m, 6 H), 1.66 (m, 2 H), 1.48–1.26 (m, 56 H), 0.88 (t, J = 5 Hz, 12 H). 13C NMR (75 MHz, CDCl3): 160.53, 156.98, 155.07, 154.48, 153.88, 152.06, 150.96, 147.25, 146.79, 146.70, 146.27, 146.19, 146.14, 146.07, 146.03, 145.90, 145.66, 145.53, 145.24, 145.19, 145.10, 145.05, 144.57, 144.52, 144.42, 144.33, 142.98, 142.94, 142.61, 142.52, 142.29, 142.23, 142.13, 142.04, 141.90, 141.74, 141.67, 141.64, 141.61, 140.12, 140.04, 139.99, 139.66, 139.45, 137.22, 136.92, 136.54, 136.27, 136.20, 136.17, 136.11, 134.70, 128.77, 127.99, 127.92, 127.05, 126.95, 126.78, 126.60, 126.43, 126.33, 125.72, 123.39, 114.76, 109.53, 107.14, 100.27, 76.10, 73.11, 70.23, 69.97, 69.60, 68.96, 68.64, 68.10, 67.19, 66.26, 56.40, 31.89, 29.53, 29.68, 29.63, 29.48, 29.34, 26.89, 26.27, 26.15, 26.09, 22.74, 22.67, 14.18, 14.10. Anal. calcd for C143H117O4NFe.CH2Cl2: C 84.29, H 5.80, N 0.68. Found: C 84.49, H 6.44, N 0.92%. FAB-MS: 1247.6 (30, [M-C60]+, calcd. for C83H117O4NFe 1247.83), 1968.6 (50, [MH]+, calcd. for C144H117O4NFe 1968.8).Electrochemistry
The electrochemical experiments were carried out in argon-purged THF (Aldrich) or CH2Cl2 (Romil Hi-Dry™) solutions at 298, 218 K with an EcoChemie Autolab 30 multipurpose instrument interfaced to a personal computer. THF was distilled according to a literature procedure.33 In the cyclic voltammetry (CV) the working electrode was a glassy carbon electrode (0.08 cm2, Amel) or a Pt disk ultramicroelectrode (r = 25 µm). In all cases, the counter electrode was a Pt spiral, separated from the bulk solution with a fine glass frit, and a silver wire was employed as a quasi-reference electrode (AgQRE). The potentials reported are referred to SCE by measuring the AgQRE potential with respect to ferrocene. The concentration of the compounds examined was of the order of 5 × 10–4 M; 0.05 M tetrabuthylammonium hexafluorophosphate (TBAPF6) was added as supporting electrolytes. Cyclic voltammograms were obtained with scan rates in the range 0.05–20 V s–1. The CV simulations were carried out by the DIGISIM program.Photophysics
The photophysical investigations were carried out in CH2Cl2 and toluene (Carlo Erba, spectrofluorimetric grade). Absorption spectra were recorded with a Perkin-Elmer λ40 spectrophotometer. Emission spectra were obtained with an Edinburgh FLS920 spectrometer (continuous 450 W Xe lamp), equipped with a Peltier-cooled Hamamatsu R928 photomultiplier tube (185–850 nm) or a Hamamatsu R5509-72 supercooled photomultiplier tube (193 K, 800–1700 nm range). Emission quantum yields were determined according to the approach described by Demas and Crosby34 using [Os(phen)3]2+ (Φ = 0.005) as standard.35 Emission lifetimes were determined with the time correlated single photon counting technique using an Edinburgh FLS920 spectrometer equipped with a laser diode head as excitation source (1 MHz repetition rate, λexc = 407 or 635 nm, 200 ps time resolution upon deconvolution) and an Hamamatsu R928 PMT as detector. Transient absorption spectra in the nanosecond-microsecond time domain were obtained by using the nanosecond flash photolysis apparatus described previously.36 Experimental uncertainties are estimated to be 8% for lifetime determinations, 20% for emission quantum yields, 10% for relative emission intensities in the NIR, 1 nm and 5 nm for absorption and emission peaks, respectively.Acknowledgements
This research was supported by the CNR (Commessa PM.P04.010 “Materiali Avanzati per la Conversione di Energia Luminosa”, MACOL), MIUR (PRIN, “Sistemi supramolecolari per la conversione dell’energia luminosa”), the CNRS (UPR 8241) and the EU (contract HPRN-CT-2002-00171, FAMOUS).References
- For reviews, see: (a) N. Martín, L. Sanchez, B. Illescas and I. Perez, Chem. Rev., 1998, 98, 2527 CrossRef CAS; (b) D. M. Guldi, Chem. Soc. Rev., 2002, 31, 22 RSC; (c) H. Imahori and Y. Sakata, Eur. J. Org. Chem., 1999, 2445 CrossRef CAS; (d) D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2001, 34, 40 CrossRef CAS; (e) H. Imahori, J. Phys. Chem. B, 2004, 108, 6130 CrossRef CAS; (f) N. Armaroli, Photochem. Photobiol. Sci., 2003, 2, 73 RSC.
- T. M. Figueira-Duarte, A. Gégout and J.-F. Nierengarten, Chem. Commun., 2007, 109 RSC.
- (a) D. M. Guldi and N. Martin, J. Mater. Chem., 2002, 12, 1978 RSC; (b) N. Armaroli and G. Accorsi, in Fullerenes. Principles and Applications, ed. F. Langa and J.-F. Nierengarten, The Royal Society of Chemistry, Cambridge, UK, 2007, p. 79 Search PubMed.
- T. M. Figueira-Duarte, J. Clifford, V. Amendola, A. Gégout, J. Olivier, F. Cardinali, M. Meneghetti, N. Armaroli and J.-F. Nierengarten, Chem. Commun., 2006, 2054 RSC.
- For reviews, see: (a) J.-F. Nierengarten, Sol. Energy Mater. Sol. Cells, 2004, 83, 187 CrossRef CAS; (b) J.-F. Nierengarten, New J. Chem., 2004, 28, 1177 RSC; (c) J. L. Segura, N. Martin and D. M. Guldi, Chem. Soc. Rev., 2005, 34, 31 RSC.
- (a) F. Giacalone, J. L. Segura, N. Martin and D. M. Guldi, J. Am. Chem. Soc., 2004, 126, 5340 CrossRef; (b) F. Giacalone, J. L. Segura, N. Martin, J. Ramey and D. M. Guldi, Chem.–Eur. J., 2005, 11, 4819 CrossRef CAS.
- G. de la Torre, F. Giacalone, J. L. Segura, N. Martin and D. M. Guldi, Chem.–Eur. J., 2005, 11, 1267 CrossRef.
- (a) H. Kanato, K. Takimiya, T. Otsubo, Y. Aso, T. Nakamura, Y. Araki and O. Ito, J. Org. Chem., 2004, 69, 7183 CrossRef CAS; (b) T. Nakamura, H. Kanato, Y. Araki, O. Ito, K. Takimiya, T. Otsubo and Y. Aso, J. Phys. Chem. A, 2006, 110, 3471 CrossRef CAS.
- (a) H. D. Sikes, J. F. Smalley, S. P. Dudek, A. R. Cook, M. D. Newton, C. E. D. Chidsey and S. W. Feldberg, Science, 2001, 291, 1519 CrossRef CAS; (b) W. B. Davis, M. A. Ratner and M. R. Wasielewski, J. Am. Chem. Soc., 2001, 123, 7877 CrossRef CAS.
- D. M. Guldi, M. Maggini, G. Scorrano and M. Prato, J. Am. Chem. Soc., 1997, 119, 974 CrossRef CAS.
- S. Campidelli, E. Vazquez, D. Milic, M. Prato, J. Barbera, D. M. Guldi, M. Marcaccio, P. Paolucci, F. Paolucci and R. Deschenaux, J. Mater. Chem., 2004, 14, 1266 RSC.
- T. Nakamura, H. Kanato, Y. Araki, O. Ito, K. Takimiya, T. Otsubo and Y. Aso, J. Phys. Chem. A, 2006, 110, 3471 CrossRef CAS.
- L. Pérez, J. C. Garcia-Martinez, E. Diez-Barra, P. Atienzar, H. Garcia, J. Rodriguez-Lopez and F. Langa, Chem.–Eur. J., 2006, 12, 5149 CrossRef CAS.
- (a) M. Even, B. Heinrich, D. Guillon, D. M. Guldi, M. Prato and R. Deschenaux, Chem.–Eur. J., 2001, 7, 2595 CrossRef CAS; (b) M. Fujitsuka, N. Tsuboya, R. Hamasaki, O. Ito, S. Onodera, M. Ito and Y. Yamamoto, J. Phys. Chem. A, 2003, 107, 1452 CrossRef CAS.
- (a) H. Imahori, K. Tamaki, Y. Araki, Y. Sekiguchi, O. Ito, Y. Sakata and S. Fukuzumi, J. Am. Chem. Soc., 2002, 124, 5165 CrossRef CAS; (b) H. Imahori, Y. Sekiguchi, Y. Kashiwagi, T. Sato, Y. Araki, O. Ito, H. Yamada and S. Fukuzumi, Chem.–Eur. J., 2004, 10, 3184 CrossRef CAS; (c) H. Imahori, Org. Biomol. Chem., 2004, 2, 1425 RSC; (d) F. D’Souza, R. Chitta, S. Gadde, D. M. S. Islam, A. L. Schumacher, M. E. Zandler, Y. Araki and O. Ito, J. Phys. Chem. B, 2006, 110, 25240 CrossRef CAS.
- M. U. Winters, E. Dahlstedt, H. E. Blades, C. J. Wilson, M. J. Frampton, H. L. Anderson and B. Albinsson, J. Am. Chem. Soc., 2007, 129, 4291 CrossRef CAS.
- N. Armaroli, G. Accorsi, J. N. Clifford, J.-F. Eckert and J.-F. Nierengarten, Chem.–Asian J., 2006, 1, 564 CrossRef CAS.
- F. Ajamaa, T. M. Figueira Duarte, C. Bourgogne, M. Holler, P. W. Fowler and J.-F. Nierengarten, Eur. J. Org. Chem., 2005, 3766 CrossRef CAS.
- (a) M. J. Gomez-Escalonilla, F. Langa, J.-M. Rueff, L. Oswald and J.-F. Nierengarten, Tetrahedron Lett., 2002, 43, 7507 CrossRef CAS; (b) F. Langa, M. J. Gomez-Escalonilla, J. M. Rueff, T. M. Figueira Duarte, J.-F. Nierengarten, V. Palermo, P. Samori, Y. Rio, G. Accorsi and N. Armaroli, Chem.–Eur. J., 2005, 11, 4405 CrossRef CAS.
- (a) R. Schenk, H. Gregorius, K. Meerholz, J. Heinze and K. Müllen, J. Am. Chem. Soc., 1991, 113, 2634 CrossRef CAS; (b) K. Meerholz, H. Gregorius, K. Müllen and J. Heinze, Adv. Mater., 1994, 6, 671 CrossRef CAS; (c) T. Gu, P. Ceroni, G. Marconi, N. Armaroli and J.-F. Nierengarten, J. Org. Chem., 2001, 66, 6432 CrossRef CAS.
- M. Goez and G. Eckert, J. Phys. Chem., 1991, 95, 1179 CAS.
- A. D. Clegg, N. V. Rees, O. V. Klymenko, B. A. Coles and R. G. Compton, J. Electroanal. Chem., 2005, 580, 78 CrossRef CAS.
- N. Armaroli, F. Barigelletti, P. Ceroni, J.-F. Eckert, J.-F. Nicoud and J.-F. Nierengarten, Chem. Commun., 2000, 599 RSC.
- Y. S. Sohn, D. N. Hendrickson and H. B. Gray, J. Am. Chem. Soc., 1971, 93, 3603 CrossRef.
- T. Gu, P. Ceroni, G. Marconi, N. Armaroli and J.-F. Nierengarten, J. Org. Chem., 2001, 66, 6432 CrossRef CAS.
- Y. Araki, Y. Yasumura and O. Ito, J. Phys. Chem. B, 2005, 109, 9843 CrossRef CAS.
- W. G. Herkstroeter, J. Am. Chem. Soc., 1975, 97, 4161 CrossRef CAS.
- V. Balzani and F. Scandola, Supramolecular Photochemistry, Ellis Horwood, Chichester, UK, 1991 Search PubMed.
- F. Giacalone, J. L. Segura, N. Martin, J. Ramey and D. M. Guldi, Chem.–Eur. J., 2005, 11, 4819 CrossRef CAS.
- (a) Y. Rio, G. Accorsi, H. Nierengarten, C. Bourgogne, J.-M. Strub, A. Van Dorsselaer, N. Armaroli and J.-F. Nierengarten, Tetrahedron, 2003, 59, 3833 CrossRef CAS; (b) M. Gutierrez-Nava, G. Accorsi, P. Masson, N. Armaroli and J.-F. Nierengarten, Chem.–Eur. J., 2004, 10, 5076 CrossRef CAS.
- Similar bimolecular quenching experiments with potential electron donors in place of Fc such as Zn(II)-tetraphenylporphyrin, [Ru(2,2′-bipyridine)3]2+, and [Cu(2,9-diphenyl-1,10-phenanthroline)2]+ gave rise to a neat fulleropyrrolidine radical anion at 980 nm, demonstrating that the lack of the signal in the Fc experiments are not due to experimental limitations.
- M. Faraggi, D. Weinraub, F. Broitman, M. R. Defelippis and M. H. Klapper, Radiat. Phys. Chem., 1988, 32, 293 CrossRef CAS.
- F. Paolucci, M. Carano, P. Ceroni, L. Mottier and S. Soffia, J. Electrochem. Soc., 1999, 146, 3357 CrossRef CAS.
- J. N. Demas and G. A. Crosby, J. Phys. Chem., 1971, 75, 991 CrossRef.
- E. M. Kober, J. V. Caspar, R. S. Lumpkin and T. J. Meyer, J. Phys. Chem., 1986, 90, 3722 CrossRef CAS.
- J. N. Clifford, T. Gu, J.-F. Nierengarten and N. Armaroli, Photochem. Photobiol. Sci., 2006, 5, 1165 RSC.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2008 |