DOI:
10.1039/B707448D
(Paper)
New J. Chem., 2008,
32, 105-114
Synthesis, structure, spectroscopic properties and cytotoxic effect of some thiosemicarbazone complexes of palladium†
Received
(in Montpellier, France)
17th May 2007
, Accepted 21st August 2007
First published on 6th September 2007
Abstract
Reaction of salicylaldehyde thiosemicarbazone (H2L1), 2-hydroxyacetophenone thiosemicarbazone (H2L2) and 2-hydroxynaphthaldehyde thiosemicarbazone (H2L3) (general abbreviation H2L, where H2 stands for the two dissociable protons, one phenolic proton and one hydrazinic proton) with Na2[PdCl4] affords a family of polymeric complexes of type [{Pd(L)}n]. Reaction of the polymeric species with two monodentate ligands (D), viz. triphenylphosphine (PPh3) and 4-picoline (pic), has yielded complexes of type [Pd(L)(D)]. These mixed-ligand complexes have also been obtained from reaction of the thiosemicarbazones with [Pd(PPh3)2Cl2] and [Pd(pic)2Cl2]. Crystal structures of [Pd(L1)(PPh3)] and [Pd(L2)(pic)] have been determined. The [Pd(L)(D)] complexes show characteristic 1H NMR spectra and intense absorptions in the visible and ultraviolet region. They also fluoresce in the visible region at ambient temperature. In vitro cytotoxicity screenings of the complexes along with four human clinical drugsviz.cisplatin, BCNU, 5-fluorouracil (5-FU) and hydroxyurea have been carried out in two human tumor cell lines, namely promyelocytic leukemia HL-60 and histiocytic lymphoma U-937. [Pd(L2)(PPh3)] shows the lowest IC50 value and is found to be much more cytotoxic than the reference anticancer drugs in both the cell lines. An apoptosis study in HL-60 with [Pd(L2)(PPh3)] confirms that at 10 µM concentration it induces apoptosis to a greater extent than cisplatin and camptothecin.
1. Introduction
The chemistry of transition metal complexes of thiosemicarbazones has received considerable attention, largely because of their bioinorganic relevance.1 Such complexes are of particular importance due to their potentially beneficial biological (viz. antibacterial, antimalarial, antiviral and antitumor) activities.2 Thiosemicarbazones usually bind to a metal ion, via dissociation of the hydrazinic proton, as bidentate N,S-donors, forming a five-membered chelate ring (1).3 If a third donor site (D) is incorporated in such ligands, linked to the carbonylic carbon via one or two intervening atoms, then D,N,S-tricoordination (2) normally takes place.4 In addition to displaying D,N,S-tricoordination, such ligands are also known to bridge a second metal ion through the sulfur.5 It has also been observed that salicylaldehyde thiosemicarbazone, in spite of having the phenolic oxygen as a potential third donor site, coordinates as a bidentate N,S-donor, forming a rather unusual four-membered chelate ring (3).6 This mode of binding (3) has also been displayed by some other thiosemicarbazones.7 Formation of such a chelate ring (3) by salicylaldehyde thiosemicarbazone, leaving some potential donor sites unused, has been utilized successfully in the synthesis of an interesting polynuclear complex.8 This variable binding mode of thiosemicarbazones has encouraged us to explore their coordination chemistry further,6,8,9 and the present work has originated from this exploration. Herein we have chosen three potentially tridentate thiosemicarbazones, viz. the thiosemicarbazones of salicylaldehyde, 2-hydroxyacetophenone and 2-hydroxynaphthaldehyde. These thiosemicarbazones are abbreviated in general as H2L, where H2 stands for the two dissociable protons, the phenolic proton and the hydrazinic proton. Individual ligand abbreviations are shown in 4. To interact with the chosen thiosemicarbazones, palladium has been selected as the metal. It may be mentioned here that, though the chemistry of palladium complexes of some thiosemicarbazones has been studied,10 that of the chosen thiosemicarbazones (4) appears to have remained unexplored. Reaction of the three selected thiosemicarbazones (4) has been carried out with three palladium starting materials, viz. Na2[PdCl4], [Pd(PPh3)2Cl2] and [Pd(pic)2Cl2] (pic = 4-picoline), which has afforded a family of interesting complexes containing the thiosemicarbazones coordinated in the tridentate fashion (5). The chemistry of these complexes is reported in this paper with special reference to their synthesis, structure and spectral properties, and cytotoxic activity in two human tumor cell lines.
Results and discussion
Syntheses
Reaction of the selected thiosemicarbazones (H2L, 4) has been carried out first with Na2[PdCl4] in refluxing ethanol in the presence of triethylamine to afford a family of polymeric complexes of type [{Pd(L)}n] in decent yields. Based on the compositions of these complexes, as well as the +2 oxidation state of palladium in them, the thiosemicarbazones are believed to coordinate the metal center in the expected dianionic O,N,S-fashion (5). The fourth coordination position on palladium in the Pd(L) fragment is assumed to be taken up by the sulfur of a coordinated thiosemicarbazone belonging to another Pd(L) fragment (6). This bridging action of the thiosemicarbazone sulfur, which is well documented in the literature,5 appears to be responsible for the formation of the polymeric species.
In order to explore the possibility of forming monomeric complexes by splitting the sulfur bridge in the [{Pd(L)}n] complexes, their reaction has been carried out with two monodentate ligands (D), viz. triphenylphosphine (PPh3) and 4-picoline (pic). From each of these reactions a monomeric complex of type [Pd(L)(D)] (D = PPh3 or pic) has been obtained. The same monomeric complexes have also been synthesized, in better yields, by reaction of the thiosemicarbazones (4) with [Pd(PPh3)2Cl2] and [Pd(pic)2Cl2] in refluxing ethanol in the presence of triethylamine. Synthetic methods for all the complexes are illustrated in Scheme 1. The observed elemental (C, H, N) analytical data of the synthesized complexes are found to be consistent with their compositions.
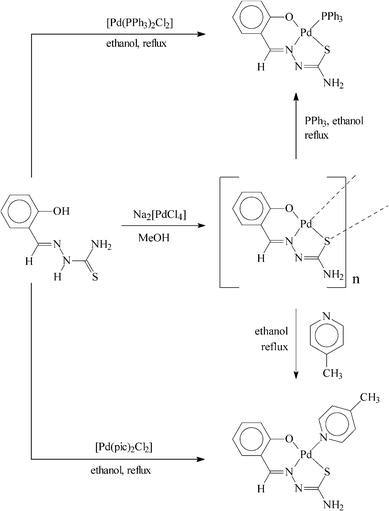 |
| | Scheme 1 | |
Crystal structures
In order to authenticate the coordination modes of the thiosemicarbazones in these monomeric [Pd(L)(D)] complexes, structures of a representative member from each family, viz. [Pd(L1)(PPh3)] and [Pd(L2)(pic)], have been determined by X-ray crystallography. The structure of the [Pd(L1)(PPh3)] complex is shown in Fig. 1 and some relevant bond parameters are listed in Table 1. The structure shows that the thiosemicarbazone ligand is coordinated to palladium in the expected tridentate fashion (5), forming six- and five-membered chelate rings with O–Pd–N and N–Pd–S bite angles of 92.82(7)° and 84.26(5)°, respectively. A triphenylphosphine is also coordinated to the metal center, which is trans to the nitrogen. Palladium is thus nested in an ONSP core, which is slightly distorted from ideal square planar geometry, as reflected in the bond parameters around the metal center. The Pd–O, Pd–N, Pd–S and Pd–P distances are all quite normal, as observed in structurally characterized complexes of palladium containing these bonds.10 Bond distances within the coordinated thiosemicarbazone are also usual.9c
![Structure of the [Pd(L1)(PPh3)] complex drawn at the 50% probability level.](/image/article/2008/NJ/b707448d/b707448d-f1.gif) |
| | Fig. 1 Structure of the [Pd(L1)(PPh3)] complex drawn at the 50% probability level. | |
Table 1 Selected bond distances and bond angles for [Pd(L1)(PPh3)] and [Pd(L2)(pic)]
| [Pd(L1)(PPh3)] |
| Bond distances/Å |
| Pd–N(3) |
2.0147(17) |
C(8)–N(1) |
1.354(3) |
| Pd–P |
2.2711(5) |
C(7)–N(3) |
1.290(3) |
| Pd–S |
2.2411(6) |
C(1)–O(1) |
1.306(3) |
| Pd–O(1) |
2.0157(16) |
N(2)–N(3) |
1.397(2) |
| |
|
C(8)–S |
1.748(2) |
| Bond angles/° |
| N(3)–Pd–P |
173.95(5) |
N(3)–Pd–O(1) |
92.82(7) |
| O(1)–Pd–S(1) |
177.07(5) |
N(3)–Pd–S |
84.26(5) |
| |
| [Pd(L2)(pic)] |
| Bond distances/Å |
| Pd(1)–N(3) |
1.9861(15) |
C(8)–N(1) |
1.359(3) |
| Pd(1)–N(4) |
2.0698(18) |
C(7)–N(3) |
1.309(2) |
| Pd(1)–S(1) |
2.2510(7) |
C(1)–O(1) |
1.321(2) |
| Pd(1)–O(1) |
1.9981(14) |
N(2)–N(3) |
1.397(2) |
| |
|
C(8)–S(1) |
1.736(2) |
| Bond angles/° |
| N(3)–Pd1–N(4) |
179.16(6) |
N(3)–Pd1–O(1) |
91.74(7) |
| O(1)–Pd1–S(1) |
175.69(5) |
N(3)–Pd1–S(1) |
85.04(5) |
The absence of any solvent of crystallization in the crystal lattice of [Pd(L1)(PPh3)] indicates the possible existence of non-covalent interaction(s) between the individual complex molecules. A closer look at the packing pattern of in the crystal reveals that hydrogen bonding interactions of two types, viz. intramolecular C–H⋯O and intermolecular N–H⋯N, are active in the lattice. A selected view of the hydrogen bonding interactions is shown in Fig. 2 and some hydrogen bond parameters are given in Table 2. One ortho C–H of a phenyl ring of the PPh3 is intramolecularly hydrogen bonded to the phenolate oxygen of the thiosemicarbazone ligand. One –NH2hydrogen of each complex molecule is hydrogen bonded to the nitrogen next to the azomethine nitrogen of a second molecule and thus a hydrogen bonded dimer is formed. Apart from these hydrogen bonding interactions, there also exist Pd–π interactions between the metal center of one complex molecule and the phenyl ring of the thiosemicarbazone ligand of a second complex molecule (Fig. S1, Table S1†). This extended Pd–π interaction seems to be responsible for holding the crystal together. It may be relevant to note here that such non-covalent interactions are of significant importance in molecular recognition processes as well as in crystal engineering.11
![Hydrogen bonding interactions in the lattice of [Pd(L1)(PPh3)].](/image/article/2008/NJ/b707448d/b707448d-f2.gif) |
| | Fig. 2
Hydrogen bonding interactions in the lattice of [Pd(L1)(PPh3)]. | |
Table 2
Hydrogen bond distances and bond angles for [Pd(L1)(PPh3)] and [Pd(L2)(pic)]a
| |
Bond distances/Å |
Bond angles/° |
|
Symmetry: i = 1 – x, 2 – y, 1 – z; ii = ½ – x, ½ + y, z.
|
| [Pd(L1)(PPh3)] |
| N1–H1A⋯N2i |
0.86 |
2.18 |
3.004(3) |
160 |
| C10–H10A⋯O1 |
0.93 |
2.35 |
3.025(4) |
130 |
| |
| [Pd(L2)(pic)] |
| N1–H2N⋯O1ii |
0.86 |
2.24 |
2.888(2) |
132 |
| C9–H9A⋯N2 |
0.96 |
2.25 |
2.621(3) |
102 |
| C10–H10⋯S1 |
0.93 |
2.66 |
3.246(2) |
122 |
| C14–H14⋯O1 |
0.93 |
2.26 |
2.829(3) |
119 |
The structure of the [Pd(L2)(pic)] complex (Fig. 3) shows that the thiosemicarbazone ligand is coordinated to palladium in the same tridentate fashion (5) as in [Pd(L1)(PPh3)], and a 4-picoline has taken up the fourth coordination site on the metal center. In this complex palladium has an ONSN coordination sphere around it. The observed Pd–N(pic) distance is normal,12 and the structural features in the Pd(L2) fragment are similar to those observed in the Pd(L1) fragment of the [Pd(L1)(PPh3)] complex. An examination of the packing pattern in this [Pd(L2)(pic)] crystal shows that three intramolecular hydrogen bonding interactions, viz. C–H⋯O, C–H⋯S and C–H⋯N, and one intermolecular hydrogen bonding interaction, viz. N–H⋯O, are active in the lattice (Fig. 4, Table 2). Two picolyl C–Hs (at the 2 and 6 positions) are intramolecularly hydrogen bonded to the phenolate oxygen and sulfur of the thiosemicarbazone ligand. One methyl C–H of the thiosemicarbazone is also intramolecularly hydrogen bonded to the nitrogen (which is not bound to the metal center) of the same ligand. One –NH2hydrogen of each complex molecule is hydrogen bonded to the phenolate oxygen of a second molecule; this intermolecular hydrogen bonding has propagated along the b-axis through the lattice. Besides these hydrogen bonding interactions, there also exist π–π interactions between the picolyl rings and also between the picolyl and phenyl rings of the thiosemicarbazone ligand (Fig. S2, Table S1†). The extended intermolecular interactions have been responsible for holding the crystal together.
![Structure of the [Pd(L2)(pic)] complex drawn at the 50% probability level.](/image/article/2008/NJ/b707448d/b707448d-f3.gif) |
| | Fig. 3 Structure of the [Pd(L2)(pic)] complex drawn at the 50% probability level. | |
![Hydrogen bonding interactions in the lattice of [Pd(L2)(pic)].](/image/article/2008/NJ/b707448d/b707448d-f4.gif) |
| | Fig. 4
Hydrogen bonding interactions in the lattice of [Pd(L2)(pic)]. | |
Spectral properties
1H NMR spectra of the [Pd(L)(PPh3)] complexes, recorded in CDCl3 solutions, show all the expected signals. Broad signals for the PPh3 ligands are observed within 7.4–7.8 ppm. In the [Pd(L1)(PPh3)] and [Pd(L3)(PPh3)] complexes the azomethine proton signal of the coordinated thiosemicarbazone is observed at 8.24 and 9.52 ppm, respectively. The methyl signal from the coordinated thiosemicarbazone in the [Pd(L2)(PPh3)] complex is observed at 2.89 ppm. The NH2 signal of the coordinated thiosemicarbazone is observed around 4.8 ppm. Most of the expected signals from the aryl fragment of the coordinated thiosemicarbazone are clearly observed in the aromatic region, while few could not be detected due to their overlap with other signals. In the 1H NMR spectra of the [Pd(L)(pic)] complexes, the methyl signal of the coordinated 4-picoline is observed at around 2.4 ppm and a distinct doublet is observed near 8.6 ppm, which is attributable to the α-proton of the picoline. The other expected doublet for the β-proton of the picoline is observed at 7.24 ppm in the [Pd(L1)(pic)] complex, but the same signal could not be detected in the other two complexes because of overlap problems. The 1H NMR spectral features for the coordinated thiosemicarbazones in these [Pd(L)(pic)] complexes are qualitatively similar to those observed for the corresponding [Pd(L)(PPh3)] complexes. The 1H NMR spectral data of the [Pd(L)(PPh3)] and [Pd(L)(pic)] complexes are therefore consistent with their compositions.
Infrared spectra of all the [Pd(L)(PPh3)] complexes show many vibrations of different intensities in the 1600–400 cm–1 region. Assignment of each individual band to a specific vibration has not been attempted. However, three strong bands displayed near 533, 694 and 743 cm–1 by these complexes, are attributed to the coordinated PPh3 ligands. Comparison with the spectrum of [Pd(PPh3)2Cl2] shows the presence of several new bands (e.g. near 811, 1130, 1229, 1285, 1346, 1562 and 1599 cm–1) in the spectra of the [Pd(L)(PPh3)] complexes, which must be due to the presence of the coordinated thiosemicarbazone ligand. Besides the absence of the three diagnostic bands for the PPh3 ligands near 533, 694 and 743 cm–1, infrared spectral properties of the [Pd(L)(pic)] complexes are qualitatively similar to those of the [Pd(L)(PPh3)] complexes.
The [Pd(L)(PPh3)] and [Pd(L)(pic)] complexes are soluble in common organic solvents such as methanol, ethanol, acetone, acetonitrile, dichloromethane, chloroform, etc., producing intense yellow solutions. Electronic spectra of these complexes have been recorded in dichloromethane solution. Spectral data are presented in Table 3 and a selected spectrum is shown in Fig. 5. All the mixed-ligand complexes show several intense absorptions in the visible and ultraviolet regions. The absorptions in the ultraviolet region are assignable to transitions within the ligand orbitals and those in the visible region are probably due to charge-transfer transitions involving both metal and ligand orbitals. To have an understanding of the nature of the transitions in the visible region, qualitative extended Hückel molecular orbital (EHMO) calculations have been performed13 on computer generated models of the [Pd(L)(PPh3)] and [Pd(L)(pic)] complexes. The results obtained are found to be similar for the three members of each group. A partial MO diagram for a representative [Pd(L)(PPh3)] complex is shown in Fig. 6 and that of a selected [Pd(L)(pic)] is deposited as Fig. S3.† The compositions of selected molecular orbitals are given in Table 4. In all the cases, the highest occupied molecular orbital (HOMO) has major contributions from the palladium d (dz2 and dxy) orbitals. The lowest unoccupied molecular orbital (LUMO) is delocalized almost entirely on the thiosemicarbazone ligand and is concentrated heavily on the imine (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) fragment. The lower energy absorption in these complexes may therefore be assigned to a charge-transfer transition taking place from the filled palladium d orbital (HOMO) to the vacant π*(imine) orbital of the coordinated thiosemicarbazone ligand (LUMO). Such charge-transfer transition in palladium complexes is, to our knowledge, rare.14 The electronic spectral properties, as well as features of the molecular orbitals, of the [Pd(L)(pic)] complexes are similar to those of the [Pd(L)(PPh3)] analogues.
N) fragment. The lower energy absorption in these complexes may therefore be assigned to a charge-transfer transition taking place from the filled palladium d orbital (HOMO) to the vacant π*(imine) orbital of the coordinated thiosemicarbazone ligand (LUMO). Such charge-transfer transition in palladium complexes is, to our knowledge, rare.14 The electronic spectral properties, as well as features of the molecular orbitals, of the [Pd(L)(pic)] complexes are similar to those of the [Pd(L)(PPh3)] analogues.
Table 3 Electronic spectral data in dichloromethane solution
| Compound |
λ
max/nm (ε/dm3 mol–1 cm–1)a |
|
s = shoulder.
|
| [Pd(L1)(PPh3)] |
398(9000), 342(11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300)s, 315(15500), 298(16100) 300)s, 315(15500), 298(16100) |
| [Pd(L2)(PPh3)] |
389(9800), 340(12600)s, 313(19000), 298(16800) |
| [Pd(L3)(PPh3)] |
430(9800), 408(9500), 389(5800)s, 348(12000), 317(19400), 261(41300) |
| [Pd(L1)(pic)] |
402(6000), 386(5400), 339(6600)s, 314(7800)s, 299(8700)s |
| [Pd(L2)(pic)] |
406(9000), 386(10100), 341(11800)s, 312(16000)s, 297(17500) |
| [Pd(L3)(pic)] |
432(3300), 409(3100), 389(2100)s, 348(3800)s, 326(4700), 258(14200) |
![Partial molecular orbital diagram of the [Pd(L1)(PPh3)] complex.](/image/article/2008/NJ/b707448d/b707448d-f6.gif) |
| | Fig. 6 Partial molecular orbital diagram of the [Pd(L1)(PPh3)] complex. | |
Table 4 Composition of selected molecular orbitals of the complexes
| Compound |
Contributing fragments |
% contribution of fragments to |
| HOMO |
LUMO |
|
The HOMO-1 has 88% contribution from the metal.
The HOMO-1 has 93% contribution from the metal.
|
| [Pd(L1)(PPh3)] |
Pd
|
89 |
— |
| L1 |
7 |
96 |
| |
|
(CN, 49) |
| [Pd(L2)(PPh3)] |
Pd
|
87 |
— |
| L2 |
8 |
93 |
| |
|
(CN, 44) |
| [Pd(L3)(PPh3)]a |
Pd
|
73 |
— |
| L3 |
21 |
97 |
| |
|
(CN, 30) |
| [Pd(L1)(pic)] |
Pd
|
92 |
— |
| L1 |
2 |
93 |
| |
|
(CN, 47) |
| pic |
— |
12 |
| [Pd(L2)(pic)] |
Pd
|
90 |
— |
| L2 |
6 |
91 |
| |
|
(CN, 26) |
| pic |
— |
44 |
| [Pd(L3)(pic)]b |
Pd
|
50 |
— |
| L3 |
41 |
95 |
| |
|
(CN, 23) |
| pic |
— |
— |
The intensities of the charge-transfer transitions in the visible region have tempted us to explore the luminescence properties of these complexes and this has been carried out in dichloromethane solution at ambient temperature (298 K). All six [Pd(L)(D)] complexes have been found to display emission in the visible region using an excitation wavelength of ∼350 nm and a selected spectrum is shown in Fig. 5. Quantum yields (φ) of these emissions have been evaluated (Table 5) with reference to [Ru(bpy)3]Cl2 (φ = 0.028 at 298 K).15 The observed quantum yields indicate that these [Pd(L)(PPh3)] and [Pd(L)(pic)] complexes are relatively poor emitters at room temperature.
Table 5 Emission spectral data for the complexes
| Compound |
Emission data |
|
λ
max/nm |
Quantum yield (φ) |
| Excitation |
Emissiona |
|
s = shoulder.
|
| [Pd(L1)(PPh3)] |
340 |
367 |
1.17 × 10–3 |
| 428 |
| [Pd(L2)(PPh3)] |
340 |
380s |
1.38 × 10–3 |
| 423 |
| [Pd(L3)(PPh3)] |
350 |
382 |
0.36 × 10–3 |
| 450 |
| [Pd(L1)(pic)] |
339 |
380 |
2.21 × 10–3 |
| 445s |
| [Pd(L2)(pic)] |
341 |
380 |
0.83 × 10–3 |
| 440 |
| 478s |
| [Pd(L3)(pic)] |
379 |
385 |
7.64 × 10–3 |
| 450 |
| 525 |
Cytotoxicity assay
The in vitrogrowth inhibitory effects of the [Pd(L)(D)] complexes were evaluated in two human cell lines viz. promyelocytic HL-60 and histiocytic lymphoma U-937. Four human clinical drugs, cisplatin, BCNU, 5-FU and hydroxyurea, were used as reference compounds for comparison. The activities are expressed in terms of IC50 value, the concentration of the respective compound required to reduce the cell survival fraction to 50% after 72 h of exposure. The lower the IC50 value is, the greater is the cytotoxicity. From the results presented in Table 6, it is found that the [Pd(L1)(PPh3)] and [Pd(L2)(PPh3)] complexes are much more active than all the clinical drugs, including cisplatin, in HL-60. In U-937, [Pd(L2)(PPh3)] is more active than cisplatin and other reference compounds, while the activity of [Pd(L1)(PPh3)] is higher than BCNU and hydroxyurea, comparable to 5-FU, and less than cisplatin. The [Pd(L3)(PPh3)] complex did not show any appreciable activity in the cell lines. In HL-60, the [Pd(L)(pic)] complexes are more active than BCNU, 5-FU and hydroxyurea. The activity of [Pd(L1)(pic)] is less than cisplatin, while that of the other two complexes is comparable to cisplatin. In U-937 the [Pd(L)(pic)] complexes are more active than BCNU and hydroxyurea, and less active than cisplatin and 5-FU.
Table 6 Cytotoxicity assay IC50 values (µM)a
| Compound |
HL-60
|
U-937
|
|
For details, see experimental.
|
| [Pd(L1)(PPh3)] |
2.5 |
4.8 |
| [Pd(L2)(PPh3)] |
0.6 |
1.3 |
| [Pd(L3)(PPh3)] |
203.0 |
231.6 |
| [Pd(L1)(pic)] |
16.2 |
7.3 |
| [Pd(L2)(pic)] |
7.1 |
6.6 |
| [Pd(L3)(pic)] |
6.5 |
7.7 |
|
Cisplatin
|
7.0 |
3.2 |
|
BCNU
|
30.5 |
12.3 |
| 5-FU |
266.0 |
4.7 |
|
Hydroxyurea
|
204.0 |
115.0 |
In vitro
apoptosis assay
Cell death may occur by one of two distinct mechanisms: necrosis or apoptosis. The most common and well-defined form of programmed cell death is apoptosis. During the past decade, cell biology as well as oncology research has focused on apoptosis. Hence, based on the results of our in vitro cytotoxicity assay in HL-60, it was thought worthwhile to further explore the apoptotic properties of [Pd(L2)(PPh3)] and [Pd(L3)(pic)], the most active members from each group. The apoptosis assay was conducted in HL-60 by the annexin V-FITC/PI method in a flow cytometer after an incubation period of 24 h. The results obtained for the complexes have been compared with those for cisplatin and the well-known apoptosis inducer camptothecin, and are shown in Fig. 7. Each drug treatment was done at 5 and 10 µM concentration. Exposure of cells to cisplatin resulted to 1.39% and 1.20% of gated cells in LR and 3.23% and 6.31% in UR, respectively (Fig. 7C and D).16Camptothecin induced 7.79% and 10.52% in LR and 5.46% and 5.30% in UR (Fig. 7E and F). The [Pd(L2)(PPh3)] complex exhibited 1.84% and 3.31% in LR and 2.48% and 27.35% in UR (Fig. 7G and H). Thus, it induces apoptosis to a greater extent than cisplatin as well as camptothecin at 10 µM concentration. On the other hand, the [Pd(L3)(pic)] complex induces 2.09% and 2.16% in LR and 2.01% and 2.02% in UR (Fig. 7I and J).
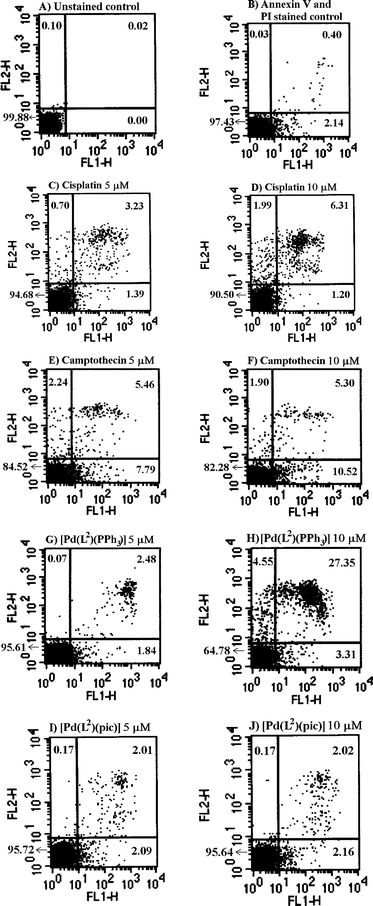 |
| | Fig. 7
Flow cytometric analysis of apoptosis and necrosis in HL-60 cells after treatment with four drugs or complexes at two different concentrations. Quadrant analysis of fluorescence intensity of gated cells in FL-1 and FL-2 channels was from 10000 events. Figures in the cytogram indicate the percent of gated cells in each quadrant. (FL1-H = annexin V, FL2-H = propidium iodide). | |
Conclusions
The present study shows that salicylaldehyde thiosemicarbazones and similar ligands can smoothly bind to metal ions as tridentate O,N,S-donors, provided an adequate number of coordination sites are readily available on the target metal ion. This study also demonstrates that [Pd(L2)(PPh3)] has remarkable cytotoxic properties among all the compounds tested, including recognized anticancer drugs, which is manifested through the IC50 values and has been further corroborated by the apoptosis study in HL-60, where it induces apoptosis to a greater extent than cisplatin and camptothecin.
Experimental
Materials
Palladium chloride was obtained from Arora Matthey, Kolkata, India, and was converted to Na2[PdCl4].17Salicylaldehyde, 2-hydroxyacetophenone and 2-hydroxynaphthaldehyde were obtained from S. D. fine-chem, Mumbai, India. Thiosemicarbazide was procured from Loba Chemie, Mumbai, India. All other chemicals and solvents were reagent grade commercial materials and were used as received. The [Pd(PPh3)2Cl2] and [Pd(pic)2Cl2] complexes were prepared starting from Na2[PdCl4] by following reported procedures.18,19 The thiosemicarbazone ligands (H2L1, H2L2 and H2L3) were prepared by condensing the respective aldehyde or ketone with thiosemicarbazide in hot ethanol.
Synthesis
[{Pd(L1)}n].
To a solution of H2L1 (64 mg, 0.33 mmol) in hot ethanol (30 ml), was added triethylamine (66 mg, 0.66 mmol) followed by Na2[PdCl4] (100 mg, 0.33 mmol). The mixture was heated at reflux for 6 h. A brown precipitate settled down on cooling, which was collected by filtration, washed with ethanol and dried in air to afford [{Pd(L1)}n] as a brown powder. Yield: 84 mg (85%). Anal. calc. for (C8H7N3SOPd)n: C 31.96; H 2.33; N 13.98. Found: C 31.77; H 2.43; N 14.11%.
The [{Pd(L2)}n] and [{Pd(L3)}n] complexes were prepared by following the same above procedure using, respectively, H2L2 and H2L3 instead of H2L1.
[{Pd(L2)}n].
Anal. calc. for (C9H9N3SOPd)n: C 34.46; H 2.87; N 13.40. Found: C 34.58; H 2.99; N 13.23%.
[{Pd(L3)}n].
Anal. calc. for (C12H9N3SOPd)n: C 48.82; H 3.62; N 12.66. Found: C 48.67; H 3.77; N 12.87%.
[Pd(L1)(PPh3)]. Method A.
The polymeric [{Pd(L1)}n] species (100 mg) and triphenylphosphine (88 mg) were taken together in ethanol (30 ml) and the solution was refluxed for 24 h to yield a yellow solution. Evaporation of this solution gave a yellow solid, which was subjected to purification by thin layer chromatography on a silica plate. With 1 : 40 acetonitrile–benzene as the eluant, a major yellow band separated, which was extracted with acetonitrile. Upon evaporation of the acetonitrile extract [Pd(L1)(PPh3)] was obtained as a crystalline yellow solid. Yield: 113 mg (60%).
Method B.
To a solution of H2L1 (28 mg, 0.14 mmol) in hot ethanol (30 ml) was added triethylamine (29 mg, 0.29 mmol) followed by [Pd(PPh3)2Cl2] (100 mg, 0.14 mmol). The mixture was heated at reflux for 5 h to yield a yellow solution. Evaporation of this solution gave a yellow solid, which was subjected to purification by thin layer chromatography on a silica plate. With benzene as the eluant, a yellow band separated, which was extracted with acetonitrile. Upon evaporation of the acetonitrile extract [Pd(L1)(PPh3)] was obtained as a crystalline yellow solid. Yield: 52 mg (65%) Anal. calc. for C26H22N3SOPd: C 55.57; H 3.92; N 7.48. Found: C 55.62; H 3.86; N 7.50%. 1H NMR in CDCl3δ/ppm: 4.76 (s, 2H), 6.62 (t, J = 7.3 Hz, 1H), 6.68 (d, J = 8.5 Hz, 1H), 7.22 (t, J = 7.7 Hz, 1H), 7.31 (d, J = 8.0 Hz, 1H), 7.41–7.7520 (PPh3), 8.24 (d, J = 12.99 Hz, 1H).
The [Pd(L2)(PPh3)] and [Pd(L3)(PPh3)] complexes were prepared by following the same above procedure using, respectively, H2L2 and H2L3 instead of H2L1.
[Pd(L2)(PPh3)].
Anal. calc. for C27H24N3SOPd: C 56.31; H 4.17; N 7.30. Found: C 56.40; H 4.23; N 7.33%. 1H NMR in CDCl3δ/ppm: 2.89 (s, 3H), 4.78 (s, 2H), 6.62 (d, J = 8.34 Hz, 1H), 6.68 (t, J = 7.6 Hz, 1H), 7.17 (t, J = 7.7 Hz, 1H), 7.43–7.7220 (PPh3), 7.62 (d, J = 1.4 Hz, 1H).
[Pd(L3)(PPh3)].
Anal. calc. for C30H24N3SOPd: C 58.88; H 3.93; N 6.87. Found: C 58.86; H 3.88; N 6.94%. 1H NMR in CDCl3δ/ppm: 4.83 (s, 2H), 6.76 (d, J = 9.2 Hz, 1H), 7.32 (t, J = 5.6 Hz, 1H), 7.50–7.8320 (m, 15H (PPh3) and t, 1H), 8.11 (d, J = 8.5 Hz, 1H), 9.52 (d, J = 13.2 Hz, 1H).
[Pd(L1)(pic)]. Method A.
The polymeric [{Pd(L1)}n] species (100 mg) and 4-picoline (31 mg) were taken in ethanol (30 ml) and the solution was refluxed for 24 h to yield a yellow solution. Evaporation of this solution gave a yellow solid, which was subjected to purification by thin layer chromatography on a silica plate. With 1 : 20 acetonitrile–benzene as the eluant, a prominent yellow band separated, which was extracted with acetonitrile. Upon evaporation of the acetonitrile extract [Pd(L1)(pic)] was obtained as a crystalline yellow solid. Yield: 66 mg (50%).
Method B.
To a solution of H2L1 (54 mg, 0.28 mmol) in hot ethanol (30 ml) was added triethylamine (56 mg, 0.55 mmol) followed by [Pd(pic)2Cl2] (100 mg, 0.28 mmol). The mixture was heated at reflux for 6 h to yield a yellow solution. Evaporation of this solution gave a yellow solid, which was subjected to purification by thin layer chromatography on a silica plate. With 1 : 20 acetonitrile–benzene as the eluant, a major yellow band separated, which was extracted with acetonitrile. Upon evaporation of the acetonitrile extract [Pd(L1)(pic)] was obtained as a crystalline yellow solid. Yield: 59 mg (55%). Anal. calc. for C14H14N4SOPd: C 42.56; H 3.42; N 14.29. Found: C 42.38; H 3.45; N 14.23%. 1H NMR in CDCl3δ/ppm: 2.44 (s, 3H), 4.87 (s, 2H), 6.67 (t, J = 7.3 Hz, 1H), 7.03 (d, J = 8.8 Hz, 1H), 7.24 (d, J = 5.7 Hz, 2H), 7.29–7.3420 (2H), 7.92 (s, 1H), 8.62 (d, J = 5.9 Hz, 2H).
The [Pd(L2)(pic)] and [Pd(L3)(pic)] complexes were prepared by following the same above procedure using, respectively, H2L2 and H2L3 instead of H2L1.
[Pd(L2)(pic)].
Anal. calc. for C15H16N4SOPd: C 44.29; H 3.94; N 13.78. Found: C 44.44; H 3.86; N 13.76%. 1H NMR in CDCl3δ/ppm: 2.43 (s, 3H), 2.75 (s, 3H), 4.84 (s, 2H), 6.68 (t, J = 7.3 Hz, 1H), 7.04 (d, J = 8.2 Hz, 1H), 7.21–7.2620 (3H), 7.68 (d, J = 8.2 Hz, 1H), 8.61 (d, J = 6.2 Hz, 1H).
[Pd(L3)(pic)].
Anal. calc. for C18H16N4SOPd: C 48.82; H 3.62; N 12.66. Found: C 48.99; H 3.78; N 12.45%. 1H NMR in CDCl3δ/ppm: 2.45 (s, 3H), 4.87 (s, 2H), 7.13–7.2620 (4H), 7.48 (d, J = 7.44 Hz, 1H), 7.69–7.5020 (2H), 8.06 (d, J = 8.5 Hz, 1H), 8.65 (d, J = 2.5 Hz, 2H), 8.93 (s, 1H).
Cytotoxicity assay
Cell lines and culture.
HL-60 and U-937 (purchased from National Center for Cell Sciences, Pune, India) were maintained in complete sterile growth medium RPMI-1640 with L-glutamine (GIBCO, Invitrogen Corporation, USA), containing 10% heat inactivated fetal bovine serum (GIBCO, Invitrogen Corporation, USA) and 1% antibiotics [10000 U penicillin ml–1 and 10000 µg streptomycin ml–1, BioWhittaker, Cambrex Bio Science, USA]. Cell cultures were maintained in log phase using a subculture ratio of 1 : 5 twice a week.
Preparation of test material.
Working stock solutions of all the compounds were prepared by dissolving 20 mg of each compound in 1 ml of DMSO [Hybri-Max grade, Sigma-Aldrich Co., USA] and filtered under sterile conditions. These were serially diluted with complete RPMI-1640 medium prior to use to obtain different drug concentrations.
MTT-based cytotoxicity test.
100 µl of cell suspension from the stock 2 × 105HL-60 or 1 × 105U-937 cells ml–1 were added to 96-well cell culture plates. Next, 10 µl of drug solutions of different concentrations were added to respective wells followed by the addition of the medium (90 µl) in triplicate (total volume 200 µl/well). All vehicle controls contained the same concentration of DMSO. Plates were incubated for 72 h at 37 °C, 5% CO2–95% air with humidity. After removal of 100 µl of media from each well, 10 µl of a 5 mg ml–1 solution of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide [MTT, Sigma, USA] in Dulbecco’s PBS (GIBCO, BRL) were added to each well and the plate was incubated for 4 h at 37 °C. Subsequently 100 µl of acidic isopropanol solution (0.04 N HCl in isopropanol) was added to the wells to dissolve formed formazan crystals. The plate was read in a microplate reader (Bio-Rad, USA, Model 550) at 540 nm. The percentage cell viability was calculated by dividing the average absorbance of the cells treated with compounds by that of the control; IC50 (drug concentration needed for 50% inhibition of cells relative to control) was determined through Curvefit software by plotting % inhibition vs. drug concentration and expressed in µM concentration.
In vitro
apoptosis assay
Induction of apoptosis
in vitro by two representative complexes of each type [Pd(L)(D)] (when D = PPh3 and pic), cisplatin and camptothecin were determined by a flow cytometric assay using an annexin V-FITC21apoptosis detection kit (BD Biosciences Pharmingen, San Diego, USA). Exponentially growing HL-60 cells in complete RPMI-1640 media (100 µl of 5 × 106) were plated in six well plates. 10 µl of each compound in DMSO (5 and 10 µM concentrations) was added to the wells followed by the addition of required media (total volume 1 ml). Plates were incubated at 37 °C for 24 h. The cells were centrifuged at room temperature at 1500 rpm for 5 min. The pellet was washed twice with cold PBS and re-suspended in the binding buffer (1X, 100 µl) provided in the kit. The cells were stained with annexin V-FITC (5 µl) and propidium iodide [PI] (5 µl) and incubated for 15 min in the dark at 25 °C. Next, 400 µl of binding buffer was added to each tube and analyzed on a FACScan flow cytometer (Becton Dickinson, Mountainview, CA) using Cell Quest software. Two controls, viz. unstained cells and cells stained with both annexin V-FITC and PI, containing DMSO as vehicle were used. Cells that were annexin+/PI– and annexin+/PI+ were considered positive for apoptosis.
Physical measurements
Microanalyses (C, H, N) were performed using a Heraeus Carlo Erba 1108 elemental analyzer. IR spectra were obtained on a Shimadzu FTIR-8300 spectrometer with samples prepared as KBr pellets. Electronic spectra were recorded on a JASCO V-570 spectrophotometer. Emission spectra were recorded on a Perkin Elmer LS55 luminescence spectrometer. 1H NMR spectra were recorded in CDCl3 solutions on a Bruker Avance DPX 300 NMR spectrometer using TMS as the internal standard.
Single crystals of [Pd(L1)(PPh3)] and [Pd(L2)(pic)] were grown by slow evaporation of 1 : 1 dichloromethane–acetonitrile solutions of the respective compounds. Selected crystal data and data collection parameters are given in Table 7. Data on the crystals were collected on a BRUKER SMART ApexCCD area detector. A total 15972 and 30571 reflections giving 5525 and 3135 unique reflections were collected (Rint = 0.0261 and 0.022) for [Pd(L1)(PPh3)] and [Pd(L2)(pic)], respectively. X-Ray data reduction, structure solution and refinement were done using the SHELXS-97 and SHELXL-97 packages.22 The structures were solved by direct methods.
CCDC reference numbers 646551 and 646552. For crystallographic data for the [Pd(L)(PPh3)] and [Pd(L)(pic)] complexes in CIF or other electronic format see DOI: 10.1039/b707448d
| |
[Pd(L1)(PPh3)] |
[Pd(L2)(pic)] |
|
R
1 = Σ||Fo∣ – ∣Fc||/Σ∣Fo∣.
wR2 = [Σ[w(Fo2 – Fc2)2]/Σ[w(Fo2)2]]½.
GOF = [Σ[w(Fo2 – Fc2)2]/(M – N)]½, where M is the number of reflections and N is the number of parameters refined.
|
| Empirical formula |
C26H22N3OPSPd |
C15H16N4OSPd |
| Formula mass |
561.9 |
406.78 |
| Crystal system |
Triclinic |
Orthorhombic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Pbca
|
|
a/Å |
7.5385(4) |
8.3942(12) |
|
b/Å |
10.2179(5) |
15.149(2) |
|
c/Å |
16.9756(9) |
25.099(4) |
|
α/° |
77.025(1) |
90 |
|
β/° |
80.819(1) |
90 |
|
γ/° |
71.567(1) |
90 |
|
V
/Å3 |
1203.27(11) |
3191.7(8) |
|
Z
|
2 |
8 |
|
D
calc/mg m–3 |
1.551 |
1.693 |
|
F(000)
|
568 |
1632 |
|
λ/Å |
0.71073 |
0.71073 |
| Crystal size/mm3 |
0.38 × 0.30 × 0.15 |
0.12 × 0.28 × 0.48 |
|
T/K |
295(2) |
298 |
|
µ/mm–1 |
0.948 |
1.3 |
| Independent reflections |
5525 |
3135 |
|
R
int
|
0.0261 |
0.022 |
| Collected reflections |
15972 |
30751 |
|
R
1
a
|
0.026 |
0.0219 |
| wR2b |
0.0702 |
0.0595 |
| GOFc |
1.159 |
1.076 |
Acknowledgements
Financial assistance received from the Council of Scientific and Industrial Research, New Delhi, India, [grant no. 01(1952)/04/EMR-II] is gratefully acknowledged. The authors thank the anonymous referees for their constructive criticisms and suggestions, which have been of great help in preparing the revised manuscript. Sincere thanks are due to Prof. Chittaranjan Sinha and Dr Asok Nath Mandal of the Department of Chemistry, Jadavpur University, for their help with the florescence and NMR spectral measurements, respectively. Sincere thanks are also due to Dr Golam Mostafa of the Department of Physics, Jadavpur University, for his help in resolving some crystallographic queries. The authors are grateful to Dr Jaydip Biswas, Director, Chittaranjan National Cancer Institute, for his encouragement and Dr R. N. Baral, In-Charge, Department of Immunoregulation & Immunodiagnostics, for helpful discussions. Sarmistha Halder thanks the University Grants Commission, New Delhi, India, for her fellowship [grant no. 10-2(5)2004(1)-E.U.II].
References
-
(a) M. J. M. Campbell, Coord. Chem. Rev., 1975, 15, 279 CrossRef CAS;
(b) S. B. Padhye and G. B. Kaffman, Coord. Chem. Rev., 1985, 63, 127 CrossRef CAS;
(c) I. Haiduc and C. Silvestru, Coord. Chem. Rev., 1990, 99, 253 CrossRef CAS;
(d) D. X. West, S. B. Padhye and P. B. Sonawane, Struct. Bonding, 1992, 76, 1;
(e) D. X. West, A. E. Liberta, S. B. Padhye, R. C. Chikate, P. B. Sonawane, A. S. Kumbhar and R. G. Yerande, Coord. Chem. Rev., 1993, 123, 49 CrossRef CAS;
(f) J. R. Dilwarth, P. Amold, D. Morales, Y. L. Wong and Y. Zheng, Mod. Coord. Chem., 2002, 217 Search PubMed;
(g) A. G. Quiroga and C. N. Raninger, Coord. Chem. Rev., 2004, 248, 119 CrossRef CAS.
-
(a) Z. Iakovidou, A. Papageorgiou, M. A. Demertzis, E. Mioglou, D. Mourelatos, A. Kotsis, P. N. Yadav and D. Kovala-Demertzi, Anti-Cancer Drugs, 2001, 12, 65 CrossRef CAS;
(b) J. Patole, S. Dutta, S. Padhye and E. Sinn, Inorg. Chim. Acta, 2001, 318, 207 CrossRef CAS;
(c) R. I. Maurer, P. J. Blower, J. R. Dilworth, C. A. Reynolds, Y. Zheng and G. E. D. Mullen, J. Med. Chem., 2002, 45, 1420 CrossRef CAS;
(d) A. R. Cowly, J. R. Dilworth, P. S. Donnely, E. Labisbal and A. Sousa, J. Am. Chem. Soc., 2002, 124, 5270 CrossRef CAS;
(e) M. B. Ferrari, F. Bisceglie, G. Pelosi, M. Sassi, P. Tarasconi, M. Cornia, S. Capacchi, R. Albertini and S. Pinelli, J. Inorg. Biochem., 2002, 90, 113 CrossRef;
(f) E. M. Jouad, X. D. Thanh, G. Bouet, S. Bonneau and M. A. Khan, Anticancer Res., 2002, 22, 1713 CAS;
(g) S. Padhye, Z. Afrasiabi, E. Sinn, J. Fok, K. Mehta and N. Rath, Inorg. Chem., 2005, 44, 1154 CrossRef CAS;
(h) J. Ruiz, N. Cutillas, C. Vicente, M. D. Villa, G. López, J. Lorenzo, F. X. Avilés, V. Moreno and D. Bautista, Inorg. Chem., 2005, 44, 7365 CrossRef CAS.
- Y. P. Tion, C. Y. Duan, Z. L. Lu, X. Z. You, H. K. Fun and S. Kandasamy, Polyhedron, 1996, 15, 2263 CrossRef.
-
(a) A. De Bolfo, T. D. Smith, J. F. Boas and J. R. Pilbrow, Aust. J. Chem., 1976, 29, 2583;
(b) Z. Lu, C. White, A. L. Rheingold and R. H. Crabtree, Inorg. Chem., 1993, 32, 3991 CrossRef CAS;
(c) D. X. West, Y.-H. Yang, T. L. Klein, K. I. Goldberg, A. E. Liberta, J. Valdés-Martínez and R. A. Toscano, Polyhedron, 1995, 14, 1681 CrossRef CAS;
(d) D. X. West, Y.-H. Yang, T. L Klein, K. I. Goldberg, A. E. Liberta, J. Valdés-Martínez and S. Hernández-Ortega, Polyhedron, 1995, 14, 3051 CrossRef CAS;
(e) P. Souza, I. A. Matesanz and V. Fernandez, J. Chem. Soc., Dalton Trans., 1996, 3011 RSC;
(f) D. Kovala-Demertzi, A. Domopoulou, M. A. Demertzis, J. Valdés-Martínez, S. Hernández-Ortega, G. Espinosa-Pérez, D. X. West, M. M. Salberg, G. A. Bain and P. D. Bloom, Polyhedron, 1996, 15, 2587 CrossRef CAS;
(g) M. A. Ali, K. K. Dey, M. Nazimuddin, F. E. Smith, R. J. Butcher, J. P. Jasinski and J. M. Jasinski, Polyhedron, 1996, 15, 3331 CrossRef CAS.
-
(a) D. Kovala-Demertzi, N. Kourkoumelis, M. A. Demertzis, J. R. Miller, C. S. Frampton, J. K. Swearingen and D. X. West, Eur. J. Inorg. Chem., 2000, 727 CrossRef CAS;
(b) P. N. Yadav, M. A. Demertzis, D. Kovala-Demertzi, A. Castiñeiras and D. X. West, Inorg. Chim. Acta, 2002, 332, 204 CrossRef CAS.
- F. Basuli, S.-M. Peng and S. Bhattacharya, Inorg. Chem., 1997, 36, 5645 CrossRef CAS.
-
(a) A. Castiñeiras, E. Bermejo, D. X. West, A. K. EI-Sawaf and J. K. Swearingen, Polyhedron, 1998, 17, 2751 CrossRef CAS;
(b) A. Castiñeiras, M. Gil, E. Bermejo and D. X. West, Z. Naturforsch., B: Chem. Sci., 2000, 55, 863 CAS;
(c) E. Bermejo, A. Castiñeiras, L. J. Ackerman, M. D. Owens and D. X. West, Z. Anorg. Allg. Chem., 2001, 627, 1966 CrossRef CAS;
(d) C. A. Brown, W. Kaminsky, K. A. Claborn, K. I. Goldberg and D. X. West, J. Braz. Chem. Soc., 2002, 13, 10 CAS.
- I. Pal, F. Basuli, T. C. W. Mak and S. Bhattacharya, Angew. Chem., Int. Ed., 2001, 40, 2923 CrossRef CAS.
-
(a) F. Basuli, M. Ruf, C. G. Pierpont and S. Bhattacharya, Inorg. Chem., 1998, 39, 6113 CrossRef;
(b) F. Basuli, S.-M. Peng and S. Bhattacharya, Inorg. Chem., 2000, 39, 1120 CrossRef CAS;
(c) S. Dutta, F. Basuli, S. M. Peng, G. H. Lee and S. Bhattacharya, New J. Chem., 2002, 26, 1607 RSC;
(d) R. Acharyya, S. Dutta, F. Basuli, S. M. Peng, G. H. Lee, L. R. Falvello and S. Bhattacharya, Inorg. Chem., 2006, 45, 1252 CrossRef CAS;
(e) S. Basu, R. Acharyya, W. S. Sheldrick, H. Mayer-Figge and S. Bhattacharya, Struct. Chem., 2007, 18, 209 CrossRef CAS.
-
(a) L. Adrio, G. Alberdi, A. Amoedo, D. Lata, A. Fernández, J. Martínez, M. T. Pereira and J. M. Vila, Z. Anorg. Allg. Chem., 2005, 631, 2204 CrossRef CAS;
(b) J. L. Neto, G. M. Lima and H. Beraldo, Spectrochim. Acta, Part A, 2006, 62, 669 CrossRef;
(c) A. R. Jalilian, M. Sadeghi, Y. Y. Kamrani and M. R. Ensaf, J. Radioanal. Nucl. Chem., 2006, 268, 605 CrossRef CAS;
(d) L. Otero, M. Vieites, L. Boiani, A. Denicola, C. Rigol, L. Opazo, C. Olea-Azar, J. D. Maya, A. Morello, R. L. Krauth-Siegel, O. E. Piro, E. Castellano, M. González, D. Gambino and H. Cerecetto, J. Med. Chem., 2006, 49, 3322 CrossRef CAS;
(e) J. Martínez, L. A. Adrio, J. M. Antelo, J. M. Ortigueira, M. T. Pereira, J. J. Fernández, A. Fernández and J. M. Vila, J. Organomet. Chem., 2006, 691, 2721 CrossRef CAS;
(f) J. Martínez, L. A. Adrio, J. M. Antelo, J. M. Ortigueira, M. T. Pereira, M. López-Torres and J. M. Vila, J. Organomet. Chem., 2006, 691, 2891 CrossRef CAS;
(g) A. Amoedo, L. A. Adrio, J. M. Antelo, J. Martínez, M. T. Pereira, A. Fernández and J. M. Vila, Eur. J. Inorg. Chem., 2006, 3016 CrossRef CAS;
(h) N. I. Dodoff, D. Kovala-Demertzi, M. Kubiak, J. Kuduk-Jaworska, A. Kochel and G. A. Gorneva, Z. Naturforsch., B: Chem. Sci., 2006, 61, 1110 CAS;
(i) M. A. Demertzis, P. N. Yadav and D. Kovala-Demertzi, Helv. Chim. Acta, 2006, 89, 1959 CrossRef CAS;
(j) J. Martínez, L. A. Adrio, J. M. Antelo, M. T. Pereira, J. J. Fernández and J. M. Vila, Polyhedron, 2006, 25, 2848 CrossRef CAS;
(k) A. R. Jalilian, M. Sadeghi and Y. Y. Kamrani, Radiochim. Acta, 2006, 94, 865 CrossRef;
(l) A. I. Matesanz, C. Pastor and P. Souza, Inorg. Chem. Commun., 2007, 10, 97 CrossRef CAS;
(m) A. I. Matesanz and P. Souza, J. Inorg. Biochem., 2007, 101, 245 CrossRef CAS.
-
(a) S. K. Burley and G. A. Petsko, Adv. Protein Chem., 1998, 39, 125 Search PubMed;
(b)
M. Nishio, M. Hirota and Y. Umezawa, The CH–π interactions (Evidence, Nature and Consequences), Wiley-VCH, New York, 1998 Search PubMed;
(c) Y. Umezawa, S. Tsuboyama, K. Honda, J. Uzawa and M. Nishio, Bull. Chem. Soc. Jpn., 1998, 71, 1207 CAS;
(d)
G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond, IUCr Monograph on Crystallography 9, Oxford University Press, Oxford, 1999 Search PubMed;
(e) M. J. Hannon, C. L. Painting and N. W. Alcock, Chem. Commun., 1999, 2023 RSC;
(f) B. J. Mcnelis, L. C. Nathan and C. J. Clark, J. Chem. Soc., Dalton Trans., 1999, 1831 RSC;
(g) K. Biradha, C. Seward and M. J. Zaworotko, Angew. Chem., Int. Ed., 1999, 38, 492 CrossRef CAS;
(h) M. J. Calhorda, Chem. Commun., 2000, 801 RSC;
(i) C. Janiak, S. Temizdemir and S. Dechert, Inorg. Chem. Commun., 2000, 3, 271 CrossRef CAS;
(j) C. Janiak, S. Temizdemir, S. Dechert, W. Deck, F. Girgsdies, J. Heinze, M. J. Kolm, T. G. Scarmann and O. M. Zipffel, Eur. J. Inorg. Chem., 2000, 1229 CrossRef CAS.
-
(a) S. B. Halligudi, K. N. Bhatt, N. H. Khan, R. I. Kurashy and K. Venkatesubramanian, Polyhedron, 1996, 15, 2093 CrossRef CAS;
(b) G. Zhao, H. Lin, P. Yu, H. Sun, S. Zhu, X. Su and Y. Chen, J. Inorg. Biochem., 1999, 73, 145 CrossRef CAS;
(c) B. Milani, A. Scarel, E. Zangrando, G. Mestroni, C. Carfagna and B. Binotti, Inorg. Chim. Acta, 2003, 350, 592 CrossRef CAS.
-
(a)
C. Mealli and D. M. Proserpio, CACAO Version 4.0, Firenze, Italy, July 1994 Search PubMed;
(b) C. Mealli and D. M. Proserpio, J. Chem. Educ., 1990, 67, 399 CrossRef CAS.
-
(a) Y. Shimazaki, M. Tashiro, T. Motoyama, S. Iwatsuki, T. Yajima, Y. Nakabayashi, Y. Naruta and O. Yamauchi, Inorg. Chem., 2005, 44, 6044 CrossRef CAS;
(b) N. D. Jones, P. Meessen, U. Losehand, B. O. Patrick and B. R. James, Inorg. Chem., 2005, 44, 3290 CrossRef CAS.
-
(a) G. A. Crosby and W. H. Elfring, Jr, J. Phys. Chem., 1976, 80, 2206 CrossRef CAS;
(b) M. J. Cook, A. P. Lewis, G. S. J. Thomson, J. L. Glasper and D. J. Robbins, J. Chem. Soc., Perkin Trans. 2, 1984, 293 RSC.
- Abbreviations used: LR (lower right quadrant, representing early apoptotic cells, which are annexin V+/PI–); UR (upper right quadrant, representing late apoptotic cells, which are annexin V+/PI+).
-
F. R. Hertley, in The Chemistry of Palladium and Platinum, ed. P. L. Robinson, Applied Science, London, 1973, vol. 14, pp. 458 Search PubMed.
-
H. L. Grube, in Handbook of Preparative Inorganic Chemistry, ed. G. Brauer, Academic Press, London, 2nd edn, 1965, vol. 2, pp. 1584 Search PubMed.
-
(a) P. Chattopadhyay and S. K. Majumdar, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 1983, 22, 1603;
(b) T. K. Thokdar, A. Sarkar, P. K. Paria and S. K. Majumdar, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 1989, 28, 181.
- Overlapping signals.
- I. Vermes, C. Hannen, H. Steffens-Nakken and C. Reutelingsperger, J. Immunol. Methods, 1995, 184, 39 CrossRef CAS.
-
G. M. Sheldrick, SHELXS-97, Program for solution of crystal structures, University of Göttingen, Germany, 1997 Search PubMed;
G. M. Sheldrick, SHELXL-97, Program for refinement of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: A diagram showing metal–π interaction in the [Pd(L)(PPh3)] complex (Fig. S1), a diagram showing π–π interaction in the [Pd(L)(pic)] complex (Fig. S2), a table showing metal–π and π–π interactions in the [Pd(L)(PPh3)] and [Pd(L)(pic)] complexes, respectively (Table S1) and a partial MO diagram of the [Pd(L)(pic)] complex (Fig. S3). See DOI: 10.1039/b707448d |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2008 |
Click here to see how this site uses Cookies. View our privacy policy here. 



![(a) Electronic spectrum and (b) emission spectrum of [Pd(L3)(PPh3)] in dichloromethane solution.](/image/article/2008/NJ/b707448d/b707448d-f5.gif)