Nanoporous materials based on heteroleptic bilayers built up from bisphosphonium, p-sulfonatocalix[4]arene ions†‡
Mohamed
Makha
*a,
Yatimah
Alias
b,
Colin L.
Raston
*a and
Alexandre N.
Sobolev
a
aSchool of Biomedical, Biomolecular and Chemical Sciences, University of Western Australia, 35 Stirling Hwy, Crawley, W.A. 6009, Australia. E-mail: mmakha@chem.uwa.edu.au; clraston@chem.uwa.edu.au; Fax: (+61) 86488 1572; Tel: (+61) 86488 1005
bDepartment of Chemistry, Faculty of Science, University of Malaya, 50603, Kuala Lumpur, Malaysia
First published on 17th September 2007
Abstract
Sulfonatocalix[4]arene, 1, and bisphosphonium cation, 2, form extensive self-assembled arrays in the presence of lanthanide metal cations, building up new materials based on both types of ions. Systematic studies using the combinatorial approach reproducibly yielded three novel structures based on these ions. Complexes 3––5, were prepared by varying the ratio of the components, organic and metal ions, in addition to varying the pH and ionic strength of the solutions. The structural motifs in these complexes were found to be lanthanide specific and the complexes were obtained from a 1 : 1 ratio of the organic ions in the presence of an excess of lanthanide metal cations at near neutral pH. Two structures based on bilayers, 3 and 4, are accessible, differing in the ratio of the organic components, 1 and 2, 7 : 6 and 1 : 2, respectively. Complexes 3 and 4 have a supersized bilayer with 3 featuring a 2D channel system associated with a scaffolding role of the bisphosphonium cation. The structural motif in complex 5 is devoid of bilayer arrangements, instead taking on a compact structure.
Introduction
Inorganic and metal-organic porous materials have found wide applications in many fields including catalysis, adsorption, electronics, and environmental technology because of their high surface area coupled with novel physical and chemical properties.1 In contrast, the design and construction of organic materials geared towards specific functions is less documented,2 and the synthesis of such materials represent a considerable synthetic challenge. Functional organic materials have exciting potential in the field of separation science, for example, as molecular sieves or as chromatographic stationary phases. Access to the latter may be possible by varying the hydrophobic/hydrophilic properties of the surface of channels within the solid. The extreme case is lining the surface with charged head groups thereby creating ion-channels. Electrostatic effects are important in ion-chromatography or charged-based filtering, which occurs in nature in the kidney where the negatively charged albumin is prevented from passing from the blood to the primary urine by the presence of negatively charged pores in the basal membrane.3Molecular assemblies can contain large channels or voids as suitable environments for controlled chemical transformations, i.e. ‘nanoreactors’.4 Further applications of organic materials abound in separation science. Specific pore sizes are in demand for nanofiltration of gaseous mixtures and in size-exclusion chromatography. New advances in the replacement of gels, membranes and sieves of random pore structure with poor guest specificity, are of interest in separation science, and catalysis.5
p-Sulfonatocalix[4]arene, 1 (shown in its protonated form in Scheme 1), is renowned for the ‘bilayer’ arrangement or ‘clay’ structure consisting of an up and down organization of the calixarenes creating a hydrophobic and hydrophilic layered domains. It is a versatile tecton in supramolecular chemistry forming a diverse range of structural motifs. Highlights of the structures include nanoscale spheroids as potential inorganic viral mimics,6 tubules7 and most recently a ‘zeolite-like’ material formed from helically arranged chains of the supermolecule [(Co(diHOsar))∩(1 – 4H+)].8 The tetraphenylphosphonium cation can self assembly, most notably through the triphenyl embrace, as described by Dance et al.9 In conjunction with 1, and also the higher more flexible sulfonated calixarenes, it can form ionic material based on the two components, as well as metal ions. In some cases there are well defined channels which are occupied by water.10 In a recent review Barbour highlighted the need for demonstrating porosity in organic solids through guest exchange.11
 | ||
| Scheme 1 Reagents and conditions for the synthesis of 3––5. The ratio of organic ions 1 and 2 are set to 1 : 1 with a threefold excess of Ln3+ metal cation. | ||
In pursuing the preparation of self assembled functional porous materials, we have investigated the interaction between 1 and a bisphosphonium cation, 1,4-bis(triphenylphosphoniomethyl)benzene, 2. The bisphosphonium cation can interact both exo and/or endo with respect to the cavity of the calixarene involving one phenyl ring, as established for tetraphenylphosphonium complexes of the same calixarene.12 We show that under specific conditions, and in association with certain lanthanide ions, it forms remarkably porous materials, as well as a more compact complex. The porous material is based on a bilayer comprised of 1 and exo-cavity cations of 2, with additional endo-cavity cations of 2 as scaffolding between bilayers. The exo-cavity cations of 2 embedded in the bilayer imparts more hydrophobic character at the two divergent surfaces of the bilayer relative to the ubiquitous bilayer based exclusively on 1.13
Complexes 3––5, Scheme 1, were prepared by varying the ratio of the components, organic and metal ions, in addition to varying the pH of the solutions. The structural motifs in these complexes were found to be lanthanide specific and accessed from a 1 : 1 ratio of the organic ions in the presence of an excess of lanthanide metal cations at near neutral pH. Mapping out the formation of the ternary complexes involved a combinatorial approach by varying the ratios of the components at different pH, as well as ionic strength of the solutions (see ESI‡).
Results and discussion
Synthesis of host–guest complexes
The host–guests complexes 3––5 were synthesised by slow evaporation of an equimolar aqueous solutions of sodium salt of p-sulfonatocalix[4]arene 1 and the phosphonium guest cation 2 in the presence of lanthanides metal chloride salts. Hot aqueous solutions (pH ∼6) of the sodium salt of p-sulfonatocalix[4]arene 1 were combined with hot aqueous solutions of phosphonium cation 2 in the presence of a threefold excess of the lanthanides. A series of experiments was attempted, in which organic anion–cation ratios of 1 : 1 and 1 : 2 were employed with various lanthanide metal at different molar ratios and pH adjustments. On combining first 1 with 2, the mixtures became turbid, and after addition of the lanthanide, the mixtures were heated until partial dissolution occurred. The solutions were then filtered hot and the supernatant allowed to slowly cool, and on further standing (2–5 days), X-ray quality colourless crystals were formed.Two structures based on bilayers, 3 and 4, are accessible. They differ in the ratio of the organic components, 1 and 2, 7 : 6 and 2 : 1, respectively. The bilayers are distinctly different, the major difference being the arrangement of the cations 2 which act as scaffolds between bilayers whereby a phenyl group attached to one phosphorus centre resides in the cavity of a calixarerne of one bilayer and the phenyl group of the other phosphorus centre resides in the cavity of a calixarene from the adjacent bilayer. The structure in complex 5 has the ratio of calixarene to the phosphonium of 1 : 2 in a compact structure devoid of the bilayer arrangement.
Crystal structure of complex 3
The complex 3 crystallises in the triclinic space groupP![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and of composition [2]7[(1)6 – 24H+]·(Yb3+)2.33·53H2O, with the asymmetric unit containing three calixarenes and 3.5 bisphosphonium cations. Three of the cations are in a ‘cis’ configuration with both the triphenyl phosphine moieties on the same side of the plane of the central aromatic ring. The half cation arises from a ‘trans’ cation residing on a centre of inversion and is the one that spans two bilayers. Aquated ytterbium and a large number of disordered water molecules are also present, and charge balance is possible through the presence of an oxonium ion or more likely the protonation of one sulfonic acid group on the calixarene, rather than having four sulfonate groups. This behaviour has been reported for other supramolecular arrays containing sulfonated calixarenes.14 The phenyl groups bound within calixarene cavities for the ‘trans’ cation is associated with CH⋯π interactions, with the aromatic ring centroid⋯H distances at 2.4 and 2.7 Å (centroid⋯C distances of 3.29 and 3.54 Å).
and of composition [2]7[(1)6 – 24H+]·(Yb3+)2.33·53H2O, with the asymmetric unit containing three calixarenes and 3.5 bisphosphonium cations. Three of the cations are in a ‘cis’ configuration with both the triphenyl phosphine moieties on the same side of the plane of the central aromatic ring. The half cation arises from a ‘trans’ cation residing on a centre of inversion and is the one that spans two bilayers. Aquated ytterbium and a large number of disordered water molecules are also present, and charge balance is possible through the presence of an oxonium ion or more likely the protonation of one sulfonic acid group on the calixarene, rather than having four sulfonate groups. This behaviour has been reported for other supramolecular arrays containing sulfonated calixarenes.14 The phenyl groups bound within calixarene cavities for the ‘trans’ cation is associated with CH⋯π interactions, with the aromatic ring centroid⋯H distances at 2.4 and 2.7 Å (centroid⋯C distances of 3.29 and 3.54 Å).
In turning to the interplay of 1 and 2 within the bilayers in 3, the ‘cis’ arrangement of 2 is associated with the pendant arms embedded in the bilayer, Fig. 1. The net effect is that the surface of the bilayers have a large hydrophobic area relative to the usual compact arrangement of bilayers based exclusively on sulfonated calix[4]arene.14 Here the hydrophobic areas are the cavities of the calixarenes. While the bilayers in the present structure have the same cavities, some of them are occupied by phenyl groups of the ‘trans’ cations, Fig. 2. The other hydrophobic areas exposed on the surface of the bilayers are the central rings of cations 2, with their triphenylphosphine groups directed into the bilayers, Fig. 2. The cavities associated with binding of phenyl rings of the trans cation represents 1/3 of all the cavities on each side of a bilayer. The other cavities form a grid which is shown in Fig. 1. Within the bilayers the calixarenes are shrouded by the bisphosphonium cations and held by C–H⋯π interactions (short contact C⋯π distance is 3.29 Å) and π ⋯π-stacking (short contact distance 3.93 Å), Fig. 1.
 | ||
| Fig. 1 Projection of 3. (a) The upper portion of the bilayer (normal to the bilayer plane). (b) A cross section of the bilayer showing the calixarenes shrouded by the bisphosphonium cations coloured in green for clarity. | ||
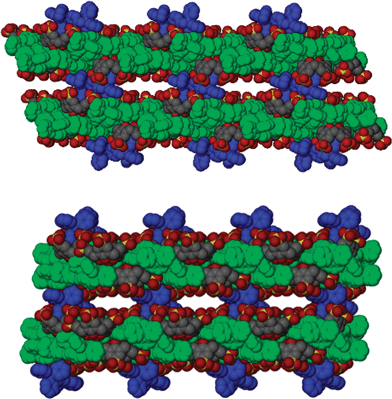 | ||
| Fig. 2 Projections along the a and b axes of the 2D channel system in complex 3: aquated ytterbium(III) cations and water molecules are omitted for clarity. The blue cations span the bilayers while the green cations are embedded within the bilayer. | ||
Cations of 2 acting as scaffolds between bilayers are inclined relative to the plane of the bilayers with the angle between the P⋯P vector and the bilayer plane being at 80°. The distance from the centre of one bilayer to the centre of the neighbouring bilayer is 18 Å, with the thickness of the bilayer estimated at 9.5 Å and the thickness of the space between them is approximately 8.5 Å. This space contains trans cations of 2. The remainder of the space is arranged into channels ranging in width from 4.0 to 24 Å which is filled with disordered water molecules and aquated ytterbium cations, [Yb(H2O)7]3+, Fig. 2. There is no coordination of the Yb3+ ions with any of the sulfonate oxygen atoms. These aquated ytterbium cations effectively fill the channels through extensive hydrogen bonding to calixarene sulfonate groups and water molecules.
Crystal structure of complex 4
The complex has a similar overall structure relative to complex 3, crystallising in the same space groupP, but with a different composition, [2][(1)2 – 8H+]·[Er3+(H2O)8]2·22H2O. Complex 4 was prepared under similar conditions as for 3 with the same ratio of components but erbium(III) chloride was used as the metal salt. The asymmetric unit contains two calixarenes and one bisphosphonium cations in a trans-configuration. Aquated erbium(III) and a large number of disordered water molecules are also present, and the electronic neutrality is achieved. The calixarenes interact through unusual and rather short CH⋯π interactions, at 2.65 Å, for a methylene bridging proton (centroid⋯C distance at 3.54 Å) to the centroid of the aromatic ring of neighbouring calixarene, Fig. 3.
![The asymmetric unit in complex 5 showing the intercalation of a [Ph3PCH2C6H4CH2PPh3]2+ dication between two calixarenes.](/image/article/2008/NJ/b706640f/b706640f-f3.gif) | ||
| Fig. 3 The asymmetric unit in complex 5 showing the intercalation of a [Ph3PCH2C6H4CH2PPh3]2+ dication between two calixarenes. | ||
Within the bilayers individual layers of calixarenes assemble into pairs held by intermolecular CH⋯π interactions (short contact C⋯π is 3.54 Å) in addition to a electrostatic stabilisation by a bisphosphonium cation. The erbium(III) cations are nona-aquated with a second coordination sphere to the sulfonate groups and remaining water molecules forming part of the hydrophilic domain with extensive hydrogen bonding (O⋯O separations: 2.60(3)–2.99 (3)Å).
The structure features the intercalation of the bisphosphonium between the layers of the calixarenes, at the interface of their hydroxyl and phenolate groups, Fig. 4. Thus the electrostatic attraction of the negatively charged lower rim of the calixarenes with the organic cations expands the bilayer arrangement forming supersized hydrophobic bilayers as has been seen for the tetraphenylphosphonium cation.13 The bisphosphonium cation forms layer arrays through continuous phenyl embraces9,15 involving CH⋯π interactions (short contact C⋯π is 3.49 Å), Fig. 4.
 | ||
| Fig. 4 (a) The mode of phenyl embraces within the bisphosphonium layer, and (b) the overall extended structure of 4 showing the expanded bilayer structure through the inclusion of the bisphosphonium cations and aquated erbium(III) cations between these layers. | ||
Crystal structure of complex 5
The complex crystallises in the monoclinic space groupP21/n, with the asymmetric unit containing one calixarene and two bisphosphonium cations in a cis-configuration and running orthogonal to each other. Aquated sodium and five disordered water molecules are also present, with the quality of the data preventing location of the hydrogen atoms in these molecules, as for complexes 3 and 4. Electronic neutrality of the system necessitates that one of the hydroxyls of the calixarene is deprotonated. One of the bisphosphonium cations undergoes inclusion within the cavity of the calixarene through a phenyl group from one end of the dication, with a weak CH⋯π interaction with the aromatic ring of the calixarene of 3.0 Å (centroid⋯C distance of 3.76 Å). The other end of the same cation is also engaged in CH⋯π interactions exo to the cavity of two other calixarenes within the layer, which are separated by the third phenyl form the same end of this cation, shortest contact CH⋯π of 3.1 Å (centroid⋯C distance of 4.01 Å), Fig. 5. The other phosphonium is engaged in the usual phenyl embraces with other phosphoniums to form a linear array as has been seen in previous structures of sulfonatocalixarenes with tetraphenylphosphonium cations.13![The asymmetric unit in complex 5 showing the inclusion and intercalation of [Ph3PCH2C6H4CH2PPh3]2+ dications in the structure induced by lanthanum or caesium metal cations, which are not included in the structure (water molecules and aquated sodium are omitted for clarity).](/image/article/2008/NJ/b706640f/b706640f-f5.gif) | ||
| Fig. 5 The asymmetric unit in complex 5 showing the inclusion and intercalation of [Ph3PCH2C6H4CH2PPh3]2+ dications in the structure induced by lanthanum or caesium metal cations, which are not included in the structure (water molecules and aquated sodium are omitted for clarity). | ||
The structure in complex 5 is not heavily hydrated, unlike the structures of 3 and 4, and the packing of the {calixarene∩[Ph3PCH2C6H4CH2PPh3]2+}·[(Ph3PCH2C6H4CH2PPh3)]2+ is more complex as the calixarenes do not assemble into the usual bilayer arrangement. The calixarenes are well separated and each is surrounded by the bisphosphonium cations. Essentially the structure is comprised of one bisphosphonium having one half residing within the cavity viaphenyl inclusion while the other half is protruding into a space separating the calixarenes inverted in the opposite direction. The orientation of the unbound bisphosphonium is orthogonal to the former forming a continuous array which dimerises with a similar chain running next to it, Fig. 6.
![Packing diagram of the compact structure in complex 5 showing the included [Ph3PCH2C6H4CH2PPh3]2+ cation in blue and the intercalated cations in green.](/image/article/2008/NJ/b706640f/b706640f-f6.gif) | ||
| Fig. 6 Packing diagram of the compact structure in complex 5 showing the included [Ph3PCH2C6H4CH2PPh3]2+ cation in blue and the intercalated cations in green. | ||
Given the open channels in complex 3, attempts were made to replace the aquated ytterbium ions, and included water molecules. Dipping crystals of 3 in 2 M HCl solution at room temperature for 30 min resulted in the crystals become slightly pitted but nevertheless they retained their crystalline morphology. After discarding the supernatant and washing the crystals with water, an aqueous solution of potassium triiodide was added to the crystals, which turned reddish upon the uptake the iodide. ICP analysis of the supernatant showed the presence of lanthanides and titration established the uptake of iodine as 30%.
TGA of complexes 3 and 4 are consistent with the degree of hydration while powder XRD for all the complexes could not be reproduced presumably because of loss of water included molecules (ESI‡).
Conclusion
We have shown, for the first time, that sulfonated calix[4]arene complexes with a bisphosphonium cation establish a structural diversity, either a compact arrangement or bilayer arrangements. We also show that the formation of these complexes requires close attention to conditions and the type of lanthanide ion, with the latter not necessarily ending up in the complex. These findings give insight into the use of organic cations and anions, and perturbations thereof in building up new materials in general. Moreover, the ability to remove the lanthanide ions from one of the bilayer structures, and the ability to then include iodine, suggests that the organic ions have a potential in building up materials with a range of inclusion properties.Experimental
p-Sulfonatocalix[4]arene 16 and [Ph3PCH2C6H4CH2PPh3]2+·2Cl–,17 were prepared according to literature procedures. Lanthanum, ytterbium and erbium trichloride hexahydrates were purchased from Sigma-Aldrich and used as received as for all other lanthanides used in this investigation.General procedure for synthesis of complexes 3––5
A hot aqueous solution of YbCl3·6H2O (13 mg, 3.35 × 10–5 mol) (0.5 ml) was added to a hot solution of [Ph3PCH2C6H4CH2PPh3]2+·2Cl– (8.2 mg, 1.1 × 10–5 mol) and sodium p-sulfonatocalix[4]arene (9.2 mg, 1.1 × 10–5 mol) in water (2 ml). The hot solutions of the mixture (pH ∼6) were filtered and left to cool and slowly evaporate, and upon standing for a week, afforded single crystals.X-Ray crystallography
The X-ray diffracted intensities were measured from single crystals at low temperature on an Oxford Diffraction Xcalibur-S (compounds 3 and 5) and Bruker ASX SMART-1000 (compound 4) diffractometers using monochromated Mo-Kα (λ = 0.71073 Å). Data were corrected for Lorentz and polarization effects and absorption correction applied using multiple symmetry equivalent reflections. The structures were solved by the direct method and refined on F2 using the SHELX-97 crystallographic package, with diagrams prepared using the X-Seed interface.18 A full matrix least-squares refinement procedure was used, minimizing w(Fo2 – Fc2), with w = [σ2(Fo2) + (AP)2 + BP]–1, where P = (Fo2 + 2Fc2)/3. Agreement factors (R = ∑||Fo| – |Fc||/∑|Fo|, wR2 = {∑[w(Fo2 – Fc2)2]/∑[w(Fo2)2]}1/2 and GOF = {∑[w(Fo2 – Fc2)2]/(n – p)}1/2 are cited, where n is the number of reflections and p the total number of parameters refined). All non-hydrogen of the non-disordered groups was refined anisotropically, while the disordered non-hydrogen atoms were refined isotropically. The positions of the hydrogen atoms partly were localized from difference Fourier maps, partly calculated from geometrical considerations. The hydrogen atomic parameters were constrained to the bonded atoms during the refinement.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 203.47, F(000) = 5297 e, triclinic, P, Z = 1, T = 153(2) K, a = 21.256(5), b = 23.536(6), c = 31.090(8) Å, α = 83.291(4), β = 71.885(4), γ = 78.756(4)°, V = 14473(6) Å3; Dc = 1.171 g cm–3; sin(θ/λ)max = 0.5946; N(unique) = 47780 (merged from 90884, Rint = 0.1425, Rsig = 0.3554), No (I > 2σ(I)) = 14 495; R = 0.2119, wR2 = 0.4677 (A, B = 0.15, 210.0), GOF = 1.118; |Δρmax| = 1.2(2) e Å–3.
, Z = 2, T = 100(2) K, a = 15.570(2), b = 21.425(3), c = 22.794(4) Å, α = 75.66(2), β = 84.20(1), γ = 77.09(1)°, V = 7172(2) Å3; Dc = 1.449 g cm–3; sin(θ/λ)max = 0.6361; N(unique) = 27624 (merged from 114182, Rint = 0.0553, Rsig = 0.1147), No (I > 2σ(I)) = 14160; R = 0.1508, wR2 = 0.3791 (A, B = 0.15, 150.0), GOF = 1.034; |Δρmax| = 4.7(2) e Å–3.
531.4(7) Å3; Dc = 1.382 g cm–3; sin(θ/λ)max = 0.6486; N(unique) = 21959 (merged from 174690, Rint = 0.105, Rsig = 0.161), No (I > 2σ(I)) = 12388; R = 0.1012, wR2 = 0.1890 (A, B = 0.092, 0), GOF = 1.122; |Δρmax| = 0.97(7) e Å–3.
203.47, F(000) = 5297 e, triclinic, P, Z = 1, T = 153(2) K, a = 21.256(5), b = 23.536(6), c = 31.090(8) Å, α = 83.291(4), β = 71.885(4), γ = 78.756(4)°, V = 14473(6) Å3; Dc = 1.171 g cm–3; sin(θ/λ)max = 0.5946; N(unique) = 47780 (merged from 90884, Rint = 0.1425, Rsig = 0.3554), No (I > 2σ(I)) = 14 495; R = 0.2119, wR2 = 0.4677 (A, B = 0.15, 210.0), GOF = 1.118; |Δρmax| = 1.2(2) e Å–3.
, Z = 2, T = 100(2) K, a = 15.570(2), b = 21.425(3), c = 22.794(4) Å, α = 75.66(2), β = 84.20(1), γ = 77.09(1)°, V = 7172(2) Å3; Dc = 1.449 g cm–3; sin(θ/λ)max = 0.6361; N(unique) = 27624 (merged from 114182, Rint = 0.0553, Rsig = 0.1147), No (I > 2σ(I)) = 14160; R = 0.1508, wR2 = 0.3791 (A, B = 0.15, 150.0), GOF = 1.034; |Δρmax| = 4.7(2) e Å–3.
531.4(7) Å3; Dc = 1.382 g cm–3; sin(θ/λ)max = 0.6486; N(unique) = 21959 (merged from 174690, Rint = 0.105, Rsig = 0.161), No (I > 2σ(I)) = 12388; R = 0.1012, wR2 = 0.1890 (A, B = 0.092, 0), GOF = 1.122; |Δρmax| = 0.97(7) e Å–3.
Because of the solvent dependency of the crystals, three additional unit cell collections were performed on randomly selected crystals to confirm the content of the bulk material for each complex. As the crystals of the complexes were solvent dependent, microanalyses were not performed.
CCDC reference numbers 642644–642646.
For crystallographic data in CIF or other electronic format see DOI: 10.1039/b706640f
Acknowledgements
We thank the Australian Research Council and the University of Western Australia for supporting this work.References
- R. Garcia-Zarracino and H. Höpfl, J. Am. Chem. Soc., 2005, 127, 3120 CrossRef CAS; O. M. Yaghi, G. Li and H. Li, Nature, 1995, 378, 703 CrossRef CAS; H. Li, M. Eddaoudi, M. O’Keeffe and O. M. Yaghi, Nature, 1999, 402, 276 CrossRef CAS; S. S.-Y. Chui, S. M.-F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148 CrossRef CAS; J. Lu, A. Mondal, B. Moulton and M. J. Zaworotko, Angew. Chem., Int. Ed., 2001, 40, 2113 CrossRef CAS; J. Lu, A. Mondal, B. Moulton and M. J. Zaworotko, Angew. Chem., Int. Ed., 2001, 113, 2171 CrossRef; M. J. Zaworotko, Nature, 1997, 386, 220 CrossRef CAS; V. Liu, V. Ch. Kravtsov, R. Larsen and M. Eddaoudi, Chem. Commun., 2006, 1488 RSC.
- S. J. Dalgarno, G. W. V. Cave and J. L. Atwood, Angew. Chem., Int. Ed., 2006, 45, 570 CrossRef CAS; S. J. Dalgarno, G. W. V. Cave and J. L. Atwood, Angew. Chem., Int. Ed., 2006, 118, 584 CrossRef; M. R. Ghadiri, J. R. Granja, R. A. Milligan, D. E. McRee and N. Khazanovich, Nature, 1993, 366, 324 CrossRef CAS; C. Valéry, M. Paternostre, B. Robert, T. Gulik-Krzywicki, T. Narayanan, J.-C. Dedieu, M.-L. Torres, R. Cherif-Cheikh, P. Calvo and F. Artzner, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10258 CrossRef CAS; V. Semetey, C. Didierjean, J.-P. Briand, A. Aubry and G. Guichard, Angew. Chem., Int. Ed., 2002, 41, 1895 CrossRef CAS; V. Semetey, C. Didierjean, J.-P. Briand, A. Aubey and G. Guichard, Angew. Chem., 2002, 114, 1975 CrossRef.
- L. Bent-Hansen, B. Feldt-Rasmussen, A. Kverneland and T. Deckert, Diabetologia, 1993, 36, 361 CrossRef CAS; T. Deckert, A. Kofoed-Enevoldsen, P. Vidal, K. Norgaard, H. B. Andreasen and B. Feldt-Rasmussen, Diabetologia, 1993, 36, 244 CrossRef CAS; A. Kverneland, B. Feldt-Rasmussen, P. Vidal, B. Welinder, L. Bent-Hansen, U. Soegaard and T. Deckert, Diabetologia, 1986, 29, 634 CrossRef CAS; H. F. G. Rennke, R. S. Cotran and M. A. Venkatachalam, J. Cell Biol., 1975, 67, 638 CrossRef CAS.
- D. M. Vriezema, M. C. Aragones, J. A. A. W. Elemans, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem. Rev., 2005, 105, 1445 CrossRef CAS.
- A. Favre-Reguillon, G. Lebuzit, J. Fooz, A. Guy, M. Draye and M. Lemaire, Ind. Eng. Chem. Res., 2003, 42, 5900 CrossRef CAS; B. Van Der Bruggen, L. Lejon and C. Vandecasteele, Environ. Sci. Technol., 2003, 37, 3733 CrossRef CAS; A. Srivastava, O. N. Srivastava, S. Talapatra, R. Vajtai and P. M. Ajayan, Nat. Mater., 2004, 3, 610 CrossRef CAS.
- J. L. Atwood, L. J. Barbour, S. J. Dalgarno, M. J. Hardie, C. L. Raston and H. R. Webb, J. Am. Chem. Soc., 2004, 126, 13170 CrossRef CAS; T. Douglas and M. Young, Nature, 1998, 393, 152 CrossRef CAS.
- G. W. Orr, L. J. Barbour and J. L. Atwood, Science, 1999, 285, 1049 CrossRef.
- C. B. Smith, L. J. Barbour, M. Makha, C. L. Raston and A. N. Sobolev, Chem. Commun., 2006, 950 RSC.
- I. Dance and M. J. Scudder, J. Chem. Soc., Dalton Trans., 1998, 3155–3165 RSC.
- M. Makha, C. L. Raston, A. N. Sobolev and A. H. White, Chem. Commun., 2005, 1962 RSC; M. Makha, A. N. Sobolev and C. L. Raston, Chem. Commun., 2006, 511 RSC; C. B. Smith, L. J. Barbour, M. Makha, C. L. Raston and A. N. Sobolev, New J. Chem., 2006, 991 RSC.
- L. J. Barbour, Chem. Commun., 2006, 1163 RSC.
- M. Makha, C. L. Raston, A. N. Sobolev and A. H. White, Chem. Commun., 2004, 1066 RSC.
- M. Makha, C. L. Raston and A. N. Sobolev, Chem. Commun., 2006, 57 RSC.
- For example, see: M. J. Hardie and C. L. Raston, J. Chem. Soc., Dalton Trans., 2000, 2483 Search PubMed; C. B. Smith, L. J. Barbour, M. Makha, C. L. Raston and A. N. Sobolev, New J. Chem., 2006, 30, 991–996 RSC; S. J. Dalgarno, M. J. Hardie, J. E. Warren and C. L. Raston, Dalton Trans., 2004, 2413 RSC; S. J. Dalgarno and C. L. Raston, Chem. Commun., 2002, 2216 RSC; J. L. Atwood, L. J. Barbour, M. J. Hardie and C. L. Raston, Coord. Chem. Rev., 2001, 222, 3 RSC.
- G. Ciani, G. D’Alfonso, P. Romiti, A. Sironi and M. Freni, J. Organomet. Chem., 1983, 244, C27 CrossRef CAS; B. Larue, L.-T. Tran, D. Luneau and C. Reber, Can. J. Chem., 2003, 81, 1168 CrossRef CAS.
- M. Makha and C. L. Raston, Chem. Commun., 2001, 2470 RSC.
- C. Stammel, R. Froehlich, C. Wolff, H. Wenck, A. de Meijere and J. Mattay, Eur. J. Org. Chem., 1999, 1709 CrossRef CAS; D. C. Harrowven, M. I. T. Nunn and D. R. Fenwick, Tetrahedron Lett., 2002, 43, 7345 CrossRef CAS; Z.-Q. Liu, Q. Fang, D.-X. Cao, D. Wang and G.-B. Xu, Org. Lett., 2004, 6, 2933 CrossRef CAS.
- G. M. Sheldrick, SHELX-97: Structure solution and refinement programs, University of Göttingen, 1997 Search PubMed; Bruker SMART, SAINT, SADABS and SHELXTL, v5.1, 1997, Bruker AXS Inc., Madison, WI, USA; L. J. Barbour, J. Supramol. Chem., 2001, 1, 189 Search PubMed; J. L. Atwood and L. J. Barbour, Cryst. Growth Des., 2003, 3, 3 Search PubMed.
Footnotes |
| † Dedicated to Prof. Jerry Atwood on the occasion of his 65 Birthday. |
| ‡ Electronic supplementary information (ESI) available: 1: Results of the combinatorial approach based on molar ratios of components: identification and characterisation of complexes 3–5. 2: Assessment of porosity in complex 3. 3: ICP records on HCl leaching of ytterbium. 4: TGA analysis on complex 3 with crystals with retained morphology at 600 °C. 5: 13C MAS, 31P MAS NMR and TGA data on complex 5. See DOI: 10.1039/b706640f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2008 |