From Spanish fly to room-temperature ionic liquids (RTILs): synthesis, thermal stability and inhibition of dynamin 1 GTPase by a novel class of RTILs†
Jie
Zhang
a,
Geoffrey A.
Lawrance
a,
Ngoc
Chau
b,
Phillip J.
Robinson
b and
Adam
McCluskey
*a
aChemistry, School of Environmental & Life Sciences, The University of Newcastle, Callaghan, NSW 2308, Australia. E-mail: Adam.McCluskey@newcastle.edu.au; Fax: 612 49215472; Tel: 612 9216486
bCell Signalling Unit, Children’s Medical Research Institute, 214 Hawkesbury Road, Westmead, NSW 2145, Australia
First published on 10th September 2007
Abstract
In a series of simple synthetic manipulations the active component of the aphrodisiac Spanish fly has resulted in the generation of a new family of room-temperature ionic liquids (RTILs). These RTILs are synthesized in high yield from readily attainable starting materials and can be generated in either meso or chiral forms dependant on the starting furan analogue. Substituted furans (2-methyl and 2-ethyl) afford chiral RTILs, furan affords a family of meso RTILs. In all cases the counterion was crucial, with CH3SO3– consistently displaying the lowest melting points. Of the RTILs synthesized, TGA plots showed most to be stable up to at least 250 °C. We had sought to use these RTILs in a series of dynamic combinatorial chemistry (DCC) assembly reactions via solubulisation of dynamin GTPases pleckstrin homology (PH) domain, as such all analogues were screened as potential inhibitors. Screening reveals that these RTILs display varying levels of dynamin GTPase inhibition with a number amongst the most potent inhibitors of dynamin GTPase yet discovered, e.g.13 IC50 = 2.3 ± 0.3 µM (4-(N,N-dimethyl-N-octadecyl-N-ethyl)-4-aza-10-oxatricyclo[5.2.1]decane-3,5-dione bromide. Accordingly these RTILs have limited utility for DCC assembly with dynamin GTPase, but may be of use with other proteins or in other fields of study.
Introduction
Over the past decade there has been an almost exponential growth in the synthesis and utility of room-temperature ionic liquids (RTILs). RTILs are defined as salts with a melting point ≤150 °C. The archetypal RTILs are the imidazolium (1), pyridinium (2) and alkyl ammonium (3) based salts (Fig. 1).1,2 They are widely regarded as viable replacements for volatile organic solvents (VOCs) due to their perceived non-toxicity, low volatility (zero), high thermal stability, tunable nature and ease of recycling, and RTILs have been studied and used in a wide variety of reactions.3,4 In a number of cases synthetic transformations occur faster, and cleaner, thus their ‘greenness’ is not limited to their own physicochemical properties but also intricately linked to their reduction in waste from cleaner reactions and lower energy consumption. To date a lack of predictably of RTIL properties is perhaps the greatest issue still to be resolved. The resultant inability to predict reaction outcomes when replacing traditional VOCs with RTILs has limited their integration into mainstream chemistry.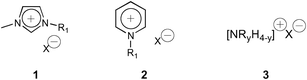 | ||
| Fig. 1 The archetypal room-temperature ionic liquids. | ||
We have developed an interest in the utility of RTILs across a wide variety of fields from precious metals recovery to sensor development with molecularly imprinted polymers.5–9 Over the past decade our group has explored three main areas, firstly the development of small molecule inhibitors of proteins such as protein phosphatase 1 (PP1) and protein phosphatase 2A (PP2A);10–15 secondly the design and synthesis of small molecule inhibitors for the inhibition of dynamin GTPase;16,17 and thirdly the synthesis and potential utility of room-temperature ionic liquids in attaining the first goal.18–21
Our recent discoveries of novel dynamin GTPase inhibitors have led us to evaluate dynamic combinatorial chemistry (DCC) approaches as a methodology to develop new classes of drugs. Pivotal to DCC approaches is access to large quantities of protein ; in this case we have access to mg quantities of dynamin’s pleckstrin homology (PH) domain. The PH domain of dynamin presents a significant challenge to a DCC approach as it is a lipid binding domain.22–25 Mutations in this domain have been implicated in Charcot–Marie–Tooth disease, a common heriditary disorder.26 The PH domain’s lipid binding role and its requirement for lipid -like compounds ensures that they are essentially mutually incompatible with the former requiring an aqueous environment to maintain protein function and the latter requiring organic solvent to ensure a homogeneous reaction mixture. We rationalised that the reported solvating powers associated with RTILs would facilitate a homogeneous, compatible environment suitable for dynamic combinatorial library assembly. We were also interested in developing new RTILs and designed such a series building on our experience with the design and synthesis of PP1 and PP2A inhibitors. In targeting PP1 and PP2A our lead compound is a modified version of the purported aphrodisiac Spanish fly, cantharidin (4) (Fig. 2). Structurally simpler norcantharidin (5) (Fig. 2) offers a rapid and green route to a number of analogues possessing noteworthy biological activity.14,15,27–29 In this instance we were more concerned with the ease of access into a rigid, stereodefined backbone that had promise for further modification and development of a series of novel ionic liquids. Furthermore, we have previously developed a number of cantharimide analogues with pendant nitrogens suitable for quaternisation, and ultimately metathesis via the addition of a variety of anions in order to develop a novel series of RTILs. The facility of our chemistry tied to the availability of substituted furans also held promise for developing chiral RTILs based on the natural product, cantharidin (4).
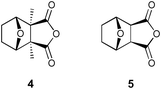 | ||
| Fig. 2 Structures of cantharidin (4) and norcantharidin (5). | ||
Results and discussion
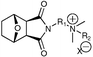
Commencing from readily available furan (6) and maleic anhydride (7), norcantharidin (8) was generated in excellent yield (two steps: Diels–Alder followed by hydrogenation). Treatment with N,N-dimethylethylenediamine or N,N-dimethylpropylenediamine afforded 9 and 10, respectively as solids. Quaternisation of 9 and 10 was achieved by alkylation with an appropriate alkyl bromide, yielding the ammonium salts 11–15. Metathesis of the bromide anion was by treatment with HBF4 (16) or CH3SO3H (17–19). All compounds were recovered in high purity in modest to good yields.Whereas the parent material norcantharidin is crystalline, we had anticipated that the introduction of a flexible side chain would permit a reduction in melting point (mp), potentially into the range associated with RTILs. This was simply and rapidly evaluated via mp determinations; this data is shown in Table 1.
|
|
||||
|---|---|---|---|---|
| Compound | R1 | R2 | X | Mp/°C |
| 11 | –C2H4 | –C4H9 | Br | 216–220 |
| 12 | –C2H4 | –C12H25 | Br | 172–175 |
| 13 | –C2H4 | –C18H37 | Br | 168–172 |
| 14 | –C3H6 | –C4H9 | Br | 199–202 |
| 15 | –C3H6 | –C12H25 | Br | 148–151 |
| 16 | –C2H4 | –C4H9 | BF4 | 192–195 |
| 17 | –C2H4 | –C4H9 | CH3SO3 | 93–95 |
| 18 | –C2H4 | –C12H25 | CH3SO3 | 46–50 |
| 19 | –C3H6 | –C4H9 | CH3SO3 | 80–83 |
| 20 | –C3H6 | –C12H25 | CH3SO3 | 28–30 |

Not surprisingly the simple bromine salts (11–15) display the highest melting points and are not RTILs, though 15 is on the cusp (assuming we adhere to the <150 °C mp definition). Insertion of a methylene bridge results in a ∼20 °C decrease in mp, cf.11vs. 14; 12vs. 15. Further elongation would undoubtedly result in a further melting point decrease; however the required N,N-dimethylbutane-1,4-diamine is not commercially available, a design limitation we considered at the outset of this investigation. Regardless anion metathesis was examined, firstly Br– to BF4– (11 ⇒ 16). However this only resulted in a ∼20 °C melting point decrease, insufficient to translate our bromine salts into RTILs and as such this metathesis route was discontinued. The corresponding CH3SO3– salts resulted in a significant depression of melting points such that the three products 17–20 are all clearly RTILs. Analogues 17 and 19 differ only by virtue of a methylene linker, with the propyl analogue 19 melting 13 °C lower than the ethyl linked 17, again suggesting that the corresponding butyl linker would be a better candidate (although we ruled out this synthesis above). Introduction of a dodecyl chain results in a ∼50 °C depression of mp, with both 18 and 20 clearly RTILs. The ethyl to propyl elongation associated with 17 and 19, respectively, also affords two novel RTILs as anticipated.
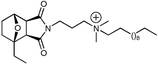
Given the positive results achieved above, we also investigated the effect of encapsulation of the terminal N,N-dimethylamino substituent within a piperidine ring. To improve the likelihood of attaining an RTIL, we limited our efforts to the generation of the CH3SO3– salt (21) (Fig. 3). Concurrently, we also explored disruption of the symmetry (chiral analogues) associated with the norcantharidin skeleton by synthesis of two 1-alkyl analogues (22 and 23) (Fig. 3). These cases limited our evaluations to the propyl linker dodecyl analogues as we have shown herein that analogues of this nature display the lowest melting points (18, 46–50 °C and 20, liquid).
 | ||
| Fig. 3 | ||
Synthesis of 21 was accomplished as described in Scheme 1 commencing with 5, via treatment under standard conditions with 1-(2-aminoethyl)piperidine. Somewhat surprisingly we were unable to alkylate the piperidine nitrogen with bromobutane even with prolonged reaction times, elevated temperatures and use of excess bromobutane. Alkylation was successful in the case of the more active methyl iodide and finally metathesis with methanesulfonic acid afforded 21 as a RTIL. Analogues 22 and 23 were synthesized as per 20 commencing with 2-methyl and 2-ethylfuran, respectively.
 | ||
| Scheme 1 Reagents and conditions: (i) diethyl ether, RT, 24 h; (ii) 10% Pd–C, H2 (50 psi), acetone; (iii) N,N-dimethylethane-1,2-diamine or N,N-dimethylpropane-1,3-diamine–THF, reflux; (iv) RBr–EtOH, reflux; (v) HBF4 or CH3SO3H. | ||
Our melting point determinations for 21–23 demonstrate our approach to the development of novel RTILs to be successful with the simpler meso21 returning a mp = 89 °C, within the range associated with RTILs. Whilst the chiral 22 and 23 deviate from our original intent, these data (both are liquids) clearly demonstrates that the ability to disrupt the crystalline matrix has a pronounced effect on melting points and in these cases returns RTILs with sub-ambient melting points.
After having a novel series of RTILs established, we next sought to investigate their working temperature range. Each new RTIL was evaluated via thermogravimetric analysis (TGA); a representative example is shown in Fig. 4.
 | ||
| Fig. 4 TGA plot of bromide salt (11). | ||
According to the TGA plot (Fig. 4), we can see the onset temperature of decomposition for bromide salt (11) are higher than 250 °C, and proceed in one major step. This is typical of all bromide salts examined. The decomposition of methanesulfonate RTILs has one more step than those above (ESI† ). The first minor step occurs near 130 °C may be attributed to some water or residual acid closely associated with the ionic liquids; loss of hydrogen-bonded or structural water invariably occurs at over 100 °C. One can estimate the amount in the ionic liquids from percent mass loss. Microanalysis confirms the presence of both water and residual ulfonic acid in these latter 17–23. The second and major decomposition step is the actual decomposition of the ionic liquid itself, which is around 300 °C.
Given our desire to utilise these RTILs in the solubilisation of dynamin’s PH domain, we felt it prudent to evaluate their effect, if any, on dynamin’s GTPase activity. The interaction of these new RTILs with dynamin was assessed via a modified Malachite Green colorimetric assay .30 The results of this screening are presented in Table 2.
|
|
||||
|---|---|---|---|---|
| Compound | R1 | R2 | X | IC50 (µM) |
| a Percentage inhibition at 300 µM compound concentration. b No observable activity at 700 µM drug concentration. | ||||
| 11 | –C2H4 | –C4H9 | Br | 15%a |
| 12 | –C2H4 | –C12H25 | Br | 8.5 ± 1.5 |
| 13 | –C2H4 | –C18H37 | Br | 2.3 ± 0.3 |
| 14 | –C3H6 | –C4H9 | Br | —b |
| 15 | –C3H6 | –C12H25 | Br | 8.4 ± 1.1 |
| 16 | –C2H4 | –C4H9 | BF4 | —b |
| 17 | –C2H4 | –C4H9 | CH3SO3 | 404 |
| 18 | –C2H4 | –C12H25 | CH3SO3 | 8.9 ± 0.9 |
| 19 | –C3H6 | –C4H9 | CH3SO3 | 18%a |
| 20 | –C3H6 | –C12H25 | CH3SO3 | 11.2 ± 1.8 |
| 21 |

|
CH3SO3 | 43.0 ± 12 | |
| 22 |

|
CH3SO3 | 11.8 ± 1.6 | |
| 23 |
|
CH3SO3 | 6.8 ± 0.5 | |
As can be clearly seen, the majority of the novel RTILs, inhibit dynamin GTPase activity. Analogues 14 and 16 are inactive. Increasing the alkyl chain length from C4 to C12 and C18 greatly enhances inhibition, as noted with 11–13 (15% inhibition at 300 µM drug concentration; IC50 = 8.5 ± 1.5, IC50 = 2.3 ± 0.3 µM, respectively) with the latter being equipotent with the most potent analogue that we have reported in this field thus far.16,17 Notwithstanding the chain length effect noted, it also appears that the incorporation of a more sterically demanding piperidine core is beneficial to the inhibition of dynamin GTPase activity (21, IC50 = 43.0 ± 12 µM). Manipulation of the counter anion (Br– ⇒ BF4– ⇒ CH3SO3–) had little effect in the analogues inhibitory potential, but as mentioned previously has a pronounced effect on analogue melting points. Introduction of bridgehead substituents to the bicyclo[2.2.1]heptane core has no effect on inhibitory potential.
It had been our belief, based on modelling data (not shown) that the introduction of the bulky norcantharidin subunit would remove any dynamin inhibition previously noted for long chain alkyl ammonium salts.16 These data suggest that inhibition is independent of the norcantharimide subunit.
Conclusions
We have generated a new family of RTILs in which to suspend the pleckstrin homology domain of dynamin 1 and found they are also dynamin inhibitors. Significantly, these data indicate that where RTILs are to be utilised in the presence of proteins that a pre-screen should be conducted to ensure that they do not interfere with the proteins intrinsic function. Whilst we have shown that these RTILs inhibit dynamin GTPase activity, it does not directly follow that they will adversely interact with other proteins . Therefore these RTILs are potentially important for the study of other proteins or domains thereof. The varying levels of dynamin inhibition noted indicate that we have serendipitously generated a new series of dynamin inhibitors. The data clearly highlights a rarely conceded fact—that in certain limited cases RTILs may adversely affect certain protein functions.Notwithstanding this adverse interaction with dynamin, alkyl ammonium norcantharimides represents a novel class of RTILs that are amenable to further manipulations to fine tune their properties.
Experimental
Materials
All reagents were of commercial quality and were used as received from Aldrich. Solvents were dried and purified using standard techniques. Both 1H and 13C NMR spectra were recorded using a Bruker Avance DPX-300 spectrometer, calibrated using the residual 1H peak associated with the deuterated solvent. MALDI-TOF mass spectra were recorded on an Ettan MALDI-TOF Pro Mass Spectrometer (Amersham Biosciences). Samples were dissolved in CH3CN (∼5 mg per mL), and then diluted 1 : 1 with the matrix solution: 1.5 mg ml–1 4-HCCA (α-cyano-4-hydroxycinnamic acid) in 50% ACN–0.1% TFA (trifluoroacetic acid). In each MS run 2 µL of sample was injected (MALDI-TOF traces are available in the ESI† ). Microanalysis were conducted at the Microanalyis Unit at the Australian National University, Canberra, Australia.
1H NMR (DMSO-d6): δ (ppm): 6.54 (2H, s, CH(–O)–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 5.31 (2H, s, CH–CH(–O)–CH), 3.27 (2H, s, CH(–O)–CH(–CH)–CO).
CH), 5.31 (2H, s, CH–CH(–O)–CH), 3.27 (2H, s, CH(–O)–CH(–CH)–CO).
13C NMR (DMSO-d6): δ (ppm): 171.5 (2 × CH–C(O)–O), 136.8 (CH(–O)–CHCH), 81.5 (CH–CH(–O)–CH), 49.0 (CH(–O)–CH(–CH)–CO).
1H NMR (DMSO-d6): δ (ppm): 4.82 (2H, d, J = 2.0 Hz, CH2–CH(–O)–CH), 3.36 (2H, s, CH(–O)–CH(–CH)–CO), 1.61 (4H, d, J = 1.4 Hz, CH(–O)–CH2–CH2).
13C NMR (DMSO-d6): δ (ppm): 172.8 (2 × CH–C(O)–O), 79.5 (CH2–CH(–O)–CH), 50.6 (CH(–O)–CH(–CH)–CO), 27.4 (CH(–O)–CH2–CH2).
1H NMR (CDCl3): δ (ppm): 4.85 (2H, dd, J = 2.2 Hz, 1.0 Hz, CH2–CH(–O)–CH), 3.59 (2H, t, J = 6.7 Hz, (OC–)N–CH2–CH2), 2.94 (2H, s, CH(–O)–CH(–CH)–CO), 2.49 (2H, t, J = 6.8 Hz, (OC–)N–CH2–CH2), 2.28 (6H, s, CH2–CH2–N(–CH3)2), 1.84 (2H, m, CH(–O)–CH2–CH2), 1.62 ( 2H, m, CH(–O)–CH2–CH2).
13C NMR (CDCl3): δ (ppm): 177.1 (2 × CH–C(O)–N), 78.7 (CH2–CH(–O)–CH), 55.7 ((OC–)N–CH2–CH2), 49.7 (CH(–O)–CH(–CH)–CO), 45.0 (CH2–CH2–N(–CH3)2), 36.4 ((OC–)N–CH2–CH2), 28.2 (CH(–O)–CH2–CH2).
1H NMR (CDCl3): δ (ppm): 4.88 (2H, dd, J = 2.3 Hz, 0.8 Hz, CH2–CH(–O)–CH), 3.53 (2H, t, J = 7.2 Hz, (OC–)N–CH2–CH2), 2.87 (2H, s, CH(–O)–CH(–CH)–CO), 2.26 (2H, t, J = 7.3 Hz, CH2–CH2–N(–CH3)2), 2.19 (6H, s, CH2–CH2–N(–CH3)2), 1.87 (2H, m, CH(–O)–CH2–CH2), 1.72 (2H, m, N–CH2–CH2–CH2–N), 1.60 (2H, m, CH(–O)–CH2–CH2).
13C NMR (CDCl3): δ (ppm): 176.6 (2 × CH–C(O)–N), 78.5 (CH2–CH(–O)–CH), 56.2 ((OC–)N–CH2–CH2), 49.3 (CH(–O)–CH(–CH)–CO), 44.8 (CH2–CH2–N(–CH3)2), 36.8 (CH2–CH2–N(–CH3)2), 28.0 (CH(–O)–CH2–CH2), 25.1 (N–CH2–CH2–CH2–N).
1H NMR (CDCl3): δ (ppm): 4.86 (2H, dd, J = 2.7 Hz, 2.1 Hz, CH2–CH(–O)–CH), 3.97 (4H, overlapping, (OC–)N–CH2–CH2), 3.58 (2H, t, J = 8.6 Hz, N+–CH2–CH2–CH2–CH3), 3.41 (6H, s, CH2–CH2–N(–C(–CH3)2), 3.12 (2H, s, CH(–O)–CH(–CH)–CO), 1.83 (2H, m, CH(–O)–CH2–CH2), 1.74 (2H, m, N+–CH2–CH2–CH2–CH3), 1.67 (2H, m, CH(–O)–CH2–CH2), 1.43 (2H, m, N+–CH2–CH2–CH2–CH3), 1.01 (3H, t, J = 7.3 Hz, N+–CH2–CH2–CH2–CH3).
13C NMR (CDCl3): δ (ppm): 177.1 (2 × CH–C(O)–N), 79.1 (CH2–CH(–O)–CH), 64.7 ((OC–)N–CH2–CH2), 60.8 (CH2–CH2–N(–CH3)2), 51.2 (CH2–CH2–N(–CH3)2), 50.4 (CH(–O)–CH(–CH)–CO), 33.1 (N+–CH2–CH2–CH2–CH3), 28.5 (CH(–O)–CH2–CH2), 24.7 (N+–CH2–CH2–CH2–CH3), 19.6 (N+–CH2–CH2–CH2–CH3), 13.7 (N+–CH2–CH2–CH2–CH3).
1H NMR (CDCl3): δ (ppm): 4.80 (2H, d, J = 2.0 Hz, CH2–CH(–O)–CH), 3.92 (4H, overlapping, (OC–)N–CH2–CH2), 3.50 (2H, t, J = 8.3 Hz, N+–CH2–CH2–(CH2)9–CH3), 3.37 (6H, s, CH2–CH2–N(–C(–CH3)2), 3.08 (2H, s, CH(–O)–CH(–CH)–CO), 1.79 (2H, m, CH(–O)–CH2–CH2), 1.70 (2H, m, N+–CH2–CH2–(CH2)9–CH3), 1.63 (2H, m, CH(–O)–CH2–CH2), 1.32–1.22 (18H, overlapping, N+–CH2–CH2–(CH2)9–CH3), 0.84 (3H, t, J = 7.1 Hz, N+–CH2–CH2–(CH2)9–CH3).
13C NMR (CDCl3): δ (ppm): 177.0 (2 × CH–C(O)–N), 79.1 (CH2–CH(–O)–CH), 64.8 ((OC–)N–CH2–CH2), 60.7 (CH2–CH2–N(–CH3)2), 51.2 (CH2–CH2–N(–CH3)2), 50.4 (CH(–O)–CH(–CH)–CO), 33.0 (N+–CH2–(CH2)10–CH3), 31.9 (CH(–O)–CH2–CH2), 29.5–22.6 (N+–CH2–(CH2)10–CH3), 14.1 (N+–CH2–(CH2)10–CH3).
1H NMR (CDCl3): δ (ppm): 4.80 (2H, s, CH2–CH(–O)–CH), 3.92 (4H, overlapping, (OC–)N–CH2–CH2), 3.51 (2H, t, J = 8.3 Hz, N+–CH2–CH2–(CH2)15–CH3), 3.37 (6H, s, CH2–CH2–N(–C(–CH3)2), 3.08 (2H, s, CH(–O)–CH(–CH)–CO), 1.78 (2H, m, CH(–O)–CH2–CH2), 1.70 (2H, m, N+–CH2–CH2–(CH2)15–CH3), 1.62 (2H, m, CH(–O)–CH2–CH2), 1.31–1.21 (30H, overlapping, N+–CH2–CH2–(CH2)15–CH3), 0.83 (3H, t, J = 7.2 Hz, N+–CH2–CH2–(CH2)15–CH3).
13C NMR (CDCl3): δ (ppm): 177.0 (2 × CH–C(O)–N), 79.1 (CH2–CH(–O)–CH), 65.0 ((OC–)N–CH2–CH2), 60.6 (CH2–CH2–N(–CH3)2), 51.2 (CH2–CH2–N(–CH3)2), 50.4 (CH(–O)–CH(–CH)–CO), 33.0 (N+–CH2–(CH2)16–CH3), 31.9 (CH(–O)–CH2–CH2), 29.6–22.6 (N+–CH2–(CH2)14–CH3), 14.1 (N+–(CH2)16–CH3).
1H NMR (CDCl3): δ (ppm): 4.81 (2H, d, J = 2.0 Hz, CH2–CH(–O)–CH), 3.64 (4H, overlapping, (OC–)N–CH2–CH2–CH2), 3.46 (2H, t, J = 8.3 Hz, N+–CH2–CH2–CH2–CH3), 3.35 (6H, s, CH2–CH2–N(–CH3)2), 3.22 (2H, s, CH(–O)–CH(–CH)–CO), 2.08 (2H, m, (OC–)N–CH2–CH2–CH2), 1.81 (2H, m, CH(–O)–CH2–CH2), 1.68 (4H, overlapping, N+–CH2–CH2–CH2–CH3 and CH(–O)–CH2–CH2), 1.41 (2H, m, N+–CH2–CH2–CH2–CH3), 1.00 (3H, t, J = 7.3 Hz, N+–CH2–CH2–CH2–CH3).
13C NMR (CDCl3): δ (ppm): 178.0 (2 × CH–C(O)–N), 79.2 (CH2–CH(–O)–CH), 64.3 ((OC–)N–CH2–CH2–CH2), 61.3 ((OC–)N–CH2–CH2–CH2), 51.1 (CH2–CH2–N(–CH3)2), 51.0 (CH(–O)–CH(–CH)–CO), 35.4 (N+–CH2–CH2–CH2–CH3), 28.5 (CH(–O)–CH2–CH2), 24.6 (N+–CH2–CH2–CH2–CH3), 21.3 ((OC–)N–CH2–CH2–CH2), 19.7 (N+–CH2–CH2–CH2–CH3), 13.7 (N+–CH2–CH2–CH2–CH3).
1H NMR (CDCl3): δ (ppm): 4.73 (2H, s, CH2–CH(–O)–CH), 3.54 (4H, overlapping, (OC–)N–CH2–CH2–CH2), 3.35 (2H, t, J = 8.5 Hz, N+–CH2–CH2–(CH2)9–CH3), 3.26 (6H, s, CH2–CH2–N(–CH3)2), 3.14 (2H, s, CH(–O)–CH(–CH)–CO), 2.04 (2H, m, (OC–)N–CH2–CH2–CH2), 1.73 (2H, m, CH(–O)–CH2–CH2), 1.60 (4H, overlapping, N+–CH2–CH2–(CH2)9–CH3 & CH(–O)–CH2–CH2), 1.25–1.18 (18H, overlapping, N+–CH2–CH2–(CH2)9–CH3), 0.80 (3H, t, J = 6.4 Hz, N+–CH2–CH2–(CH2)9–CH3).
13C NMR (CDCl3): δ (ppm): 177.8 (2 × CH–C(O)–N), 79.0 (CH2–CH(–O)–CH), 64.3 ((OC–)N–CH2–CH2–CH2), 61.1 ((OC–)N–CH2–CH2–CH2), 51.0 (CH2–CH2–N(–CH3)2), 50.5 (CH(–O)–CH(–CH)–CO), 35.3 (N+–CH2–(CH2)10–CH3), 31.7 (CH(–O)–CH2–CH2), 29.4–21.4 (N+–CH2–(CH2)10–CH3 and (OC–)N–CH2–CH2–CH2), 14.0 (N+–CH2–(CH2)10–CH3).
1H NMR (DMSO-d6): δ (ppm): 4.69 (2H, s, CH2–CH(–O)–CH), 3.75 (2H, t, J = 7.3 Hz, (OC–)N–CH2–CH2), 3.35 (4H, overlapping, (OC–)N–CH2–CH2 and N+–CH2–CH2–CH2–CH3), 3.09 (2H, s, CH(–O)–CH(–CH)–CO), 3.06 (6H, s, CH2–CH2–N(–CH3)2), 1.65 (6H, overlapping, CH(–O)–CH2–CH2 and N+–CH2–CH2–CH2–CH3), 1.28 (2H, m, N+–CH2–CH2–CH2–CH3), 0.93 (3H, t, J = 7.5 Hz, N+–CH2–CH2–CH2–CH3).
13C NMR (DMSO-d6): δ (ppm): 177.3 (2 × CH–C(O)–N), 78.5 (CH2–CH(–O)–CH), 62.8 ((OC–)N–CH2–CH2), 58.5 (CH2–CH2–N(–CH3)2), 50.3 (CH2–CH2–N(–CH3)2), 49.7 (CH(–O)–CH(–CH)–CO), 31.8 (N+–CH2–CH2–CH2–CH3), 27.9 (CH(–O)–CH2–CH2), 23.7 (N+–CH2–CH2–CH2–CH3), 19.1 (N+–CH2–CH2–CH2–CH3), 13.5 (N+–CH2–CH2–CH2–CH3).
1H NMR (CDCl3): δ (ppm): 4.80 (2H, t, J = 2.5 Hz, CH2–CH(–O)–CH), 3.88 (2H, t, J = 5.2 Hz, (OC–)N–CH2–CH2), 3.80 (2H, t, J = 5.4 Hz, (OC–)N–CH2–CH2), 3.47 (2H, t, J = 8.5 Hz, N+–CH2–CH2–CH2–CH3), 3.30 (6H, s, CH2–CH2–N(–CH3)2)), 3.06 (2H, s, CH(–O)–CH(–CH)–CO), 2.82 (3H, s, CH3SO3), 1.79 (2H, m, CH(–O)–CH2–CH2), 1.70 (2H, m, N+–CH2–CH2–CH2–CH3), 1.63 (2H, m, CH(–O)–CH2–CH2), 1.38 (2H, m, N+–CH2–CH2–CH2–CH3), 0.97 (3H, t, J = 7.4 Hz, N+–CH2–CH2–CH2–CH3).
13C NMR (CDCl3): δ (ppm): 177.1 (2 × CH–C(O)–N), 79.1 (CH2–CH(–O)–CH), 64.7 ((OC–)N–CH2–CH2), 60.6 (CH2–CH2–N(–CH3)2), 51.3 (CH2–CH2–N(–CH3)2), 50.3 (CH(–O)–CH(–CH)–CO), 39.4 (CH3SO3), 33.0 (N+–CH2–CH2–CH2–CH3), 28.4 (CH(–O)–CH2–CH2), 24.6 (N+–CH2–CH2–CH2–CH3), 19.5 (N+–CH2–CH2–CH2–CH3), 13.6 (N+–CH2–CH2–CH2–CH3).
1H NMR (CDCl3): δ (ppm): 4.82 (2H, s, CH2–CH(–O)–CH), 3.90 (2H, t, J = 5.3 Hz, (OC–)N–CH2–CH2), 3.78 (2H, t, J = 5.5 Hz, (OC–)N–CH2–CH2), 3.42 (2H, t, J = 8.6 Hz, N+–CH2–CH2–(CH2)9–CH3), 3.29 (6H, s, CH2–CH2–N(–CH3)2), 3.07 (2H, s, CH(–O)–CH(–CH)–CO), 2.86 (3H, s, CH3SO3), 1.80 (2H, m, CH(–O)–CH2–CH2), 1.71 (2H, m, N+–CH2–CH2–(CH2)9–CH3), 1.64 (2H, m, CH(–O)–CH2–CH2), 1.33–1.23 (18H, overlapping, N+–CH2–CH2–(CH2)9–CH3), 0.85 (3H, t, J = 7.0 Hz, N+–CH2–CH2–(CH2)9–CH3).
13C NMR (CDCl3): δ (ppm): 177.2 (2 × CH–C(O)–N), 79.2 (CH2–CH(–O)–CH), 65.0 ((OC–)N–CH2–CH2), 60.1 (CH2–CH2–N(–CH3)2), 51.5 (CH2–CH2–N(–CH3)2), 50.4 (CH(–O)–CH(–CH)–CO), 39.5 (CH3SO3), 33.0 (N+–CH2–(CH2)10–CH3), 31.9 (CH(–O)–CH2–CH2), 29.6–22.7 (N+–CH2–(CH2)10–CH3), 14.1 (N+–CH2–(CH2)10–CH3).
1H NMR (CDCl3): δ (ppm): 4.82 (2H, t, J = 2.2 Hz, CH2–CH(–O)–CH), 3.63 (2H, t, J = 5.8 Hz, (OC–)N–CH2–CH2–CH2), 3.37 (4H, overlapping, (OC–)N–CH2–CH2–CH2 and N+–CH2–CH2–CH2–CH3), 3.22 (6H, s, CH2–CH2–N(–CH3)2)), 3.14 (2H, s, CH(–O)–CH(–CH)–CO), 3.00 (3H, s, CH3SO3), 1.82 (2H, m, (OC–)N–CH2–CH2–CH2), 1.69 (4H, overlapping, N+–CH2–CH2–CH2–CH3 and CH(–O)–CH2–CH2), 1.41 (2H, m, N+–CH2–CH2–CH2–CH3), 1.00 (3H, t, J = 7.3 Hz, N+–CH2–CH2–CH2–CH3).
13C NMR (CDCl3): δ (ppm): 177.8 (2 × CH–C(O)–N), 79.2 (CH2–CH(–O)–CH), 64.5 ((OC–)N–CH2–CH2–CH2), 61.5 ((OC–)N–CH2–CH2–CH2), 51.2 (CH2–CH2–N(–CH3)2), 50.3 (CH(–O)–CH(–CH)–CO), 39.6 (CH3SO3), 35.5 (N+–CH2–CH2–CH2–CH3), 28.5 (CH(–O)–CH2–CH2), 24.4 (N+–CH2–CH2–CH2–CH3), 21.2 ((OC–)N–CH2–CH2–CH2), 19.6 (N+–CH2–CH2–CH2–CH3), 13.6 (N+–CH2–CH2–CH2–CH3).
1H NMR (CDCl3): δ (ppm): 4.78 (2H, s, CH2–CH(–O)–CH), 3.59 (2H, t, J = 5.6 Hz, (OC–)N–CH2–CH2–CH2), 3.49 (2H, t, J = 7.9 Hz, (OC–)N–CH2–CH2–CH2), 3.33 (2H, t, J = 8.5 Hz, N+–CH2–CH2–(CH2)9–CH3), 3.24 (6H, s, CH2–CH2–N(–CH3)2), 3.13 (2H, s, CH(–O)–CH(–CH)–CO), 2.89 (3H, s, CH3SO3), 2.07 (2H, m, (OC–)N–CH2–CH2–CH2), 1.78 (2H, m, CH(–O)–CH2–CH2), 1.62 (4H, overlapping, N+–CH2–CH2–(CH2)9–CH3 and CH(–O)–CH2–CH2), 1.30–1.22 (18H, overlapping, N+–CH2–CH2–(CH2)9–CH3), 0.85 (3H, t, J = 6.4 Hz, N+–CH2–CH2–(CH2)9–CH3).
13C NMR (CDCl3): δ (ppm): 177.9 (2 × CH–C(O)–N), 79.2 (CH2–CH(–O)–CH), 64.3 ((OC–)N–CH2–CH2–CH2), 61.1 ((OC–)N–CH2–CH2–CH2), 51.2 (CH2–CH2–N(–CH3)2), 50.5 (CH(–O)–CH(–CH)–CO), 39.6 (CH3SO3), 35.3 (N+–CH2–(CH2)10–CH3), 31.9 (CH(–O)–CH2–CH2), 29.5–22.7 (N+–CH2–(CH2)10–CH3 and (OC–)N–CH2–CH2–CH2), 14.1 (N+–CH2–(CH2)10–CH3).
Firstly 5 (1.000 g, 6.0 mmol) was treated as per the synthesis of 9 with 1-(2-aminoethyl)piperidine (0.928 g, 98%, 7.1 mmol) to afford 4-(N-ethylpiperidine)-4-aza-10-oxatricyclo[5.2.1]decane-3,5-dione as a white solid (1.405 g, 85%).
1H NMR (CDCl3): δ (ppm): 1.35 (2H, m, (CH2–)N–CH2–CH2–CH2), 1.48 (4H, m, 2 × (CH2–)N–CH2–CH2–CH2), 1.55 (2H, m, CH(–O)–CH2–CH2), 1.81 (2H, m, CH(–O)–CH2–CH2), 2.35–2.43 (6H, overlapping, (OC–)N–CH2–CH2–N(–CH2)–CH2), 2.82 (2H, s, CH(–O)–CH(–CH)–CO), 3.55 (2H, t, J = 7.1 Hz, (OC–)N–CH2–CH2–N), 4.81 (2H, dd, J = 2.2 Hz, 1.0 Hz, CH2–CH(–O)–CH).
13C NMR (CDCl3): δ (ppm): 24.2 ((CH2–)N–CH2–CH2–CH2), 25.9 (2 ×(CH2–)N–CH2–CH2–CH2), 28.6 (CH(–O)–CH2–CH2), 36.4 (OC–)N–CH2–CH2–N), 50.0 (CH(–O)–CH(–CH)–CO), 54.4 (CH2–)N–CH2–CH2–CH2), 55.4 (OC–)N–CH2–CH2–N), 79.0 (CH2–CH(–O)–CH), 177.1 (2 × CH–C(O)–N).
4-(N-ethylpiperidine)-4-aza-10-oxatricyclo[5.2.1]decane-3,5-dione (1.405 g, 5.0 mmol) was dissolved in ethanol (10 cm3). To this was added 1-methyl iodide (0.745 g, 99%, 5.2 mmol) and the mixture refluxed for 16 hours. The ethanol was removed in vacuo. Recrystallization from ethanol and chloroform afforded an off-white solid 4-(N-methyl-N-ethylpiperidine)-4-aza-10-oxatricyclo[5.2.1]decane-3,5-dione iodide (0.98 g, 47%).
1H NMR (D2O): δ (ppm): 1.63–1.88 (10H, overlapping, (CH2–)N+–CH2–CH2–CH2–CH2 and CH(–O)–CH2–CH2), 3.13 (3H, s, CH3–(CH2–)N+(–CH2)–CH2), 3.25 (2H, s, CH(–O)–CH(–CH)–CO), 3.39 (4H, m, CH3–(CH2–)N+(–CH2)–CH2), 3.54 (2H, t, J = 6.9 Hz, (OC–)N–CH2–CH2–N+), 3.97 (2H, t, J = 7.1 Hz, (OC–)N–CH2–CH2–N+), 4.94 (2H, d, J = 2.3 Hz, 1.0 Hz, CH2–CH(–O)–CH).
13C NMR (D2O): δ (ppm): 19.0 (2 ×(CH2–)N+–CH2–CH2–CH2), 19.9 ((CH2–)N+–CH2–CH2–CH2), 27.4 (CH(–O)–CH2–CH2), 31.6 (OC–)N–CH2–CH2–N+), 47.6 (CH3–(CH2–)N+(–CH2)–CH2), 50.0 (CH(–O)–CH(–CH)–CO), 57.9 ((OC–)N–CH2–CH2–N+), 61.4 (CH2–)N+–CH2–CH2–CH2), 79.2 (CH2–CH(–O)–CH), 179.0 (2 × CH–C(O)–N).
4-(N-methyl-N-ethylpiperidine)-4-aza-10-oxatricyclo[5.2.1]decane-3,5-dione iodide (0.420 g, 1.0 mmol) was dissolved in water (15 cm3) and cooled to 0 °C. To this was added methanesulfonic acid (0.146 g, 70% w/w solution, 1.1 mmol) and the resultant solution stirred for 1 h. The solution was rotary evaporated to remove residual HI, water and any excess methanesulfonic acid present to afford a pale yellow waxy solid (0.375 g, 96%). Mp 89–91 °C; MS (MALDI-TOF): m/z 293.0 (M+ – CH3SO3H). Calc. for C17H28N2O6: C 52.56, H 7.26, N 7.21. Found: C 52.62, H 7.70, N 7.57%.
1H NMR (CDCl3): δ (ppm): 1.63–1.91 (10H, overlapping, (CH2–)N+–CH2–CH2–CH2–CH2, and CH(–O)–CH2–CH2), 2.89 (3H, s, CH3SO3), 3.10 (2H, s, CH(–O)–CH(–CH)–CO), 3.29 (3H, s, CH3–(CH2–)N+(–CH2)–CH2), 3.59 (4H, m, CH3–(CH2–)N+(–CH2)–CH2), 3.88–3.92 (4H, overlapping, (OC–)N–CH2–CH2–N+ and (OC–)N–CH2–CH2–N+), 4.81 (2H, d, J = 2.3 Hz, 1.0 Hz, CH2–CH(–O)–CH).
13C NMR (CDCl3): δ (ppm): 20.1 (2 ×(CH2–)N+–CH2–CH2–CH2), 20.6 ((CH2–)N+–CH2–CH2–CH2), 28.5 (CH(–O)–CH2–CH2), 32.6 (OC–)N–CH2–CH2–N+), 39.5 (CH3SO3), 48.5 (CH3–(CH2–)N+(–CH2)–CH2), 50.5 (CH(–O)–CH(–CH)–CO), 59.7 ((OC–)N–CH2–CH2–N+), 61.9 (CH2–)N+–CH2–CH2–CH2), 79.2 (CH2–CH(–O)–CH), 177.2 (2 × CH–C(O)–N).
A solution of maleic anhydride (15.075 g, 0.15 mol) in anhydrous diethyl ether (50 cm3) was heated under reflux and treated dropwise with freshly distilled 2-methylfuran (15.484 g, 0.189 mol). The mixture was refluxed for 2 h, and then stirred at room temperature overnight. The white precipitate produced was then filtered off and dried under suction (18.473 g, 67%).
The resultant Diels–Alder adduct, 4,10-dioxa-7,8-dehydro-6-methyltricyclo[5.2.1]decane-3,5-dione (10.013 g, 0.06 mol) was dissolved in acetone (150 cm3) and degassed by using ultrasonic cleaner for 20 min, then was cooled to ∼0 °C. To this, palladium catalyst (1.008 g, 10%-Pd/C) was added and hydrogenated at 50 psi overnight at room temperature. After this time, the catalyst was filtered off through a Celite pad, the pad washed with ice-cold acetone (3 × 30 cm3), and the solvent was removed by rotary evaporation. Recrystallisation from ethyl acetate afforded an off-white solid (7.975 g, 81%).
1H NMR (CDCl3): δ (ppm): 4.91 (1H, d, J = 5.3 Hz, CH2–CH(–O)–CH–(CO)), 3.25 (1H, d, J = 7.4 Hz, (O–)CH–CH–(CO)), 3.02 (1H, d, J = 7.4 Hz, (O–)C(–CH3)–CH–(CO)), 2.00 (2H, m, (O–)CH–CH2–CH2–(CH3–)C(–O)), 1.67 (2H, m, (O–)CH–CH2–CH2–(CH3–)C(–O)), 1.63 (3H, s, (O–)C(–CH2)–CH3).
13C NMR (CDCl3): δ (ppm): 171.4 ((CH–)CH–C(O)–O), 170.0 ((C–)CH–C(O)–O), 86.8 ((CH2–)C(–O)–CH3), 79.7 ((CH2–)CH(–O)–CH(CO)), 52.7 ((O–)CH–CH–C(O)), 52.3 ((O–)C(CH3)–CH–C(O)), 35.7 ((O–)CH–CH2–CH2–(CH3–)C(–O)), 30.0 ((O–)CH–CH2–CH2–(CH3–)C(–O)), 17.5 ((O–)C(CH2)–CH3).
4.10-Dioxa-6-methyltricyclo[5.2.1]decane-3,5-dione (0.891 g, 4.9 mmol) was dissolved in THF (15 cm3). To this, N,N-dimethylethylenediamine (0.623 g, 6.1 mmol) was added and the mixture refluxed overnight, cooled and poured onto water (100 cm3) and extracted with dichloromethane (3 × 40 cm3). The organic layer was washed with saturated NaHCO3 (2 × 40 cm3), dried over MgSO4 and filtered, and the solvent removed in vacuo to afford a yellow oil (1.185 g, 91%).
1H NMR (CDCl3): δ (ppm): 4.76 (1H, d, J = 5.3 Hz, CH2–CH(–O)–CH–(CO)), 3.50 (2H, t, J = 7.1 Hz, (OC–)N–CH2–CH2–CH2), 2.91 (1H, d, J = 7.4 Hz, (O–)CH–CH–(CO)), 2.71 (1H, d, J = 7.4 Hz, (O–)C(–CH3)–CH–(CO)), 2.25 (2H, t, J = 7.4 Hz, (OC–)N–CH2–CH2–CH2), 2.18 (6H, s, CH2–CH2–N(–CH3)2), 1.98 (2H, m, (O–)CH–CH2–CH2–(CH3–)C(–O)), 1.70 (2H, m, (OC–)N–CH2–CH2–CH2), 1.64 (2H, m, (O–)CH–CH2–CH2–(CH3–)C(–O)), 1.57 (3H, s, (O–)C(–CH2)–CH3).
13C NMR (CDCl3): δ (ppm): 177.3 ((CH–)CH–C(O)–O), 176.1 ((C–)CH–C(O)–O), 85.7 ((CH2–)C(–O)–CH3), 78.8 ((CH2–)CH(–O)–CH(CO)), 56.8 ((OC–)N–CH2–CH2–CH2), 51.9 ((O–)CH–CH–C(O)), 51.6 ((O–)C(CH3)–CH–C(O)), 45.3 (CH2–CH2–N(–CH3)2), 37.2 (CH2–CH2–CH2–N(–CH3)2), 36.4 ((O–)CH–CH2–CH2–(CH3–)C(–O)), 30.3 ((O–)CH–CH2–CH2–(CH3–)C(–O)), 25.6 (N–CH2–CH2–CH2–N), 17.7 ((O–)C(CH2)–CH3).
4-(N,N-dimethylamino-N-propyl)-4-aza-6-methyl-10-oxatricyclo[5.2.1]decane-3,5-dione (1.161 g, 4.36 mmol) was dissolved in ethanol (10 cm3). To this was added 1-bromododecane (1.123 g, 4.37 mmol) and the mixture refluxed for 16 h. The ethanol was removed in vacuo, and purified viacolumn chromatography (DCM–MeOH–AcOH 90 : 5 : 5 solvent), afforded a clear waxy solid 4-(N,N-dimethylamino-N-butyl-N-propyl)-4-aza-6-methyl-10-oxatricyclo[5.2.1]decane-3,5-dione (1.506 g, 67%).
1H NMR (CDCl3): δ (ppm): 4.59 (1H, d, J = 5.2 Hz, CH2–CH(–O)–CH–(CO)), 3.49 (4H, m, overlapping, (OC–)N–CH2–CH2–CH2), 3.28 (2H, t, J = 8.3 Hz, N+–CH2–CH2–(CH2)9–CH3), 3.20 (6H, s, CH2–CH2–N(–CH3)2), 3.16 (1H, d, J = 6.9 Hz, (O–)CH–CH–(CO)), 2.93 (1H, d, J = 6.9 Hz, (O–)C(–CH3)–CH–(CO)), 2.07 (2H, m, (OC–)N–CH2–CH2–CH2), 1.82 (2H, m, CH(–O)–CH2–CH2), 1.61 (4H, overlapping, N+–CH2–CH2–(CH2)9–CH3 and CH(–O)–CH2–CH2), 1.42 (3H, s, (O–)C(–CH2)–CH3), 1.20–1.13 (18H, overlapping, N+–CH2–CH2–(CH2)9–CH3), 0.75 (3H, t, J = 6.6 Hz, N+–CH2–CH2–(CH2)9–CH3).
13C NMR (CDCl3): δ (ppm): 177.8 ((CH–)CH–C(O)–O), 176.4 ((C–)CH–C(O)–O), 85.4 ((CH2–)C(–O)–CH3), 78.6 ((CH2–)CH(–O)–CH(CO)), 64.4 ((OC–)N–CH2–CH2–CH2), 61.2 ((OC–)N–CH2–CH2–CH2), 52.2 ((O–)CH–CH–C(O)), 51.9 ((O–)C(CH3)–CH–C(O)), 51.0 (CH2–CH2–N(–CH3)2), 35.9 (N+–CH2–(CH2)10–CH3), 31.6 (CH(–O)–CH2–CH2), 29.3–22.4 (N+–CH2–(CH2)10–CH3 and (OC–)N–CH2–CH2–CH2), 17.5 ((O–)C(CH2)–CH3), 13.8 (N+–CH2–(CH2)10–CH3).
N-Butyl-N-ethylpiperidine-10-oxa-4-azatricyclo[5.2.1]decane-3,5-dione (0.286 g, 0.55 mmol) was dissolved in water (15 cm3) and cooled to 0 °C. To this was added methanesulfonic acid (0.104 g, 70% w/w water solution) and the resultant solution stirred for 1 h. The solution was rotary evaporated to remove residual HBr and any excess ulfonic acid present to afford a pale yellow oil (0.290 g, 99%). MS (MALDI-TOF): m/z 435.1 (M+ – CH3SO3H). Calc. for C27H50N2O6S·2H2O: C 57.21, H 9.60, N 4.94. Found: C 57.67, H 9.14, N 4.59%.
1H NMR (CDCl3): δ (ppm): 4.70 (1H, d, J = 5.0 Hz, CH2–CH(–O)–CH–(CO)), 3.60 (2H, m, overlapping, (OC–)N–CH2–CH2–CH2), 3.30 (2H, t, J = 8.3 Hz, N+–CH2–CH2–(CH2)9–CH3), 3.20 (6H, s, CH2–CH2–N(–CH3)2), 3.16 (1H, d, J = 6.9 Hz, (O–)CH–CH–(CO)), 2.93 (1H, d, J = 6.9 Hz, (O–)C(–CH3)–CH–(CO)), 2.89 (3H, s, CH3SO3), 2.07 (2H, m, (OC–)N–CH2–CH2–CH2), 1.82 (2H, m, CH(–O)–CH2–CH2), 1.61 (4H, overlapping, N+–CH2–CH2–(CH2)9–CH3 and CH(–O)–CH2–CH2), 1.42 (3H, s, (O–)C(–CH2)–CH3), 1.31–1.23 (18H, overlapping, N+–CH2–CH2–(CH2)9–CH3), 0.85 (3H, t, J = 6.6 Hz, N+–CH2–CH2–(CH2)9–CH3).
13C NMR (CDCl3): δ (ppm): 178.0 ((CH–)CH–C(O)–O), 176.6 ((C–)CH–C(O)–O), 85.7 ((CH2–)C(–O)–CH3), 78.9 ((CH2–)CH(–O)–CH(CO)), 65.0 ((OC–)N–CH2–CH2–CH2), 61.7 ((OC–)N–CH2–CH2–CH2), 52.4 ((O–)CH–CH–C(O)), 52.1 ((O–)C(CH3)–CH–C(O)), 51.6 (CH2–CH2–N(–CH3)2), 39.5 (CH3SO3), 36.2 (N+–CH2–(CH2)10–CH3), 31.9 (CH(–O)–CH2–CH2), 29.6–22.6 (N+–CH2–(CH2)10–CH3 and (OC–)N–CH2–CH2–CH2), 17.8 ((O–)C(CH2)–CH3), 14.0 (N+–CH2–(CH2)10–CH3).
1H NMR (CDCl3): δ (ppm): 4.71 (1H, d, J = 5.0 Hz, CH2–CH(–O)–CH–(CO)), 3.60 (2H, m, overlapping, (OC–)N–CH2–CH2–CH2), 3.30 (2H, t, J = 8.3 Hz, N+–CH2–CH2–(CH2)9–CH3), 3.20 (6H, s, CH2–CH2–N(–CH3)2), 3.16 (1H, d, J = 6.9 Hz, (O–)CH–CH–(CO)), 2.93 (1H, d, J = 6.9 Hz, (O–)C(–C2H5)–CH–(CO)), 2.90 (3H, s, CH3SO3), 2.07 (2H, m, (OC–)N–CH2–CH2–CH2), 1.82 (2H, m, CH(–O)–CH2–CH2), 1.92–1.52 (6H, overlapping, N+–CH2–CH2–(CH2)9–CH3, CH(–O)–CH2–CH2 and (O–)C(–CH2)–CH2–CH3), 1.31–1.23 (18H, overlapping, N+–CH2–CH2–(CH2)9–CH3), 1.09 (3H, t, J = 7.5 Hz, (O–)C(–CH2)–CH2–CH3), 0.84 (3H, t, J = 5.0 Hz, N+–CH2–CH2–(CH2)9–CH3).
13C NMR (CDCl3): δ (ppm): 178.0 ((CH–)CH–C(O)–O), 176.4 ((C–)CH–C(O)–O), 89.7 ((CH2–)C(–O)–CH2–CH3), 78.7 ((CH2–)CH(–O)–CH(CO)), 65.0 ((OC–)N–CH2–CH2–CH2), 61.7 ((OC–)N–CH2–CH2–CH2), 51.8 ((O–)CH–CH–C(O)), 51.6 ((O–)C(CH3)–CH–C(O)), 51.2 (CH2–CH2–N(–CH3)2), 39.5 (CH3SO3), 35.4 (N+–CH2–(CH2)10–CH3), 31.9 (CH(–O)–CH2–CH2), 29.7–22.6 (N+–CH2–(CH2)10–CH3, (OC–)N–CH2–CH2–CH2 and (O–)C(–CH2)–CH2–CH3), 14.1 (N+–CH2–(CH2)10–CH3), 9.43 ((O–)C(–CH2)–CH2–CH3).
Thermogravimetric analysis (TGA)
Thermogravimetric analysis was undertaken using a Perkin Elmer® Pyris Diamond TG /DTA to determine the impurity content and onset temperature of decomposition. The temperature was increased from room temperature to a maximum of 800 °C linearly at a rate of 10 °C min–1 with nitrogen used as the purge gas.Malachite Green GTPaseassay
The assay was modified from that reported earlier,30 with the main change being a reduction of dynamin I from 200 nM to 20 nM in the present study. Purified dynamin I (20 nM) (diluted in dynamin diluting buffer: 6 mM Tris-HCl, 20 mM NaCl, 0.01% Tween 80, pH 7.4) was incubated in GTPase buffer (5 mM Tris-HCl, 10 mM NaCl, 2 mM Mg2+, 0.05% Tween 80, pH 7.5, 1 µg mL–1 leupeptin and 0.1 mM PMSF) and GTP at 0.3 mM in the presence of RTIL for 30 min at 37 °C in a final assay volume of 150 µL. Plates were incubated in a dry heating block with shaking at 300 rpm (Eppendorf Thermomixer). Dynamin GTPase activity was stimulated by phosphatidylserine liposomes (2 µg/mL). The reaction was terminated with 10 µL of 0.5 M EDTA pH 8.0 followed by 40 µL of Malachite Green solution (2% w/v ammonium molybdate tetrahydrate, 0.15% w/v malachite green and 4 M HCl). Colour was developed for 5 min (stable up to 2 h), and sample absorbance was determined on a microplate spectrophotometer at 650 nm. Phosphate release was quantified by comparison with a standard curve of dry baked sodium dihydrogen orthophosphate monohydrate. GraphPad Prism 4 (GraphPad Software Inc., San Diego, CA) was used for plotting data points and calculation of IC50.Acknowledgements
This work was supported by the Australian Research Council and the National Health & Medical Research Council (Australia). J. Z. acknowledges the receipt of a University of Newcastle PhD scholarship.References
- S. A. Forsyth, J. M. Pringle and D. R. MacFarlane, Aust. J. Chem., 2004, 57, 113–119 CrossRef.
- R. D. Rodgers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef.
- K. R. Seddon, A. Stark and M.-J. Torres, Pure Appl. Chem., 2000, 72, 2275–2287 CrossRef CAS.
- T. Welton, Chem. Rev., 1999, 99, 2071–2083 CrossRef CAS.
- J. A. Whitehead, J. Zhang, N. Pereira, A. McCluskey and G. A. Lawrance, Hydrometallurgy, 2007, 88, 109–120 CrossRef CAS.
- J. A. Whitehead, G. A. Lawrance and A. McCluskey, Aust. J. Chem., 2004, 57, 151–155 CrossRef.
- J. A. Whitehead, G. A. Lawrance and A. McCluskey, Green Chem., 2004, 6, 313–315 RSC.
- K. Booker, M. C. Bowyer, C. J. Lennard, C. I. Holdsworth and A. McCluskey, Aust. J. Chem., 2007, 60, 51–56 CrossRef CAS.
- K. Booker, M. C. Bowyer, C. I. Holdsworth and A. McCluskey, Chem. Commun., 2006, 1970–1972 Search PubMed.
- A. McCluskey, A. T. R. Sim and J. A. Sakoff, J. Med. Chem., 2002, 45, 1151–1175 CrossRef CAS.
- A. McCluskey and J. A. Sakoff, Mini Rev. Med. Chem., 2001, 1, 43–45 Search PubMed.
- A. McCluskey and J. A. Sakoff, Curr. Pharm. Des., 2004, 10, 1139–1159 CrossRef.
- J. A. Sakoff, S. P. Ackland, M. L. Baldwin, M. A. Keane and A. McCluskey, Invest. New Drugs, 2002, 21, 1–11 CrossRef.
- A. McCluskey, M. C. Bowyer, C. Walkom, S. P. Ackland, E. Gardiner and J. A. Sakoff, Bioorg. Med. Chem. Lett., 2001, 11, 2941–2946 CrossRef CAS.
- M. E. Hart, A. R. Chamberlin, C. Walkom, J. A. Sakoff and A. McCluskey, Bioorg. Med. Chem. Lett., 2004, 14, 1969–1973 CrossRef CAS.
- T. A. Hill, L. R. Odell, A. Quan, R. Abagyan, G. Ferguson, P. J. Robinson and A. McCluskey, Bioorg. Med. Chem. Lett., 2004, 14, 3275–3278 CrossRef CAS.
- A. McCluskey, L. R. Odell, J. K. Edwards, J. Rusak, R. Abagyan, J. L. Scott, P. J. Robinson and T. Hill, J. Med. Chem., 2005, 48, 7781–7788 CrossRef.
- C. M. Gordon and A. McCluskey, Chem. Commun., 1999, 1431–1432 RSC.
- A. McCluskey, J. Garner, S. Caballero and D. J. Young, Tetrahedron Lett., 2000, 41, 8147–8151 CrossRef CAS.
- Z. M. A. Judeh, C. C. Bun, J. Bu and A. McCluskey, Tetrahedron Lett., 2002, 29, 5089–5091 CrossRef.
- A. McCluskey, M. C. Bowyer, C. M. Gordon, S. L. Leitch and C. Ritchie, Aust. J. Chem., 2004, 57, 135–137 CrossRef.
- S. Otto, Curr. Opin. Drug Discovery Dev., 2003, 6, 509 CAS.
- S. Otto, S. Furlan and J. K. M. Sanders, Drug Discovery Today, 2002, 7, 117–125 CrossRef CAS.
- M. Hochguertel, R. Biesinger, H. Kroth, D. Piecha, M. W. Hofmann, S. Krause, O. Schaaf, C. Nicolau and A. V. Eliseev, J. Med. Chem., 2003, 46, 356–358 CrossRef CAS.
- M. S. Congreve, D. J. Davis, L. Devine, C. Granata, M. O'Reilly, P. G. Wyatt and H. Jhoti, Angew. Chem., Int. Ed., 2003, 42, 4479–4482 CrossRef CAS.
- S. Zuchner, S. Noureddine, M. Kennerson, M. Verhoeven, K. Claeys, K. de Jonghe, P. Merory, J. Oliveira, S. A. Speer, M. C. Stenger, J. E. Walizada, G. Zhu, M. A. Pericak-Vance, G. Nicholson, V. Timmerman and J. M. Vance, Nat. Genet., 2005, 37, 289–294 CrossRef.
- J. A. Sakoff, I. J. Howitt, S. P. Ackland and A. McCluskey, Can. Chemother. Pharm., 2004, 53, 225–232 Search PubMed.
- T. Efferth, R. Rauh, S. Kahl, M. Tomicic, H. Böchzelt, M. E. Tome, M. M. Briehl, R. Bauer and B. Kaina, Biochem. Pharmacol., 2005, 69, 81–181.
- F. Massicot, H. Dutertre-Catella, C. Pham-Huy, X.-H. Liu, H. T. Duc and J. M. Warnet, Pharm. Toxicol., 2005, 96, 26–32 Search PubMed.
- A. Quan and P. J. Robinson, Methods Enzymol., 2005, 404, 556–569 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TGA plots for RTILs. See DOI: 10.1039/b707092f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2008 |