Synthesis and X-ray structures of cadmium-containing dinuclear double helicates derived from ligands containing N-oxide units
Georgios
Bokolinis
a,
T.
Riis-Johannessen†
b,
John C.
Jeffery
b and
Craig R.
Rice
*a
aDepartment of Chemical and Biological Sciences, University of Huddersfield, Huddersfield, UK HD1 3DH. E-mail: c.r.rice@hud.ac.uk; Fax: (+44)1484 472182; Tel: (+44)1484 473759
bSchool of Chemistry, University of Bristol, Cantocks Close, Bristol, UK BS8 1TS
First published on 7th September 2007
Abstract
The syntheses of two novel polydentate pyridyl-thiazolyl ligands which contain an N-oxide unit on the central pyridyl ring are reported. Reaction of either of these ligands with Cd(ClO4)2·6H2O results in the formation of a dinuclear double helicate. The solid state structures of these transition metal helicates demonstrate the ability of the N-oxide unit to partition the ligands into two separate binding domains.
1. Introduction
Formation of transition metal helicates from the self-assembly of organic ligands and transition metal ions has received considerable attention in recent years.1–6 One of the main considerations for the successful self-assembly of these species is the inherent information held within the ligand strand. If the ligand is sufficiently instructed or “pre-programmed” it will partition into different binding domains and coordinate two or more metal ions, thus preventing the formation of mononuclear complexes. We have previously shown how ligands that contain a mixture of pyridyl and thiazolyl N-donor groups form a variety of helical complexes whose geometries are determined, in part, by the natural break of the ligands into distinct binding domains imposed by the presence of the thiazole units in the oligopyridyl chain.7In this paper, we describe the use of pyridyl N-oxides to partition ligands into different binding domains. To the best of our knowledge, this is the first example of the use of N-oxide ligands for the formation of transition metal helicates, although the formation of mononuclear complexes from N-oxides of 2,2′:6′,2″-terpyridine and 2,2′:6′,2″:6″,2″′-quaterpyridine have been reported.8,9
2. Results and discussion
Both ligands L1 and L2 were synthesized as shown in Scheme 1. The starting material in both cases was 2,2′:6′,2″-terpyridine tris-N-oxide, which was converted to the 6,6″-dicyano derivative by reaction of trimethylsilyl cyanide and benzoyl chloride in refluxing CH2Cl2. Further reaction with hydrogen sulfide gas gave the 6,6″-dithioamide in excellent yield. Reaction of this compound with either chloroacetone or 2-(α-bromoacetyl)pyridine gave ligands L1 and L2, respectively.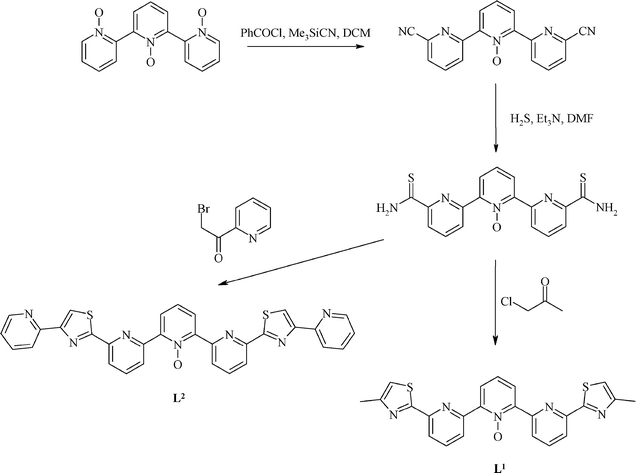
Although the ligand L1 is insoluble in common organic solvents, reaction with Cd(ClO4)2·6H2O in MeCN produced a clear pale yellow solution which gave ions in the ESI-MS at m/z 966 and 1409, corresponding to {[Cd2(L1)](ClO4)3}+ and {[Cd2(L1)2](ClO4)3}+, indicating the formation of a dinuclear double-stranded helicate. The solid-state structure was confirmed from X-ray crystallographic analysis. In the complex cation, two Cd2+ ions are coordinated by two bridging ligands in a double helicate arrangement (Fig. 1 and 2). Both ligands are partitioned into two tridentate binding domains, with the N-oxide acting as a bridging bidentate donor to both cadmium(II) ions. The NCCN torsion angles between the terminal pyridyl-thiazole domain and central pyridine N-oxide, range from 39(1)–43(1)°. As a consequence of the bridging oxygen atoms in the {Cd(µ-O2)Cd}4+ core, the metal–metal distance (i.e. the effective helical pitch length) is quite short (4.027(1) Å) when compared to other cadmium-containing helicates.14,15 The Cd–O(N-oxide) distances range from 2.327(3)–2.355(3) Å, whilst those to the N-donors lie in the range 2.256(4)–2.537(4) Å (Table 1). Each of the two cadmium centres is further coordinated by one oxygen atom of a bridging perchlorate anion. By interacting with the Cd(II) ion in the neighbouring cation, these interstitial perchlorates link adjacent helicates end-on throughout the lattice. The infinite columns propagate along the crystallographic c-axis, but unlike previously observed packing motifs,16 the helicate units alternate between having P- and M-screw senses.
![Structure of the complex cation [Cd2(L1)2(ClO4)2]2+.](/image/article/2008/NJ/b711039a/b711039a-f1.gif)
![Structure of the complex cation [Cd2(L1)2(ClO4)2]2+ showing the {Cd(µ-O2)Cd} core.](/image/article/2008/NJ/b711039a/b711039a-f2.gif)
| Cd(1)–N(61) | 2.256(4) | Cd(2)–N(51) | 2.297(5) |
| Cd(1)–N(11) | 2.277(4) | Cd(2)–N(101) | 2.300(4) |
| Cd(1)–O(81) | 2.335(3) | Cd(2)–O(31) | 2.327(3) |
| Cd(1)–O(31) | 2.355(3) | Cd(2)–O(81) | 2.332(3) |
| Cd(1)–N(71) | 2.452(4) | Cd(2)–N(41) | 2.480(4) |
| Cd(1)–N(21) | 2.456(4) | Cd(2)–N(91) | 2.537(4) |
| Cd(1)–O(1B) | 2.644(4) | Cd(2)–O(1C) | 2.713(4) |
| N(61)–Cd(1)–N(11) | 145.7(1) | N(51)–Cd(2)–N(101) | 146.9(2) |
| N(61)–Cd(1)–O(81) | 126.0(1) | N(51)–Cd(2)–O(31) | 122.3(2) |
| N(11)–Cd(1)–O(81) | 85.8(1) | N(101)–Cd(2)–O(31) | 89.1(1) |
| N(61)–Cd(1)–O(31) | 88.0(1) | N(51)–Cd(2)–O(81) | 86.7(1) |
| N(11)–Cd(1)–O(31) | 122.6(1) | N(101)–Cd(2)–O(81) | 120.9(1) |
| O(81)–Cd(1)–O(31) | 60.8(1) | O(31)–Cd(2)–O(81) | 61.2(1) |
| N(61)–Cd(1)–N(71) | 72.7(1) | N(51)–Cd(2)–N(41) | 72.6(2) |
| N(11)–Cd(1)–N(71) | 116.4(1) | N(101)–Cd(2)–N(41) | 116.9(1) |
| O(81)–Cd(1)–N(71) | 66.7(1) | O(31)–Cd(2)–N(41) | 66.6(1) |
| O(31)–Cd(1)–N(71) | 92.8(1) | O(81)–Cd(2)–N(41) | 97.5(1) |
| N(61)–Cd(1)–N(21) | 111.9(1) | N(51)–Cd(2)–N(91) | 111.0(1) |
| N(11)–Cd(1)–N(21) | 72.5(1) | N(101)–Cd(2)–N(91) | 70.8(1) |
| O(81)–Cd(1)–N(21) | 95.6(1) | O(31)–Cd(2)–N(91) | 97.5(1) |
| O(31)–Cd(1)–N(21) | 66.7(1) | O(81)–Cd(2)–N(91) | 64.9(1) |
| N(71)–Cd(1)–N(21) | 158.3(1) | N(41)–Cd(2)–N(91) | 161.1(1) |
| N(61)–Cd(1)–O(1B) | 76.1(1) | O(1C)–Cd(2)–N(101) | 78.0(1) |
| N(11)–Cd(1)–O(1B) | 69.8(1) | O(1C)–Cd(2)–N(51) | 73.2(1) |
| O(81)–Cd(1)–O(1B) | 143.0(1) | O(1C)–Cd(2)–N(44) | 77.4(1) |
| O(31)–Cd(1)–O(1B) | 156.1(1) | O(1C)–Cd(2)–N(91) | 121.4(1) |
| N(71)–Cd(1)–O(1B) | 99.2(1) | O(1C)–Cd(2)–O(31) | 130.7(1) |
| N(21)–Cd(1)–O(1B) | 102.5(1) | O(1C)–Cd(2)–O(81) | 159.9(1) |
In a similar manner to L1, in its non-complexed form L2 is insoluble in common organic solvents. However, reaction with Cd(ClO4)2·6H2O in MeNO2 produces a clear pale yellow solution, the ESI-MS of which shows ions at m/z 1662, 1352 and 782, corresponding to {[Cd2(L2)2](ClO4)3}+, {[Cd(L2)2](ClO4)}+ and {[Cd(L2)](ClO4)}+ species, respectively. The formation of a dinuclear double helicate [Cd2(L2)2]4+ in the solid-state was confirmed from single crystal X-ray diffraction studies (Fig. 3). The structure shows two distorted octahedral cadmium cations coordinated by two ligands. Each of the ligands coordinate via the central N-oxide and the two terminal bidentate pyridyl-thiazole domains. The N-oxide again acts as a bridging bidentate donor coordinating both cadmium cations, with Cd–O bond lengths ranging from 2.373(3)–2.395(3) Å (Table 2). The Cd–N distances involving the terminal pyridyl-thiazole domains range from 2.299(4) to 2.593(4) Å (Fig. 4). However, the pyridyl units adjacent to the central N-oxide of the terpyridyl domain have Cd–N distances between 2.732(3)–2.772(3) Å; interactions which are rather too long to be considered bonding. These units may thus be considered as non-bonding or hypodentate. Again the “twist” caused by partitioning of the ligand strands about the central pyridine N-oxide evokes NCCN torsion angles in the range 32(1)–38(1)°. The pitch length of this helicate (4.347(1) Å) is slightly longer than that of [Cd2(L1)2]4+ but is still quite short when compared to other cadmium-containing helicates.14,15
![Structure of the complex cation [Cd2(L2)2]4+.](/image/article/2008/NJ/b711039a/b711039a-f3.gif)
![Structure of the complex cation [Cd2(L2)2]4+ showing the {Cd(µ-O2)Cd} core.](/image/article/2008/NJ/b711039a/b711039a-f4.gif)
| Cd(1)–N(91) | 2.299(4) | Cd(2)–N(61) | 2.326(3) |
| Cd(1)–N(21) | 2.315(4) | Cd(2)–N(131) | 2.331(4) |
| Cd(1)–O(111) | 2.373(3) | Cd(2)–O(41) | 2.388(3) |
| Cd(1)–O(41) | 2.377(3) | Cd(2)–O(111) | 2.395(3) |
| Cd(1)–N(11) | 2.558(4) | Cd(2)–N(141) | 2.478(4) |
| Cd(1)–N(81) | 2.593(4) | Cd(2)–N(71) | 2.580(4) |
| N(91)–Cd(1)–N(21) | 153.1(2) | N(61)–Cd(2)–N(131) | 153.2(1) |
| N(91)–Cd(1)–O(111) | 116.4(1) | N(61)–Cd(2)–O(41) | 117.1(1) |
| N(21)–Cd(1)–O(111) | 88.4(1) | N(131)–Cd(2)–O(41) | 87.6(1) |
| N(91)–Cd(1)–O(41) | 87.4(1) | N(61)–Cd(2)–O(111) | 88.1(1) |
| N(21)–Cd(1)–O(41) | 114.7(1) | N(131)–Cd(2)–O(111) | 114.1(1) |
| O(111)–Cd(1)–O(41) | 62.6(1) | O(41)–Cd(2)–O(111) | 62.1(1) |
| N(91)–Cd(1)–N(11) | 99.6(1) | N(61)–Cd(2)–N(141) | 98.5(1) |
| N(21)–Cd(1)–N(11) | 67.9(1) | N(131)–Cd(2)–N(141) | 68.4(1) |
| O(111)–Cd(1)–N(11) | 91.3(1) | O(41)–Cd(2)–N(141) | 92.6(1) |
| O(41)–Cd(1)–N(11) | 153.1(1) | O(111)–Cd(2)–N(141) | 153.9(1) |
| N(91)–Cd(1)–N(81) | 67.4(1) | N(61)–Cd(2)–N(71) | 66.8(1) |
| N(21)–Cd(1)–N(81) | 93.3(1) | N(131)–Cd(2)–N(71) | 95.2(1) |
| O(111)–Cd(1)–N(81) | 159.0(1) | O(41)–Cd(2)–N(71) | 156.0(1) |
| O(41)–Cd(1)–N(81) | 98.0(1) | O(111)–Cd(2)–N(71) | 95.3(1) |
| N(11)–Cd(1)–N(81) | 108.7(1) | N(141)–Cd(2)–N(71) | 110.6(1) |
Comparison of these complexes with others derived from similar ligands and Cd(II) demonstrate that the N-oxide unit is instrumental in partitioning of the ligand. For example, reaction of a ligand strand identical to L2 (but which does not have a central N-oxide unit) results in the formation of a trinuclear double helicate.14 In this case, the ligand partitions into three different binding domains consisting of two terminal pyridyl-thiazole bidentate units and a central terdentate terpyridyl unit. Thus, the three Cd(II) ions occupy two different coordination environments, with the unique central cadmium ion coordinated by two terpyridyl units (one from each ligand) and the two identical terminal cadmium ions coordinated by a bidentate pyridyl-thiazole unit. In the case of L2, the central terpyridyl unit cannot act as a terdentate unit due to the N-oxide, which effectively separates the ligand into two distinct binding domains.
Cadmium complexes of the non-functionalised analogue of L1 (i.e. the same potentially pentadentate ligand but without the N-oxide unit) have not been reported to date. However, studies carried out on a similar potentially pentadentate pyridyl-thiazole ligand, containing a pyridyl–thiazole–pyridyl–thiazole–pyridyl chain, showed the formation of a mononuclear complex in which the cadmium ion is coordinated by a near-planar pentadentate “equatorial” ligand.15 Although it cannot be assumed that the non-functionalised analogue of L1 would act in a similar fashion, it may be reasonable to expect that, as with L2, the N-oxide ring of the central terpyridyl unit will prevent the ligand from efficiently coordinating to one metal ion.
3. Experimental
3.1 General details
All reagents and starting materials were purchased from Aldrich and/or Lancaster and were used without further purification. 1H NMR spectra were obtained on a Bruker 400 MHz NMR instrument and mass spectrometry was performed on a Micromass Quattro VG quadrupole and a Bruker MicroTOF LC mass spectrometer. 2,2′,6′,2″-terpyridine-1,1′,1″-trioxide was prepared via oxidation with H2O2 in acetic acid.103.2 Synthesis
3.3 Crystallography‡
Diffraction intensity data were collected on a Bruker SMART CCD area-detector diffractometer with Mo-Kα radiation (λ = 0.71073 Å). Intensities were integrated11 from several series of exposures, each exposure covering 0.3° in ω, and the total data set being a sphere. Absorption corrections were applied based on multiple and symmetry-equivalent measurements.12 The structure was solved by direct methods and refined by least squares on weighted F2 values for all reflections.13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 281 reflections were measured in the range 1.83 ≤ θ ≤ 27.52° (hkl range indices –30 ≤ h ≤ 30, –16 ≤ k ≤ 16, –27 ≤ l ≤ 27), 13984 independent reflections (Rint = 0.0539). The structure was refined to F2 to wR2 = 0.1615, R1 = 0.0548 [8783 reflections with I > 2σ(I)] and GOF = 0.992 on F2 for 812 refined parameters, largest difference peak and hole 1.603 and –1.600 eÅ–3.
281 reflections were measured in the range 1.83 ≤ θ ≤ 27.52° (hkl range indices –30 ≤ h ≤ 30, –16 ≤ k ≤ 16, –27 ≤ l ≤ 27), 13984 independent reflections (Rint = 0.0539). The structure was refined to F2 to wR2 = 0.1615, R1 = 0.0548 [8783 reflections with I > 2σ(I)] and GOF = 0.992 on F2 for 812 refined parameters, largest difference peak and hole 1.603 and –1.600 eÅ–3.
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 11.4892(8), b = 13.6419(9), c = 23.7577(16) Å, α = 93.2420(10), β = 99.9830(10), γ = 97.9550(10)°, V = 3619.6(4) Å3, Z = 2, Dcalc = 1.785 Mg m–3, F(000) = 1952, µ = 0.944 mm–1, crystal dimensions 0.4 × 0.2 × 0.2 mm, T = 173(2) K. A total of 27891 reflections were measured in the range 1.51 ≤ θ ≤ 27.62° (hkl range indices –14 ≤ h ≤ 14, –17 ≤ k ≤ 14, –30 ≤ l ≤ 30), 15653 independent reflections (Rint = 0.0411). The structure was refined to F2 to wR2 = 0.0927, R1 = 0.0439 [9053 reflections with I > 2σ(I)] and GOF = 0.906 on F2 for 1111 refined parameters, largest difference peak and hole 1.902 and –1.011 eÅ–3.
, a = 11.4892(8), b = 13.6419(9), c = 23.7577(16) Å, α = 93.2420(10), β = 99.9830(10), γ = 97.9550(10)°, V = 3619.6(4) Å3, Z = 2, Dcalc = 1.785 Mg m–3, F(000) = 1952, µ = 0.944 mm–1, crystal dimensions 0.4 × 0.2 × 0.2 mm, T = 173(2) K. A total of 27891 reflections were measured in the range 1.51 ≤ θ ≤ 27.62° (hkl range indices –14 ≤ h ≤ 14, –17 ≤ k ≤ 14, –30 ≤ l ≤ 30), 15653 independent reflections (Rint = 0.0411). The structure was refined to F2 to wR2 = 0.0927, R1 = 0.0439 [9053 reflections with I > 2σ(I)] and GOF = 0.906 on F2 for 1111 refined parameters, largest difference peak and hole 1.902 and –1.011 eÅ–3.
4. Conclusions
The syntheses of two symmetric polydentate pyridyl-thiazolyl ligands, whose central unit comprises a pyridine N-oxide moiety, have been described. Reaction of either of these ligands with Cd(ClO4)2·6H2O results in the formation of a dinuclear double-stranded helicate complex. Comparison of these complexes with those formed from analogous and/or related non-functionalised ligands, demonstrates how N-oxide functionalisation of the central pyridine ring induces the ligand to partition into two discreet binding domains.Acknowledgements
This work was supported by both EPSRC and the University of Huddersfield.References
- (a) J.-M. Lehn, Supramolecular Chemistry, VCH, Weinheim, 1995 Search PubMed; (b) J. W. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley and Sons, Chichester, 2000 Search PubMed.
- M. J. Hannon and L. J. Childs, Supramol. Chem., 2004, 16, 7.
- (a) M. Albrecht, Chem. Rev., 2001, 101, 3547; (b) M. Albrecht, Chem. Soc. Rev., 1998, 27, 281 RSC.
- C. Piguet, G. Bernardinelli and G. Hopfgartner, Chem. Rev., 1997, 97, 2005 CrossRef CAS.
- E. C. Constable, Polynuclear Transition Metal Helicates, in Comprehensive Supramolecular Chemistry, ed. J.-P. Sauvage, Elsevier, Oxford, 1996, vol. 9, p. 213 Search PubMed.
- For recent examples of metallo-helicates, see: (a) S. P. Argent, H. Adams, L. P. Harding, T. Riis-Johannessen, J. C. Jeffery and M. D. Ward, New J. Chem., 2005, 29, 904 RSC; (b) Q. Z. Sun, Y. Bai, G. J. He, C. Y. Duan, Z. H. Lin and Q. J. Meng, Chem. Commun., 2006, 2777 RSC; (c) M. Cantuel, F. Gumy, J. C. G. Bunzli and C. Piguet, Dalton Trans., 2006, 2647 RSC; (d) M. Hutin, R. Frantz and J. R. Nitschke, Chem.–Eur. J., 2006, 12, 4077 CrossRef CAS.
- (a) C. R. Rice, S. Wörl, J. C. Jeffery, R. L. Paul and M. D. Ward, Chem. Commun., 2000, 1529 RSC; (b) C. R. Rice, S. Wörl, J. C. Jeffery, R. L. Paul and M. D. Ward, J. Chem. Soc., Dalton Trans., 2001, 550 RSC; (c) C. R. Rice, C. J. Baylies, J. C. Jeffery, R. L. Paul and M. D. Ward, Inorg. Chim. Acta, 2001, 324, 331 CrossRef CAS.
- A. J. Amoroso, M. W. Burrows, T. Gelbrich, R. Haigh and M. B. Hursthouse, J. Chem. Soc., Dalton Trans., 2002, 2415 RSC.
- A. J. Amoroso, M. W. Burrows, A. A. Dickinson, C. Jones, D. J. Willock and W.-T. Wong, J. Chem. Soc., Dalton Trans., 2001, 255 Search PubMed.
- R. P. Thummel and Y. Jahng, J. Org. Chem., 1985, 50, 3635 CrossRef CAS.
- SAINT integration software, Siemens Analytical X-ray Instruments Inc., Madison, WI, 1994 Search PubMed.
- G. M. Sheldrick, SADABS, Program for absorption correction with the Siemens SMART system, Institute for Inorganic Chemistry, University of Göttingen, Germany, 1996 Search PubMed.
- SHELXTL program system version 5.1, Bruker Analytical X-ray Instruments Inc., Madison, WI, 1998 Search PubMed.
- Previous cadmium-containing helicates derived from pyridyl-thiazole ligands have pitch lengths ranging from 4.259–4.632 Å; C. R. Rice, C. J. Baylies, H. J. Clayton, J. C. Jeffery, R. L. Paul and M. D. Ward, Inorg. Chim. Acta, 2003, 351, 207 Search PubMed.
- C. R. Rice, C. J. Baylies, L. P. Harding, J. C. Jeffery, R. L. Paul and M. D. Ward, Polyhedron, 2003, 22, 755 CrossRef CAS.
- J. C. Jeffery, T. Riis-Johannessen, C. J. Anderson, C. J. Adams, A. Robinson, S. P. Argent, M. D. Ward and C. R. Rice, Inorg. Chem., 2007, 46, 2417 CrossRef CAS.
Footnotes |
| † Current address: Section de Chimie et Biochimie, Université de Genève, Sciences II, 30 quai Ernest-Ansermet, 1211 Genève 4, Switzerland. |
| ‡ CCDC 654463, [Cd2(L1)2]4+, and 654464, [Cd2(L2)2]4+. For crystallographic data in CIF or other electronic format, see DOI: 10.1039/b711039a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2008 |