Inclusion of electrochemically active guests by novel oxacalixarene hosts†
David
Sobransingh
,
Mahender B.
Dewal
,
Jacob
Hiller
,
Mark D.
Smith
and
Linda S.
Shimizu
*
Department of Chemistry and Biochemistry, University of South Carolina, 631 Sumter St. Columbia, SC 29208, USA. E-mail: shimizul@mail.chem.sc.edu; Fax: +1 803 777 9521; Tel: +1 803 777 2066
First published on 6th September 2007
Abstract
We demonstrate for the first time the utility of oxacalixarenes as hosts and investigate the forces that influence the thermodynamics of binding.
Introduction
Calixarenes are useful host molecules in supramolecular systems and have a wide range of industrial applications such as metal sequestration and waste remediation.1 Classical calixarenes employ carbon atoms to bridge their aromatic moieties; however, the incorporation of non-carbon bridging atoms within the calixarene framework is an interesting yet under-utilized method of increasing structural diversity and conferring new chemical and physical properties upon this class of molecule.2 The calixarene skeleton has been modified with heteroatoms including sulfur3 and nitrogen,4 but incorporation of bridging oxygen atoms has been scarce.5Oxygen bridged calixarenes (oxacalixarenes) first appeared in the literature in 19665 but only a handful of papers were published on them prior to 2005. Recent advances by Katz et al.6 has led to a doubling of the number of published papers6,7 on the synthesis and structure of oxacalixarenes. The exploration of the host properties of oxacalixarenes however, has been conspicuously absent from the literature. In this paper we probe the utility of oxacalixarenes as hosts using the electrochemically active guest molecules ferrocene and cobaltocenium. We report the synthesis of two new oxacalixarene based hosts (2, 3) (Fig. 1) and demonstrate for the first time that oxacalixarenes (1, 3) show host–guest interactions with the ferrocene (F)/ferrocenium (F+) and cobaltocenium (C+)/cobaltocene (C) redox couples. Both host 1 and 3 show a significant thermodynamic preference for positively charged guests over the neutral species and display an unusually large negative shift of the half-wave potential (E1/2) in voltammetric experiments.
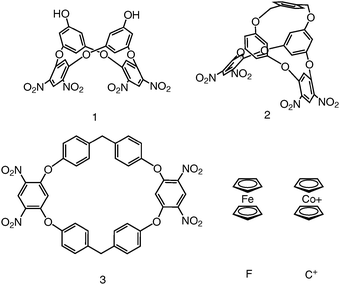
Results and discussion
Functionalized oxacalix[4]arenes such as 1, adopt a 1,3-alternate or saddle conformation in which the hydroxyl substituted rings are nearly parallel.6 Oxacalixarene 1 has the unique characteristic of having two electron-rich aromatic rings by virtue of the electron donating hydroxyl substituents alternating with two electron deficient nitro substituted aromatic rings. Analysis of the geometry and electrostatic surface potential (Fig. 2) of 1 reveals that the two hydroxyl substituted aromatics in the saddle conformation are capable of including complementary guest species by non-covalent interactions. The steric requirements of F and C+ on the energy minimized structure were investigated using space filling models.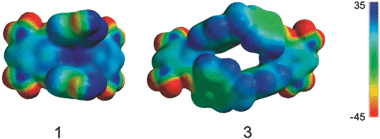 | ||
| Fig. 2 Electrostatic surface potential of hosts 1 and 3 after energy minimization in a PM3 force field using Spartan. | ||
We investigated the guest binding properties of 1 in solution by 1H NMR spectroscopy between 1 and neutral F in a wide range of polar and non-polar solvents including CD2Cl2, however solubility issues hampered these experiments and no interactions could be observed. In contrast, cyclic voltammetric experiments in CH2Cl2 showed a shift of the E1/2 associated with the one-electron oxidation of F to less positive values by –114 mV in the presence of 8 equiv. of host 1 (Fig. 3). This reveals that the host has a significant thermodynamic preference for the positively charged F+ over the neutral species and is strong evidence of complexation between the electrochemically generated F+ and 1. This negative shift corresponds to a 85 fold enhancement of binding upon electrochemical generation of the positive charge as calculated from eqn (1). 8 The negative shift was quantified as a 2.6 kcal mol–1 increase in stability of the complex in going from F to F+. The recorded voltammograms also showed some quasi-reversibility upon host addition. This is an expected consequence of complexation as the electrode has less access to F/F+ and electron transfer takes place at a greater average distance than when uncomplexed.
| Kred/Kox = exp(nFΔE1/2/RT) | (1) |
| E1/2(complex) = E1/20(free) + (RT/F)ln(1 + Kox[H]) | (2) |
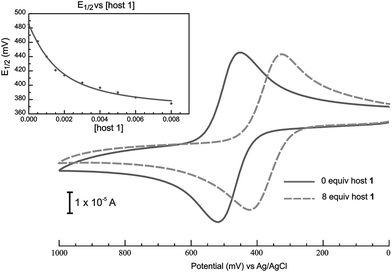 | ||
| Fig. 3 Cyclic voltammetric response on a glassy carbon electrode (0.071 cm2) of F (1.0 mM) in CH2Cl2 in the absence (solid line) and in the presence of 8 equiv. host 1 (broken line) at a scan rate of 0.1 V s–1 with 0.1 M tetrabutylammonium hexafluorophosphate supporting electrolyte. Inset shows shifting of E1/2 as a function of host concentration. | ||
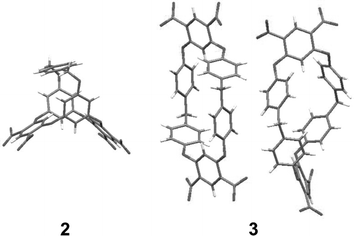
In order to verify that the chopsticks formed by the hydroxyl substituted electron dense aromatic rings of 1 is the binding site we synthesized the control host molecule 2 which contains a p-xylene group bridging the two hydroxyls. The X-ray crystal structure of 2, obtained by vapor diffusion of hexane into an ethyl acetate solution, has crystallographically imposed mirror symmetry. The proposed binding site is blocked, hindering the approach of any guest. In sharp contrast to electrochemistry of F in the presence of host 1, the cyclic voltammetric experiments of F with host 2 showed no shifting of the E1/2 associated with the one-electron oxidation of F. This observation is consistent with no complexation between host 2 and F provides further evidence that the 1,3-alternating electron-rich rings of 1 are involved in complexation.
1H NMR experiments with C+ were performed and upon addition of cobaltocenium hexafluorophosphate to a CD2Cl2 solution of 1 a upfield shift of the proton resonance corresponding to the protons para to the hydroxyl groups on 1 was observed. This observation is in agreement with the formation of a complex between these two species and shows that the hydroxyl substituted rings are involved in complexation. As with F+ , the interaction between 1 and C+ is driven by chopstick type π-coordination of host to cationic guest. Control experiments using quaternary ammonium salts produced a similar shift of the host proton resonances supporting the hypothesis that cation–π interactions drive the complexation. 1H NMR experiments with control host 2 with both F and C+ guests also showed no complexation induced shifting of any proton resonances demonstrating that control host 2 has very little interaction with these guests.
The electron deficient dinitro substituted rings of 1 can be reduced in cyclic voltammetric experiments. The reduction of 1 however, has slow electrochemical kinetics and also caused precipitation on the electrode surface which results in distortion of the voltammetric waves. These distortions mask the voltammetric response of C+ at higher concentrations and prevented binding saturation from being observed in cyclic voltammetric experiments. Despite these complications, the E1/2 associated with the one-electron reduction of C+ was observable at lower concentrations of 1 which underwent a shift to less positive values upon addition of 1 (ESI†). This is also evidence that 1 prefers the positively charged C+ over the reduced neutral species and strengthens the contention that cation–π interactions drive this complexation. Voltammetric experiments showed no shifts of the E1/2 associated with the one-electron reduction of C+ when host 2 was added. This observation consistent with no complexation between these species, which is expected as host 2 has xylene blocking the proposed binding site.
Building upon these observations we synthesized the new macrocycle 3, that has an oxacalixarene framework with a larger cavity for guest inclusion. 3 was synthesized by reaction of bis(4-hydroxyphenyl)methane with 1,5-difluoro-2,4-dinitrobenzene using K2CO3 as the base in DMSO (ESI†). Crystals were obtained by vapor diffusion of hexane into an ethyl acetate solution of 3. The crystal structure of 3 (Fig. 4) reveals two crystallographically independent molecules, one located on a position of general symmetry and the other situated about an inversion center. Both conformations display a sizeable cavity filled with disordered solvent molecules. Although these conformations differ from host 1, analysis of the electrostatic surface potential (Fig. 2) reveals that they also have an electron-rich inner cavity formed by four aromatic rings which can bind cationic guests.
1H NMR experiments showed complexation induced downfield shifting and broadening of the F proton resonances (Fig. 5) upon addition of host 3 in CD2Cl2. Voltammetric experiments show that host 3 itself can also undergo electrochemical reduction of the electron deficient nitro substituted rings. Upon complexation with F the reduction of host 3 underwent a shift of –22.5 mV indicating that the electron-poor rings of the host are afforded thermodynamic stability by interaction with the more electron-rich F. This electrochemical evidence suggests that the driving force of this interaction is electrostatic in nature between the electron-rich F and the electron deficient nitro substituted rings of 3.
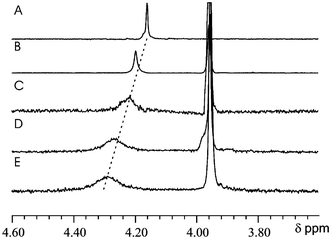 | ||
| Fig. 5 1H NMR (300 MHz) spectra of F in CD2Cl2, in the presence of (A) 0 equiv., (B) 3 equiv., (C) 4 equiv., (D) 5 equiv. and (E) 6 equiv. of 3. Complexation induced broadening and downfield shifting of F proton resonances are observed as more host is added. | ||
The E1/2 associated with the one-electron oxidation of ferrocene was also influenced by host 3. Voltammetric experiments showed that this E1/2 was shifted to less positive values by –27 mV upon addition of 8 equiv. of host 3. Despite observed complexation with neutral F, this host cavity displays a thermodynamic preference for the positively charged F+ as cation–π interactions would be possible with the oxidized guest which would be stronger than any electrostatic attraction in the 3·F complex. The E1/2 shift reflects a 2.9 fold increase of the association constant upon electrochemical oxidation of F to the cationic F+ and corresponds to a 0.6 kcal mol–1 increase in stability from the 3·F to the 3·F+ complex. We estimated the association constants to be 107 M–1 for the 3·F complex and 310 M–1 for the 3·F complex. When the 3·F+ association constant is contrasted to the association constant for the 1·F+ complex, it becomes evident that 1 is capable of stronger cation–π interactions. This is probably due to the higher electron density (Fig. 2) of the hydroxyl substituted saddle of 1 compared to the unsubstituted rings of 3.
1H NMR experiments also confirmed that 3 formed a complex with C+ which was observed by a small upfield shift of the C+ proton resonances. The exchange was slow in the NMR timescale as observed by two sets of proton resonances, for complexed and uncomplexed C+, with the complexed proton resonances growing at the expense of the uncomplexed until binding saturation. The driving force for this complexation is cation–π interactions. Host 3 displays a reduction wave very close to that of C+, which made voltammetric experiments difficult, however, a negative shift of the E1/2 associated with the one-electron reduction of C+ was observed, a phenomenon consistent with cation–π interactions.
Conclusion
Overall, this work sets a precedent for oxacalixarene binding and demonstrates that oxacalixarenes can function as effective hosts by using ferrocene (F) and cobaltocenium (C+) and their redox partners as guests. Although the association constants reported herein are modest, this study is relevant and necessary for advancing the field of oxacalixarenes. Electrochemical and other data suggest that the strongest complexes are formed with cationic guests and as this affords the opportunity for cation–π interactions which is typical and characteristic of classical calixarene hosts. More elaborate molecular designs involving electron donating groups are being investigated to improve binding characteristics.Acknowledgements
We would like to thank the NSF (CHE-071817), the Petroleum Research Fund and the University of South Carolina, Office of Research and Health Sciences for financial support of this work.References
- (a) C. D. Gutsche, Calixarenes Revisited, Royal Society of Chemistry, Cambridge, 2000 Search PubMed; (b) V. Böhmer, Angew. Chem., Int. Ed. Engl., 1995, 34, 713 CrossRef.
- B. König and M. H. Fonseca, Eur. J. Inorg. Chem., 2000, 2303 CrossRef CAS.
- (a) H. Kumagai, M. Hasegawa, S. Miyanari, Y. Sugawa, Y. Sato, T. Hori, S. Ueda, H. Kamiyama and S. Miyano, Tetrahedron Lett., 1997, 38, 3971 CrossRef CAS; (b) N. Kon, N. Iki and S. Miyano, Tetrahedron Lett., 2002, 43, 2231 CrossRef CAS; (c) P. Lhotak, Eur. J. Org. Chem., 2004, 1675 CrossRef CAS.
- (a) A. Ito, Y. Ono and K. Tanaka, J. Org. Chem., 1999, 64, 8236 CrossRef CAS; (b) T. D. Selby and S. C. Blackstock, Org. Lett., 1999, 1, 2053 CrossRef CAS; (c) M.-X. Wang, X.-H. Zhang and Q.-Y. Zheng, Angew. Chem., Int. Ed., 2004, 43, 838 CrossRef CAS; (d) Y. Suzuki, T. Yanagi, T. Kanbara and T. Yamamoto, Synlett, 2005, 263 CAS; (e) H. Tsue, K. Ishibashi, H. Takahashi and R. Tamura, Org. Lett., 2005, 7, 2165 CrossRef CAS.
- (a) N. Sommer and H. A. Staab, Tetrahedron Lett., 1966, 25, 2837 CrossRef; (b) F. P. A. Lehmann, Tetrahedron, 1974, 30, 727 CrossRef; (c) E. E. Gilbert, J. Heterocycl. Chem., 1974, 11, 899 CrossRef CAS; (d) F. Bottino, S. Foti and S. Papalardo, Tetrahedron, 1976, 32, 2567 CrossRef CAS; (e) X. Li, T. G. Upton, C. L. D. Gibb and B. C. Gibb, J. Am. Chem. Soc., 2003, 125, 650 CrossRef CAS.
- (a) J. L. Katz, M. B. Feldman and R. R. Conry, Org. Lett., 2005, 7, 91 CrossRef CAS; (b) J. L. Katz, K. J. Selby and R. R. Conry, Org. Lett., 2005, 7, 3505 CrossRef CAS; (c) J. L. Katz, B. J. Geller and R. R. Conry, Org. Lett., 2006, 8, 2755 CrossRef CAS; (d) J. L. Katz, B. J. Geller and P. D. Foster, Chem. Commun., 2007, 1026 RSC.
- (a) W. Maes, W. Van Rossum, K. Van Hecke, L. Van Meervelt and W. Dehaen, Org. Lett., 2006, 8, 4161 CrossRef CAS; (b) C. Zhang and C. -F. Chen, J. Org. Chem., 2007, 72, 3880 CrossRef CAS; (c) F. Yang, L. Yan, K. Ma, L. Yang, J. Li, L. Chen and J. You, Eur. J. Org. Chem., 2006, 12, 1109 CrossRef.
- (a) A. E. Kaifer and M. Gómez-Kaifer, Supramolecular Electrochemistry, Wiley-VCH, Weinheim, Germany, 2000 Search PubMed; (b) C. Bourgel, A. S. F. Boyd, G. Cooke, H. A. de Cremiers, F. M. A. Duclairoir and V. M. Rotello, Chem. Commun., 2001, 1954 RSC.
- (a) N. Gupta and H. Linschitz, J. Am. Chem. Soc., 1997, 119, 6384 CrossRef CAS; (b) J. -M. Savéant, J. Phys. Chem. B, 2001, 105, 8995 CrossRef CAS; (c) M. Gómez, C. Z. Gómez-Castro, I. I. Padilla-Martínez, F. J. Martínez-Martínez and F. J. González, J. Electroanal. Chem., 2004, 567, 269 CrossRef CAS.
- L. Avram and Y. Cohen, J. Am. Chem. Soc., 2002, 124, 15148 CrossRef CAS.
- (a) A. B. Rozhenko, W. W. Schoeller, M. C. Letzel, B. Decker, C. Agena and J. Mattay, Chem.–Eur. J., 2006, 12, 8995 CrossRef CAS; (b) S. Ishihara and S. Takeoka, Tetrahedron Lett., 2005, 47, 181; (c) H. Konishi, K. Takahashi, N. Nakamura, H. Sakamoto and K. Kimura, J. Inclusion Phenom. Macrocycl. Chem., 2006, 54, 147 Search PubMed; (d) R. Arnecke, V. Böhmer, R. Cacciapaglia, A. D. Cort and L. Mandolini, Tetrahedron, 1997, 53, 4901 CrossRef CAS; (e) D. A. Dougherty, Science, 1996, 271, 163 CrossRef CAS.
- J. Y. Lee, J. Kwon, C. S. Park, J.-E. Lee, W. Sim, J. S. Kim, J. Seo, I. Yoon, J. H. Jung and S. S. Lee, Org. Lett., 2007, 9, 493 CrossRef CAS.
-
Crystallography: 2: C32H18N4O14, M = 682.50, monoclinic, space group P21/m, a = 8.8759(8), b = 12.7053(12), c = 16.6618(15) Å, β = 103.925(2)°, V = 1823.7(3) Å3, Z = 2, T = 150 K, 2533 reflections, 1861 with I > 2σ(I); R(all) = 0.0648, R(I > 2σ(I)) = 0.0480, wR(all) = 0.1269, wR(I > 2σ(I)) = 0.1206. 3: C38H24N4O12, M = 728.61, triclinic, space group P
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 12.1115(4), b = 14.0678(4), c = 17.4915(5) Å, α = 79.9180(10), β = 86.4530(10), γ = 74.7700(10)°, V = 2830.77(15) Å3, Z = 3, T = 150 K, 10008 reflections, 7259 with I > 2σ(I); R(all) = 0.0530, R(I > 2σ(I)) = 0.0377, wR(all) = 0.0827, wR(I > 2σ(I)) = 0.07820. CCDC reference numbers 659154 (2) and 659155 (3). For crystallographic data in CIF or other electronic format see DOI: 10.1039/b709806e.
, a = 12.1115(4), b = 14.0678(4), c = 17.4915(5) Å, α = 79.9180(10), β = 86.4530(10), γ = 74.7700(10)°, V = 2830.77(15) Å3, Z = 3, T = 150 K, 10008 reflections, 7259 with I > 2σ(I); R(all) = 0.0530, R(I > 2σ(I)) = 0.0377, wR(all) = 0.0827, wR(I > 2σ(I)) = 0.07820. CCDC reference numbers 659154 (2) and 659155 (3). For crystallographic data in CIF or other electronic format see DOI: 10.1039/b709806e.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of hosts, estimation of binding constants, and X-ray structures of hosts 2 and 3. See DOI: 10.1039/b709806e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2008 |