Post-coordination functionalisation of pyrazolyl-based ligands as a route to polynuclear complexes based on an inert RuIIN6 core
Qiao-Hua Wei, Stephen P. Argent, Harry Adams and Michael D. Ward*
Department of Chemistry, University of Sheffield, Sheffield, UK S3 7HF. E-mail: m.d.ward@sheffield.ac.uk
First published on 10th September 2007
Abstract
The simple mononuclear complex [Ru(H2bpp)2][PF6]2 [H2bpp = 2,6-bis(pyrazol-3-yl)pyridine] contains four coordinated pyrazolyl ligands which each have a reactive NH site at the position adjacent to the coordinated N atom. Alkylation of these with either 2-[1-{4-(bromomethyl)benzyl}-1H-pyrazol-3-yl]pyridine or 4′-[(4-bromomethyl)phenyl]terpyridine allows attachment of four additional chelating groups, either bidentate pyrazolyl–pyridine and terdentate terpyridyl units, respectively, which are pendant from the central kinetically inert RuIIN6 complex core. These functionalised mononuclear complexes [Ru(L1)2][PF6]2 (with four pendant pyrazolyl–pyridine bidentate sites) and [Ru(L2)2][PF6]2 (with four pendant terpyridyl sites) can be used as the starting point for polynuclear assemblies by attachment of additional labile metal ions as the secondary sites. As examples of this we prepared and structurally characterised the trinuclear complex [RuAg2(L1)2][ClO4]4, an unusual example of a polynuclear helicate containing a kinetically inert metal centre, and the pentanuclear complex [RuCu4(MeCN)5(H2O)1.5(L2)2](SbF6)6(BF4)4 in which each of the pendant terpyridyl sites of the [Ru(L2)2]2+ core is coordinated to a Cu(II) ion.
Introduction
For practitioners of modern coordination chemistry it is a common requirement to be able to synthesise polynuclear complexes in which different metal ions occupy different sites of a multinucleating ligand which contains different binding domains. The problems under investigation can be highly varied (small molecule activation or catalysis by cooperative binding at a polynuclear site;1 photoinduced energy or electron transfer from one metal complex fragment to another;2 electrochemical interactions across a conjugated bridging ligand3) but the basic synthetic problem is the same in every case. There are two general methods which may be employed. Firstly, if kinetically labile metal ions are used, a multi-topic ligand may be treated with a mixture of metal ions and a single product results in which each ion has sought out the binding domain which given the most thermodynamically stable product. This is illustrated by our recent one-pot preparation of the heteronuclear helicate [FeAg2(L1)2]4+, in which the propensity of Fe(II) for six-fold coordination and of Ag(I) for four-fold coordination led to these ions occupying the central and peripheral sites, respectively of the double helical structure.4 Alternatively, if kinetically inert metal ions are used, a stepwise ‘complexes as ligands’ approach has to be employed in which the binding sites of a multi-topic ligand are occupied sequentially.2,3,5 It can be difficult to put a single kinetically inert ion at one site of a multi-topic ligand, because mixtures of mononuclear and polynuclear complexes inevitably result which require careful separation.Consequently, a better method is to make a coordinatively saturated mononuclear complex with a reactive peripheral site. Attachment of a second ligand fragment to this complex core, or even attachment of an entire additional complex fragment, provides an efficient stepwise method for assembly of heteropolynuclear complexes. This method is illustrated by work from the groups of Constable6 and Tor.7 Constable has shown how complexes of functionalised 2,2′:6′,2″-terpyridines can be used as building blocks to make polynuclear dendrimer-like complexes of Ru(II) and Os(II), by reaction together of {M(terpy)2}2+ species with complementary reactive functional groups (e.g. hydroxyl, halogen) at their peripheries.6 Tor has shown how two complexes, one containing a 5-bromophenanthroline ligand and the other containing an ethynylphenanthroline ligand, can be coupled using conventional organic synthetic methodology to give a heterodinuclear complex containing an alkyne-linked bis-phenanthroline ligand.7
In this paper we illustrate a new variant on this theme, in which alkylation of the pyrazole rings of a coordinated terdentate ligand allows additional ligand sites to be added around the periphery of a kinetically inert Ru(II) core. The resulting mononuclear Ru(II) complexes then contain four peripheral bidentate or tridentate sites, and these species can be used as the basis for assembly of polynuclear complexes which would not be accessible from the pre-formed complete ligands.
Results and discussion
Preparation of mononuclear complexes with pendant binding sites
For this work we employed the terdentate ligand 2,6-bis(pyrazol-3-yl)pyridine (H2bpp) which coordinates as a simple tridentate chelate, but which has reactive pyrazolyl NH groups that can be functionalised further (Scheme 1). We reported the synthesis of this ligand a while ago and described some of its lanthanide(III) complexes.8 Reaction of commercial ruthenium(III) chloride with two equivalents of H2bpp in ethylene glycol at reflux afforded, after chromatographic purification and anion metathesis with ammonium hexafluorophosphate, the mononuclear complex [Ru(H2bpp)2][PF6]2 in good yield. The crystal structure of the complex (Fig. 1, Table 1) reveals a conventional slightly distorted octahedral coordination geometry. The Ru–N(pyridyl) bond distances to the central rings of the ligands are slightly shorter (average 2.019 Å) than the Ru–N(pyrazolyl) bond distances, which average 2.072 Å; this follows the normal pattern for complexes of terpyridine in which the central pyridyl ring generally has the shortest metal–ligand bond for steric reasons.9 The N(pyrazolyl)–Ru–N(pyrazolyl) bite angles are both 154°, and the two terdentate ligands are essentially mutually perpendicular.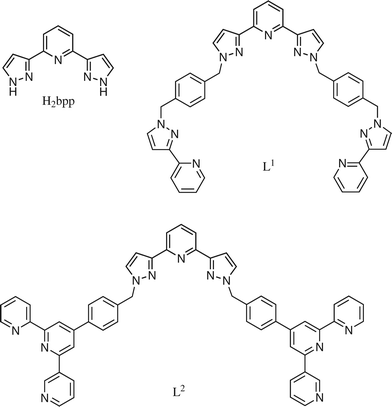 | ||
| Scheme 1 Structural formulae of the ligands described in this paper. | ||
![Structure of the complex cation of [Ru(H2bpp)2][PF6]2·2Et2O·2MeCN (H atoms, except for pyrazolyl N–H atoms, removed for clarity).](/image/article/2008/NJ/b708572a/b708572a-f1.gif) | ||
| Fig. 1 Structure of the complex cation of [Ru(H2bpp)2][PF6]2·2Et2O·2MeCN (H atoms, except for pyrazolyl N–H atoms, removed for clarity). | ||
| Ru(1)–N(8) | 2.016(3) | Ru(1)–N(7) | 2.068(3) |
| Ru(1)–N(3) | 2.021(3) | Ru(1)–N(2) | 2.076(3) |
| Ru(1)–N(9) | 2.066(3) | Ru(1)–N(4) | 2.076(3) |
| N(8)–Ru(1)–N(3) | 177.74(13) | N(9)–Ru(1)–N(2) | 90.60(13) |
| N(8)–Ru(1)–N(9) | 77.19(13) | N(7)–Ru(1)–N(2) | 93.77(13) |
| N(3)–Ru(1)–N(9) | 102.77(13) | N(8)–Ru(1)–N(4) | 100.80(13) |
| N(8)–Ru(1)–N(7) | 77.13(13) | N(3)–Ru(1)–N(4) | 76.94(14) |
| N(3)–Ru(1)–N(7) | 102.99(13) | N(9)–Ru(1)–N(4) | 93.98(13) |
| N(9)–Ru(1)–N(7) | 154.20(14) | N(7)–Ru(1)–N(4) | 93.15(13) |
| N(8)–Ru(1)–N(2) | 105.19(13) | N(2)–Ru(1)–N(4) | 153.98(14) |
| N(3)–Ru(1)–N(2) | 77.07(13) | ||
The UV/Vis spectroscopic and redox properties of [Ru(H2bpp)2][PF6]2 can be understood with reference to the known electronic properties of pyrazole groups as ligands compared to pyridyl groups, viz. pyrazolyl ligands are poorer σ-donors than pyridyl ligands (cf. the pKa values for protonation of 5.2 for pyridine and 2.6 for pyrazole), and are also much poorer π-acceptors with high-energy π* orbitals compared to polypyridines.10 The latter effect dominates such that compared to [Ru(terpy)2]2+,11 for example, the metal centre of [Ru(H2bpp)2][PF6]2 is more electron-rich and accordingly has a less positive Ru(II)/Ru(III) redox potential (+0.54 V vs. Fc/Fc+, cf. +0.89 V for [Ru(terpy)2]2+). This effect is formalised in Lever’s electrochemical parameters for the different ligand types in Ru(II)/Ru(III) complexes.12 The poor π-accepting ability of the H2bpp ligands compared to terpy is reflected in a higher π* energy, resulting in a blue-shifted 1MLCT absorption compared to [Ru(terpy)2]2+ (418 nm vs. 470 nm,11 respectively).
From the structural point of view of further functionalisation at the pyrazolyl NH sites, the geometry of the complex is ideal with the two pyrazolyl N–H vectors of one H2bpp ligand being essentially parallel to the plane of the other ligand between them. Thus, any substituents attached at these sites will not diverge too much from the complex core, and will not interfere sterically with the other ligand. This situation may be contrasted with the situation when a [Ru(terpy)2]2+ core is used as the basis for further functionalisation (Scheme 2). In this case, the pinching of the terpy ligand on coordination to Ru(II) means that substituents at the C6 and C6″ positions converge on the other ligand and interfere with it sterically, resulting in the complexes becoming particularly labile;13 the C5 and C5″ positions however are still divergent, and are notably difficult positions at which to introduce substituents.14 Thus [Ru(H2bpp)2][PF6]2 provides a geometrically ideal scaffold for synthetically facile further functionalisation of an octahedral metal complex core.
 | ||
| Scheme 2 Illustration of the different extents to which ligand substituents (R) converge on, and sterically interfere with, the alternate ligand using (a) terpyridine and (b) H2bpp, coordinated to a six-coordinate Ru(II) centre. The second ligand is shown edge-on as a thick black bar. | ||
Reaction of [Ru(H2bpp)2][PF6]2 with >4 equivalents of 2-[1-{4-(bromomethyl)benzyl}-1H-pyrazol-3-yl]pyridine to afford [Ru(L1)2][PF6]2 (Scheme 3) was investigated under a variety of conditions. In the end the best conditions were found to be MeCN–CH2Cl2 (3 : 1) as solvent at 80 °C, using NaH as base to deprotonate the pyrazole groups of [Ru(H2bpp)2][PF6]2 (use of alternative bases such as Et3N or Bu4NOH was much less successful), and using a catalytic amount of Bu4NI to speed up the nucleophilic substitution at the CH2Br sites. Under these conditions we could effect the tetraalkylation of [Ru(H2bpp)2][PF6]2 to give [Ru(L1)2][PF6]2 in 26% yield, as confirmed by the ESI mass spectrum and 1H NMR spectrum (see Experimental section). The ligand L1 is the same one that we described earlier and used to form polynuclear helicates;4 however, it is clear that reaction of pre-formed L1 with a source of Ru(II) would be very unlikely to have given significant quantities of [Ru(L1)2][PF6]2 but would have generated a complicated mixture of products.
![Syntheses and structural formulae of the complexes [Ru(L1)2]2+ and [Ru(L2)2]2+, bearing four pendant bipyridyl or terpyridyl sites, respectively.](/image/article/2008/NJ/b708572a/b708572a-s3.gif) | ||
| Scheme 3 Syntheses and structural formulae of the complexes [Ru(L1)2]2+ and [Ru(L2)2]2+, bearing four pendant bipyridyl or terpyridyl sites, respectively. | ||
A small amount of [Ru(L1)2][PF6]2 was converted to the nitrate salt to grow X-ray quality crystals. The structure of [Ru(L1)2](NO3)2 (Fig. 2, Table 2) has some interesting features. The complex crystallises in the space group I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) such that only one quarter of the cation is unique. The phenyl ring of each pendant arm is almost perpendicular to the pyrazolyl ring from which it is pendant (angle between mean planes of 88.5°), and is involved in an aromatic π-stacking interaction with a coordinated pyrazolyl ring from the alternate ligand; the stacked phenyl and pyrazolyl rings are ca. 3.5 Å apart and nearly parallel (angle of 9.3° between their mean planes). This stacking interaction is facilitated by the ideal geometry of the complex core as described above, which results in each pendant phenyl group neither diverging from nor converging with the other ligand, but lying parallel to it at an appropriate distance for π-stacking.
such that only one quarter of the cation is unique. The phenyl ring of each pendant arm is almost perpendicular to the pyrazolyl ring from which it is pendant (angle between mean planes of 88.5°), and is involved in an aromatic π-stacking interaction with a coordinated pyrazolyl ring from the alternate ligand; the stacked phenyl and pyrazolyl rings are ca. 3.5 Å apart and nearly parallel (angle of 9.3° between their mean planes). This stacking interaction is facilitated by the ideal geometry of the complex core as described above, which results in each pendant phenyl group neither diverging from nor converging with the other ligand, but lying parallel to it at an appropriate distance for π-stacking.
2. (a) A view showing the atom labeling scheme. The symmetry operations invoked by the additional labels A, B, C are as follows: A (–1 + y, 1 – x, 2 – z), B (–x, 2 – y, z), C (1 – y, 1 + x, 2 – z). (b) A view down the 4̄ axis with the two ligands shaded separately for clarity.](/image/article/2008/NJ/b708572a/b708572a-f2.gif) | ||
| Fig. 2 Two views of the complex cation of [Ru(L1)2](NO3)2. (a) A view showing the atom labeling scheme. The symmetry operations invoked by the additional labels A, B, C are as follows: A (–1 + y, 1 – x, 2 – z), B (–x, 2 – y, z), C (1 – y, 1 + x, 2 – z). (b) A view down the axis with the two ligands shaded separately for clarity. | ||
| Symmetry transformations used to generate equivalent atoms. A: y–1, –x + 1, –z + 2. B: –x, –y + 2, z. | |
|---|---|
| Ru(1)–N(51) | 2.034(2) |
| Ru(1)–N(41) | 2.0894(19) |
| N(51A)–Ru(1)–N(51) | 180.0 |
| N(51A)–Ru(1)–N(41) | 103.46(6) |
| N(51)–Ru(1)–N(41) | 76.54(6) |
| N(41B)–Ru(1)–N(41) | 153.08(11) |
| N(41)–Ru(1)–N(41A) | 93.11(3) |
Under similar conditions, we could prepare [Ru(L2)2][PF6]2 by alkylation of the four pyrazolyl rings of [Ru(H2bpp)2][PF6]2 with an excess of 4′-[(4-bromomethyl)phenyl]terpyridine (Scheme 3) in 49% yield. Again, ESI mass spectrometry and 1H NMR spectroscopy confirmed the formulation of the product with four pendant terpyridyl groups. The crystal structure of [Ru(L2)2][PF6]2·C6H6 is shown in Fig. 3 with selected parameters in Table 3. In this case two of the four pendant groups have their phenyl groups [C(41)–C(46) and its symmetry equivalent] involved in aromatic stacking interactions with a coordinated pyrazolyl ring from the alternate ligand [N(71)–C(75) and its symmetry equivalent], and the two ligands are entwined around one another in a pseudo-helical manner.
![Structure of the complex cation of [Ru(L2)2][PF6]2·C6H6, with the two ligands shaded separately for clarity.](/image/article/2008/NJ/b708572a/b708572a-f3.gif) | ||
| Fig. 3 Structure of the complex cation of [Ru(L2)2][PF6]2·C6H6, with the two ligands shaded separately for clarity. | ||
| Symmetry transformations used to generate equivalent atoms. A: –x + 1/2, –y, z. | |||
|---|---|---|---|
| Ru(1)–N(61) | 2.026(3) | Ru(1)–N(71) | 2.102(3) |
| Ru(1)–N(51) | 2.094(3) | ||
| N(61)–Ru(1)–N(61A) | 175.67(15) | N(51)–Ru(1)–N(71A) | 99.47(11) |
| N(61)–Ru(1)–N(51A) | 106.43(11) | N(61)–Ru(1)–N(71) | 77.00(11) |
| N(61)–Ru(1)–N(51) | 76.84(11) | N(61A)–Ru(1)–N(71) | 99.80(11) |
| N(51A)–Ru(1)–N(51) | 85.80(15) | N(51)–Ru(1)–N(71) | 153.75(11) |
| N(61)–Ru(1)–N(71A) | 99.80(11) | N(71A)–Ru(1)–N(71) | 87.15(17) |
[Ru(L1)2][PF6]2 and [Ru(L2)2][PF6]2 have very similar redox potentials (+0.75 and +0.77 V vs. Fc/Fc+, respectively) and identical 1MLCT absorption maxima (402 nm). The positive shift of the Ru(II)/Ru(III) redox couple of ca. 200 mV on alkylation of the four pyrazole rings of [Ru(H2bpp)2][PF6]2 implies a decrease in electron density at the metal centre compared to [Ru(H2bpp)2][PF6]2 because L1 and L2 are poorer σ-donors or better π-acceptors (or a combination of both). If the alkylated ligands L1 and L2 were better π-acceptors than H2bpp, the 1MLCT absorption maximum would be expected to be red-shifted compared to [Ru(H2bpp)2][PF6]2 as the ligand-based π* energy is reduced. In fact the converse is true, implying that the main effect of alkylation of the pyrazole groups is to make L1 and L2 poorer σ-donors, because the pyrazole rings have less pz–⋯H+ character when the potentially acidic protons are replaced by alkyl groups.
Polynuclear complexes
Since [Ru(L1)2][PF6]2 has pendant bidentate chelating units, reaction with an excess of AgClO4 was carried out to see if four-coordinate (bis-bidentate) Ag(I) centres could form as part of a polynuclear assembly. Assuming that two of the four pendant bidentate units coordinate to each Ag(I) centre, this could afford one of two types of structure. If the two pendant pyrazolyl–pyridine units from the same ligand coordinated to each Ag(I) ion, the result would be a catenated structure in which two metallamacrocycles [Ag/L1 loops] are connected by coordination of each of their terdentate sites to the central Ru(II).15 Alternatively, if the two pyrazolyl–pyridine units which bind to each Ag(I) ion come from different ligands, the result will be a complexes in which each ligand spans all three metal ions, as we saw before in trinuclear double helicates with L1.4It is the second possibility which pertains here. The structure of [RuAg2(L1)2][ClO4]4 (Fig. 4 and 5) is a trinuclear double helicate containing six-coordinate Ru(II) in the central cavity, and four-coordinate Ag(I) ions at the two peripheral sites in irregular four-coordinate geometries. The helical assembly is characterised by an extensive degree of stacking between the two intertwined ligands, which form a five-layer stack on one side of the complex involving terminal pyridyl/pyrazoles [coordinated to Ag(1)] of one ligand (denoted A), phenyl spacer groups of ligand B, and the central Ru(II)-bound pyridyl ring of ligand A. The Ru⋯Ag separation is 8.65 Å. This structure is generally similar to that of the FeIIAgI2 trinuclear helicate that we prepared recently in a one-pot reaction from labile Fe(II) and Ag(I) starting materials.4 However the important difference is that this stepwise strategy has allowed the preparation of a helical scaffold based around a kinetically inert Ru(II) core, of which there are very few examples.16 It is notable that in the 1H NMR spectrum the two doublets arising from the central phenylene spacer groups are at low chemical shifts (6.47 and 5.64 ppm) which are indicative of the aromatic stacking involving these rings that is apparent in the crystal structure being retained in solution. However the presence of only two signals (an AX doublet-of-doublets) implies that all phenyl rings are in the same environment in solution, i.e. the complex has a more symmetric structure than that observed in the solid state. Given the lability of Ag(I) ions, and the distorted coordination environment about the Ag(I) ions with relatively long and weak Ag–N bonds (up to 2.5 Å, see Table 4), it is possible that the Ag(I) is undergoing a dissociation/reassociation equilibrium facilitated by the strong donor solvent which allows the phenylene groups to become equivalent in solution. The fact that the complex was only significantly soluble in dmso at the concentrations required for NMR spectroscopy unfortunately precluded low-temperature studies.
![Structure of the double helical complex cation of [RuAg2(L1)2][ClO4]4·H2O·2dmf, showing the atom labeling scheme.](/image/article/2008/NJ/b708572a/b708572a-f4.gif) | ||
| Fig. 4 Structure of the double helical complex cation of [RuAg2(L1)2][ClO4]4·H2O·2dmf, showing the atom labeling scheme. | ||
![Alternative view of the structure of the complex cation of [RuAg2(L1)2][ClO4]4·H2O·2dmf, with the ligands coloured separately for clarity.](/image/article/2008/NJ/b708572a/b708572a-f5.gif) | ||
| Fig. 5 Alternative view of the structure of the complex cation of [RuAg2(L1)2][ClO4]4·H2O·2dmf, with the ligands coloured separately for clarity. | ||
| Symmetry transformations used to generate equivalent atoms. A: –x, y, –z + 1/2. | |||
|---|---|---|---|
| Ru(1)–N(41) | 2.005(7) | Ag(1)–N(1) | 2.226(6) |
| Ru(1)–N(91) | 2.035(7) | Ag(1)–N(61) | 2.305(5) |
| Ru(1)–N(31) | 2.090(5) | Ag(1)–N(51) | 2.321(6) |
| Ru(1)–N(81) | 2.109(4) | Ag(1)–N(11) | 2.498(5) |
| N(41)–Ru(1)–N(91) | 180.000(2) | N(1)–Ag(1)–N(61) | 135.0(2) |
| N(41)–Ru(1)–N(31) | 77.27(13) | N(1)–Ag(1)–N(51) | 151.7(2) |
| N(91)–Ru(1)–N(31) | 102.73(13) | N(61)–Ag(1)–N(51) | 72.82(19) |
| N(31)–Ru(1)–N(31A) | 154.5(3) | N(1)–Ag(1)–N(11) | 71.4(2) |
| N(41)–Ru(1)–N(81) | 102.89(13) | N(61)–Ag(1)–N(11) | 121.29(16) |
| N(91)–Ru(1)–N(81) | 77.11(13) | N(51)–Ag(1)–N(11) | 99.66(18) |
| N(31)–Ru(1)–N(81) | 98.50(16) | ||
| N(31)–Ru(1)–N(81A) | 87.17(16) | ||
| N(81)–Ru(1)–N(81A) | 154.2(3) | ||
With [Ru(L2)2][PF6]2 as the starting unit, there are likewise numerous possibilities for different architectures to form depending on how the pendant terpyridyl sites arrange themselves on coordination to the secondary metal ion. In principle trinuclear catenates or helicates could form, as with the previous example, if two of the terpyridyl sites can converge on the same metal centre. However this is not sterically likely given their divergent spatial arrangement. Simple attachment of one metal ion (plus ancillary ligands) at each terpyridyl site would give a pentanuclear species; coordination of two terpyridyl units from separate complexes to the same metal ion could easily result in infinite cross-linked chain structures with a fourfold-connected node at the Ru(II) centre.
Reaction of [Ru(L2)2][PF6]2 with Zn(BF4)2 in dmf, followed by precipitation of the product using NaSbF6 and recrystallisation, afforded substantial and well-formed red crystals of a material whose elemental analysis indicated the formulation [RuZn2(L2)2](BF4)3(SbF6)3, i.e. two Zn(II) ions per Ru(II) centre, implying that the Zn(II) ions are each coordinated by two terpyridyl sites. From inspection of Fig. 3 it is clear that it is not possible for two terpyridyl sites from the same Ru(II) complex unit to bind to the same Zn(II) ion, so the structure is likely to be a cross-linked network of some sort in which each Zn(II) ion is coordinated by pendant terpyridyls from two separate [Ru(L2)2]2+ units. Frustratingly these crystals hardly scattered at all, even using synchrotron radiation, despite being large and well-formed, and it has not been possible to determine the structure of this network.
We had more success by reaction of [Ru(L2)2][PF6]2 with four equivalents of Cu(BF4)2, followed by anion metathesis with NaSbF6. Crystallisation of the crude product with MeNO2/ether afforded in 25% yield green-brown crystals of the pentanuclear cluster [RuCu4(MeCN)5(H2O)1.5(L2)2](SbF6)6(BF4)4·5H2O in which each of the four pendant terpyridyl sites of the central [Ru(L2)2]2+ core is occupied by a Cu(II) centre which is either four or five coordinate (from one terpyridyl ligand plus one or two monodentate solvent molecules). The crystal structure is shown in Fig. 6 and 7 and structural parameters are given in Table 5. The complex cation has approximate fourfold rotational symmetry, with each of the four Cu(II)–terpyridyl fragments being perpendicular to the plane of the pyrazolyl ring from which it is pendant. Each pendant phenyl ring is therefore parallel to and stacked with a pyrazolyl ring of the alternate ligand, with all four coordinated pyrazolyl rings of the Ru(II) core each interacting with one phenyl ring of a pendant arm. The coordination geometries around the Cu(II) centres are approximately square planar [Cu(1), with one MeCN ligand in addition to the terpy ligand]; square pyramidal [Cu(2), with equatorial MeCN and axial water ligands]; square pyramidal [Cu(3), with two MeCN ligands]; and a mixture of four- and five-coordinate [Cu(4), with an equatorial MeCN ligand and an axial water ligand which has a site occupancy of 0.5].
![Structure of the pentanuclear complex cation of [RuCu4(MeCN)5(H2O)1.5(L2)2] (SbF6)6(BF4)4·5H2O, showing the atom labeling scheme.](/image/article/2008/NJ/b708572a/b708572a-f6.gif) | ||
| Fig. 6 Structure of the pentanuclear complex cation of [RuCu4(MeCN)5(H2O)1.5(L2)2] (SbF6)6(BF4)4·5H2O, showing the atom labeling scheme. | ||
![Alternative view of the structure of the complex cation of [RuCu4(MeCN)5(H2O)1.5(L2)2] (SbF6)6(BF4)4·5H2O, with the ligands coloured separately for clarity.](/image/article/2008/NJ/b708572a/b708572a-f7.gif) | ||
| Fig. 7 Alternative view of the structure of the complex cation of [RuCu4(MeCN)5(H2O)1.5(L2)2] (SbF6)6(BF4)4·5H2O, with the ligands coloured separately for clarity. | ||
| Ru(1)–N(61) | 2.026(17) | Cu(1)–N(101) | 1.908(18) | Cu(2)–N(121) | 2.00(2) | Cu(3)–N(306) | 2.15(2) |
| Ru(1)–N(171) | 2.035(15) | Cu(1)–N(91) | 1.98(3) | Cu(2)–O(2) | 2.27(4) | Cu(4)–N(211) | 1.927(16) |
| Ru(1)–N(52) | 2.064(16) | Cu(1)–N(111) | 1.99(3) | Cu(3)–N(21) | 1.934(18) | Cu(4)–N(315) | 1.926(19) |
| Ru(1)–N(161) | 2.085(15) | Cu(2)–N(131) | 1.923(17) | Cu(3)–N(31) | 1.98(2) | Cu(4)–N(201) | 2.000(17) |
| Ru(1)–N(71) | 2.097(16) | Cu(2)–N(303) | 1.95(3) | Cu(3)–N(309) | 1.99(2) | Cu(4)–N(221) | 2.022(17) |
| Ru(1)–N(181) | 2.100(17) | Cu(2)–N(141) | 1.98(3) | Cu(3)–N(11) | 2.00(2) | Cu(4)–O(1) | 2.239(13) |
| Cu(1)–N(312) | 1.74(3) | ||||||
| N(61)–Ru(1)–N(171) | 177.7(7) | N(161)–Ru(1)–N(181) | 154.7(7) | N(141)–Cu(2)–N(121) | 158.9(9) | N(309)–Cu(3)–N(306) | 91.5(8) |
| N(61)–Ru(1)–N(52) | 74.8(7) | N(71)–Ru(1)–N(181) | 93.2(6) | N(131)–Cu(2)–O(2) | 97.9(12) | N(11)–Cu(3)–N(306) | 94.2(10) |
| N(171)–Ru(1)–N(52) | 103.9(6) | N(312)–Cu(1)–N(101) | 175.9(13) | N(303)–Cu(2)–O(2) | 101.1(17) | N(211)–Cu(4)–N(315) | 164.6(8) |
| N(61)–Ru(1)–N(161) | 101.0(6) | N(312)–Cu(1)–N(91) | 101.5(14) | N(141)–Cu(2)–O(2) | 92.5(12) | N(211)–Cu(4)–N(201) | 80.2(8) |
| N(171)–Ru(1)–N(161) | 77.2(7) | N(101)–Cu(1)–N(91) | 82.3(11) | N(121)–Cu(2)–O(2) | 98.3(11) | N(315)–Cu(4)–N(201) | 99.3(9) |
| N(52)–Ru(1)–N(161) | 92.5(5) | N(312)–Cu(1)–N(111) | 94.7(14) | N(21)–Cu(3)–N(31) | 79.3(9) | N(211)–Cu(4)–N(221) | 81.0(8) |
| N(61)–Ru(1)–N(71) | 77.4(8) | N(101)–Cu(1)–N(111) | 81.6(11) | N(21)–Cu(3)–N(309) | 153.7(8) | N(315)–Cu(4)–N(221) | 98.3(9) |
| N(171)–Ru(1)–N(71) | 104.0(7) | N(91)–Cu(1)–N(111) | 163.7(12) | N(31)–Cu(3)–N(309) | 98.2(9) | N(201)–Cu(4)–N(221) | 161.0(8) |
| N(52)–Ru(1)–N(71) | 152.1(7) | N(131)–Cu(2)–N(303) | 160.9(14) | N(21)–Cu(3)–N(11) | 79.6(9) | N(211)–Cu(4)–O(1) | 104.6(6) |
| N(161)–Ru(1)–N(71) | 92.3(6) | N(131)–Cu(2)–N(141) | 81.2(11) | N(31)–Cu(3)–N(11) | 158.9(8) | N(315)–Cu(4)–O(1) | 90.8(7) |
| N(61)–Ru(1)–N(181) | 104.4(7) | N(303)–Cu(2)–N(141) | 96.4(14) | N(309)–Cu(3)–N(11) | 100.2(9) | N(201)–Cu(4)–O(1) | 94.5(6) |
| N(171)–Ru(1)–N(181) | 77.5(7) | N(131)–Cu(2)–N(121) | 79.5(11) | N(21)–Cu(3)–N(306) | 114.8(8) | N(221)–Cu(4)–O(1) | 92.2(6) |
| N(52)–Ru(1)–N(181) | 94.1(6) | N(303)–Cu(2)–N(121) | 99.1(14) | N(31)–Cu(3)–N(306) | 95.5(9) | ||
Conclusions
The functionalisation of the pyrazolyl groups of [Ru(H2bpp)2][PF6]2 with bipyridyl or terpyridyl fragments provides a kinetically inert Ru(II) complex core which can be used as the basis for preparation of polynuclear complexes and assemblies by addition of labile metal ions at the peripheral sites. This is a simple and effective route into multimetallic assemblies which would not be available by conventional self-assembly methods. Possibilities in the areas of photophysically active assemblies and coordination networks/crystal engineering are obvious and under investigation.Experimental
General details
The following instrumentation was used for routine spectroscopic and electrochemical analyses: 1H NMR spectra, Bruker AC-250, AMX2-400 or DRX-500 MHz spectrometers; EI and FAB mass spectra, a VG AutoSpec magnetic sector instrument; electrospray mass spectra, a Waters LCT instrument; IR spectra, a Perkin-Elmer Spectrum One instrument; UV/Vis spectra, a Cary-50 spectrometer. Cyclic voltammetric measurements were performed with an Ecochimie Autolab 100 potentiostat using a conventional three-electrode cell with a Pt wire working electrode; the base electrolyte was 0.1 M Bu4NPF6 and the solvent was MeCN.All reactions were performed under an atmosphere of N2 using standard Schlenk-line procedures. 2,6-bis(pyrazol-3-yl)pyridine,8 2-[1-{4-(bromomethyl)benzyl}-1H-pyrazol-3-yl]pyridine,4 and 4′-[(4-bromomethyl)phenyl]terpyridine17 were prepared according to the published methods. Other organic reagents and metal salts were obtained from the usual commercial sources and used as received.
Syntheses
The second (minor) red band was evaporated to dryness in vacuo and the residue was dissolved in MeCN–EtOH (10 cm3, 1 : 1 v/v), which was filtered to remove KNO3. Diffusion of diethyl ether into the concentrated red filtrate gave [Ru(H2bpp)2][NO3]2 as red crystals. Yield: 20%. IR (KBr pellet, cm–1): ν (cm–1) 1384 (s, NO3–). ESI-MS: m/z 262 {M – 2(NO3–)}2+. This could be simply converted to the hexafluorophosphate salt by dissolving it in water and adding NH4PF6 to precipitate it.
X-Ray crystallography
X-ray crystallographic data are summarised in Table 6. In each case a suitable crystal was coated with hydrocarbon oil and attached to the tip of a glass fibre and transferred to a Bruker APEX-2 CCD diffractometer (graphite monochromated Mo-Kα radiation, λ = 0.71073 Å) under a stream of cold N2. After collection and integration the data were corrected for Lorentz and polarisation effects and for absorption by semi-empirical methods (SADABS)18 based on symmetry-equivalent and repeated reflections. The structures were solved by direct methods or heavy atom Patterson methods and refined by full matrix least squares methods on F2. Hydrogen atoms were placed geometrically and refined with a riding model and with Uiso constrained to be 1.2 (1.5 for methyl groups) times Ueq of the carrier atom. Structures were solved and refined using the SHELX suite of programs.19 Significant bond distances and angles for the structures of the metal complexes are in Tables 1–5. Particular problems associated with the refinements are as follows.| Complex | [Ru(H2bpp)2][PF6]2·2Et2O·2MeCN | [Ru(L1)2](NO3)2·4H2O | [Ru(L2)2][PF6]2·C6H6 | [RuAg2(L1)2][ClO4]4·H2O·2dmf | [RuCu4(MeCN)5(H2O)1.5(L2)2](SbF6)6(BF4)4·5H2O |
|---|---|---|---|---|---|
| a R1 = ∑||Fo| – |Fc||/∑|Fo| for ‘observed data’; wR2 = [∑w(Fo2 – Fc2)2/∑w(Fo2)2]1/2 for all unique data. | |||||
| Formula | C34H44F12N12O2P2Ru | C86H78N24O10Ru | C116H84F12N22P2Ru | C92H84Ag2Cl4N24O19Ru | C120H107B4Cu4F52N26O6.5Ru Sb6 |
| M | 1043.82 | 1708.79 | 2177.06 | 2288.44 | 4134.29 |
| T/K | 150(2) | 150(2) | 100(2) | 100(2) | 150(2) |
| Crystal system | Monoclinic | Tetragonal | Orthorhombic | Monoclinic | Monoclinic |
| Space group | C2/c | I | Pnna | C2/c | P21/c |
| a/Å | 25.6147(16) | 16.7252(5) | 27.3364(11) | 30.736(4) | 16.1145(8) |
| b/Å | 10.1935(6) | 16.7252(5) | 15.9057(6) | 14.284(2) | 43.921(2) |
| c/Å | 36.361(2) | 16.1385(10) | 24.5589(9) | 21.992(3) | 24.9915(12) |
| β/° | 105.085(4) | 90 | 90 | 98.756(9) | 96.158(3) |
| V/Å3 | 9166.8(10) | 4514.5(3) | 10 678.3(7) | 9543(2) | 17 586.1(15) |
| Z | 8 | 2 | 4 | 4 | 4 |
| Dc/Mg m–3 | 1.513 | 1.257 | 1.354 | 1.593 | 1.561 |
| µ/mm–1 | 0.506 | 0.242 | 0.258 | 0.757 | 1.568 |
| Reflections collected | 53 604 | 62 954 | 175 242 | 64 337 | 127 534 |
| Independent reflections | 10 549 | 8235 | 9418 | 8394 | 23 020 |
| Rint | 0.0827 | 0.0315 | 0.0560 | 0.0759 | 0.0517 |
| Data/restraints/parameters | 10 549/17/565 | 8235/0/249 | 9418/4/723 | 8394/32/642 | 23 020/311/1904 |
| Final R1, wR2 indicesa | 0.0553, 0.1475 | 0.0582, 0.1532 | 0.0599, 0.1837 | 0.0675, 0.1943 | 0.1281, 0.3847 |
symmetry axis; thus only one quarter of the molecule (i.e. half of a ligand) lies in the asymmetric unit, which also contains two water molecules in general positions (site occupancy 0.5 for each). The nitrate anions could not be located, presumably due to extensive disorder, and are absent from the refinement; a large number of small residual electron density peaks was apparent. Application of the SQUEEZE function in PLATON was used to remove these. Note that the pendant pyrazolyl–pyridine ring has the N atoms N(11) and N(21) syn to one another, rather than the more usual anti conformation which might be expected if these N atoms were to avoid each other. This is because both of these N atoms are involved in a hydrogen-bonding interaction with a nearby water molecule in an adjacent asymmetric unit [O(100)], with non-bonded distances N(11)⋯O(100A) and N(21)⋯O(100A) of 3.12 and 3.10 Å, respectively. Although the H atoms were not located for this water molecule, it is reasonable to assume a chelating H-bonding interaction in which both N(11) and N(21) interact with the same proton.
CCDC reference numbers 659047–659051.
For crystallographic data in CIF or other electronic format see DOI: 10.1039/b708572a
Acknowledgements
We thank the Royal Society for a post-doctoral fellowship to Q.-H. W.References
- (a) A. Jansco, S. Mikkola, H. Lonnberg, K. Hegetschweiler and T. Gajda, Chem.–Eur. J., 2003, 9, 5404 CrossRef CAS; (b) H. Ohtsu, Y. Shimazaki, A. Odani, O. Yamauchi, W. Mori, S. Itoh and S. Fukuzumi, J. Am. Chem. Soc., 2000, 122, 5733 CrossRef CAS; (c) B. M. Trost and S. Hisaindee, Org. Lett., 2006, 8, 6003 CrossRef CAS; (d) M. Lanznaster, A. Neves, A. J. Bortoluzzi, B. Szpoganicz and E. Schwingel, Inorg. Chem., 2002, 41, 5641 CrossRef CAS.
- (a) S. Goeb, A. De Nicola, R. Ziessel, C. Sabatini, A. Barbieri and F. Barigelletti, Inorg. Chem., 2006, 45, 1173 CrossRef CAS; (b) T. Lazarides, T. L. Easun, C. Veyne-Marti, W. Z. Alsindi, M. W. George, N. Deppermann, C. A. Hunter, H. Adams, G. M. Davies and M. D. Ward, J. Am. Chem. Soc., 2007, 129, 4014 CrossRef CAS; (c) K. Senechal-David, S. J. A. Pope, S. Quinn, S. Faulkner and T. Gunnlaugsson, Inorg. Chem., 2006, 45, 10040 CrossRef CAS; (d) S. Encinas, L. Flamigni, F. Barigelletti, E. C. Constable, C. E. Housecroft, E. R. Schofield, E. Figgemeier, D. Fenske, M. Neuburger, J. G. Vos and M. Zehnder, Chem.–Eur. J., 2002, 8, 137 CrossRef CAS.
- D. M. D’Alessandro, M. S. Davies and F. R. Keene, Inorg. Chem., 2006, 45, 1656 CrossRef CAS.
- S. P. Argent, H. Adams, L. P. Harding, T. Riis-Johannessen, J. C. Jeffery and M. D. Ward, New J. Chem., 2005, 29, 904 RSC.
- M. Sommovigo, G. Denti, S. Serroni, S. Campagna, C. Mingazzini, C. Mariotti and A. Juris, Inorg. Chem., 2001, 40, 3318 CrossRef CAS.
- (a) E. C. Constable, C. E. Housecroft and I. Poleschak, Inorg. Chem. Commun., 1999, 2, 565 CrossRef CAS; (b) E. C. Constable, C. E. Housecroft, M. Neuburger, S. Schaffner and L. J. Scherer, Dalton Trans., 2004, 2635 RSC; (c) E. C. Constable, R. W. Handel, C. E. Housecroft, A. F. Morales, B. Ventura, L. Flamigni and F. Barigelletti, Chem.–Eur. J., 2005, 11, 4024 CrossRef CAS; (d) E. C. Constable, A. M. W. Cargill Thompson, P. Harveson, L. Macko and M. Zehnder, Chem.–Eur. J., 1995, 360 CrossRef CAS.
- (a) Y. Tor, Synlett, 2002, 1043 CrossRef CAS; (b) P. J. Connors, D. Tzalis, A. L. Dunnick and Y. Z. Tor, Inorg. Chem., 1998, 37, 1121 CrossRef CAS; (c) D. Tzalis and Y. Tor, Angew. Chem., Int. Ed. Engl., 1997, 36, 2666 CrossRef CAS.
- D. A. Bardwell, J. C. Jeffery, P. L. Jones, J. A. McCleverty, E. Psillakis, Z. Reeves and M. D. Ward, J. Chem. Soc., Dalton Trans., 1997, 2079 RSC.
- E. C. Constable, Adv. Inorg. Chem. Radiochem., 1986, 30, 69 CAS.
- D. L. Jameson, J. K. Blaho, K. T. Kruger and K. A. Goldsby, Inorg. Chem., 1989, 28, 4312 CrossRef , and references therein.
- A. Juris, V. Balzani, F. Barigeletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85 CrossRef CAS.
- A. B. P. Lever, Inorg. Chem., 1990, 29, 1271 CrossRef CAS.
- (a) C. O. Dietrich-Buchecker, P. A. Marnot, J.-P. Sauvage, J.-P. Kintinger and P. Maltese, New J. Chem., 1984, 8, 573 Search PubMed; (b) J. R. Kirchhoff, D. R. McMillin, P. A. Marnot and J.-P. Sauvage, J. Am. Chem. Soc., 1985, 107, 1138 CrossRef CAS.
- J. P. Sauvage and M. Ward, Inorg. Chem., 1991, 30, 3869 CrossRef CAS.
- (a) S. P. Argent, H. Adams, T. Riis-Johannessen, J. C. Jeffery, L. P. Harding, W. Clegg, R. W. Harrington and M. D. Ward, Dalton Trans., 2006, 4996 RSC; (b) C. Piguet, G. Bernardinelli, A. F. Williams and B. Bocquet, Angew. Chem., Int. Ed. Engl., 1995, 34, 582 CrossRef CAS.
- (a) P. K. K. Ho, K. K. Cheung and C.-M. Che, Chem. Commun., 1996, 1197 RSC; (b) A. C. G. Hotze, B. M. Kariuki and M. J. Hannon, Angew. Chem., Int. Ed., 2006, 45, 4839 CrossRef; (c) S. L. Larson, S. M. Hendrickson, S. Ferrere, D. L. Derr and C. M. Elliott, J. Am. Chem. Soc., 1995, 117, 5881 CrossRef CAS; (d) J. D. Crane and J.-P. Sauvage, New J. Chem., 1992, 16, 649 Search PubMed; (e) M. H. W. Lam, S. T. C. Cheung, K.-M. Fung and W.-T. Wong, Inorg. Chem., 1997, 36, 4618 CrossRef CAS; (f) N. C. Fletcher, R. T. Brown and A. P. Doherty, Inorg. Chem., 2006, 45, 6132 CrossRef CAS; (g) S. Torelli, S. Delahaye, A. Hauser, G. Bernardinelli and C. Piguet, Chem.–Eur. J., 2004, 10, 3503 CrossRef CAS; (h) G. I. Pascu, A. C. G. Hotze, C. Sanchez Cano, M. M. Kariuki and M. J. Hannon, Angew. Chem., Int. Ed., 2007, 46, 4374 CrossRef CAS.
- (a) J.-P. Collin, S. Guillerez, J.-P. Sauvage, F. Barigelletti, L. De Cola, L. Flamigni and V. Balzani, Inorg. Chem., 1991, 30, 4230 CrossRef CAS; (b) W. Spahni and G. Calzaferri, Helv. Chim. Acta, 1984, 67, 450 CrossRef CAS.
- G. M. Sheldrick, SADABS version 2.10, University of Göttingen, 2003 Search PubMed.
- G. M. Sheldrick, SHELXS-97 and SHELXL-97, University of Göttingen, 1997 Search PubMed.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2008 |